VEGFR2 Blockade Improves Renal Damage in an Experimental Model of Type 2 Diabetic Nephropathy


 ,
,  and
and
Abstract
1. Introduction
2. Experimental Section
2.1. Design of the Experimental Model of Diabetic Nephropathy and Characterization
2.2. VEGFR2 Pharmacological Treatment
2.3. Mice Model of Gremlin-Induced Renal Damage
2.4. Protein Studies
2.5. Histological Analysis and Immunohistochemistry
2.6. Gene Expression Studies
2.7. RNA Sequencing Studies
3. Results
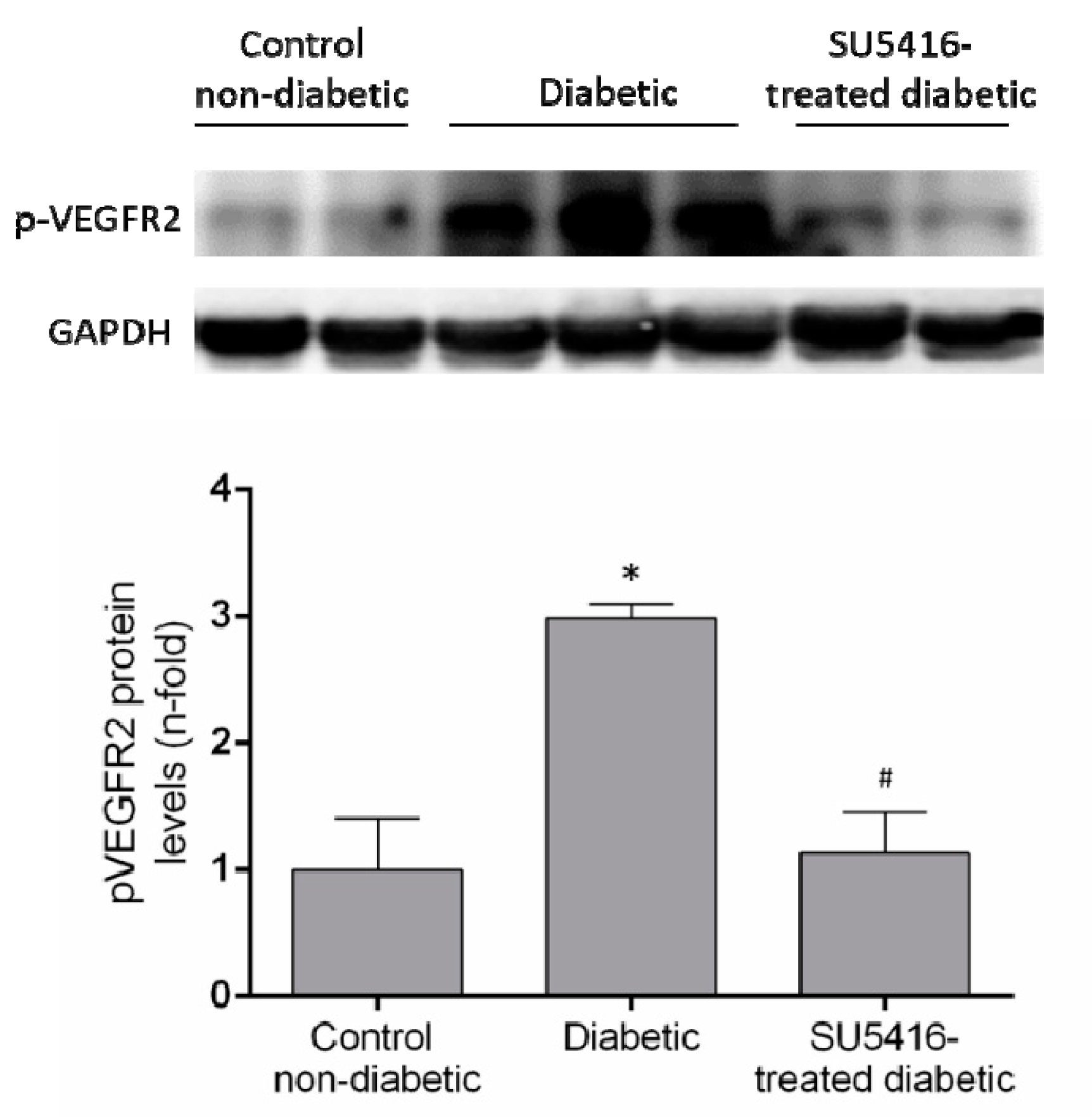
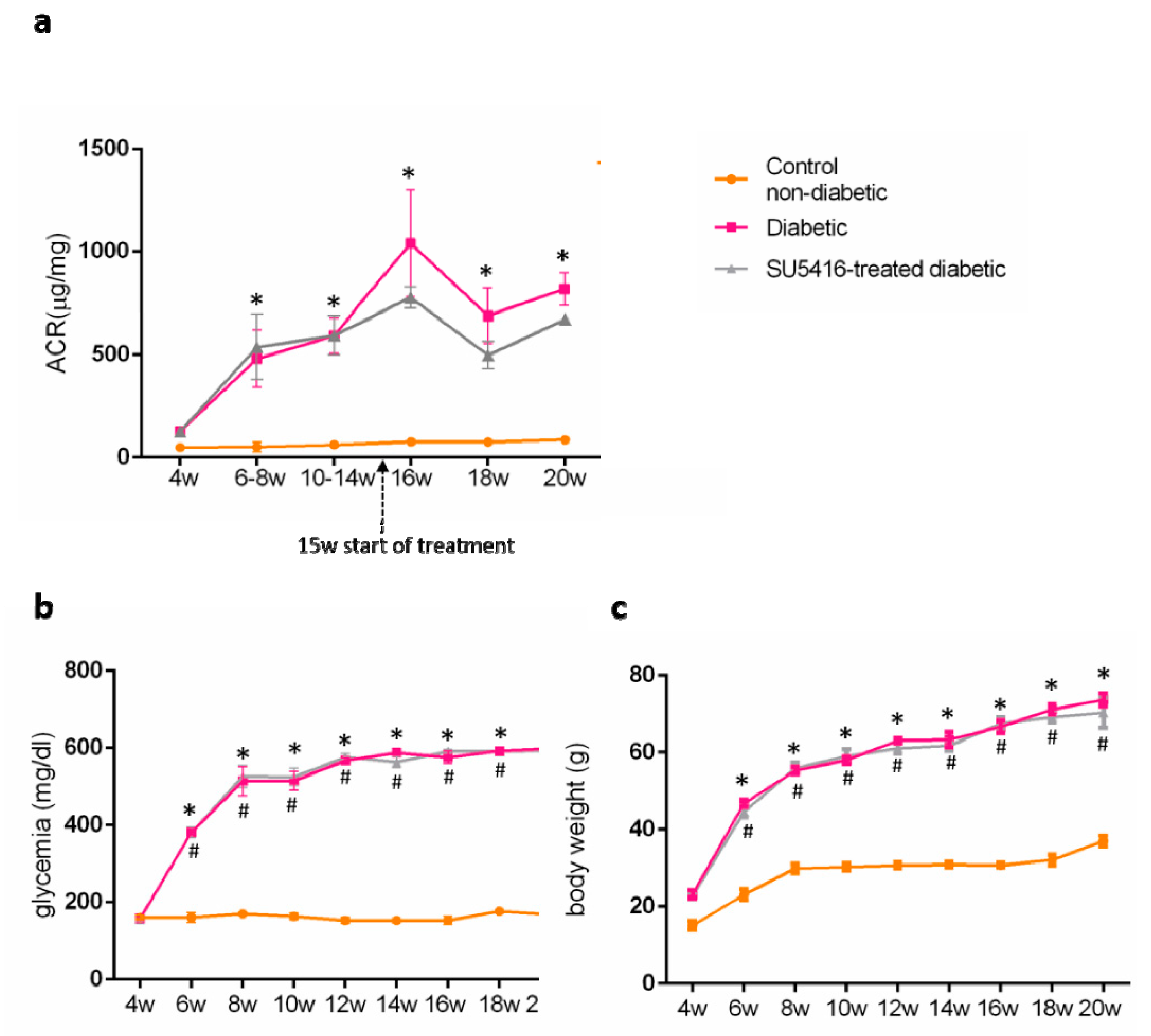
3.1. VEGFR2 Kinase Inhibition Improves Renal Function in Experimental Diabetic Nephropathy
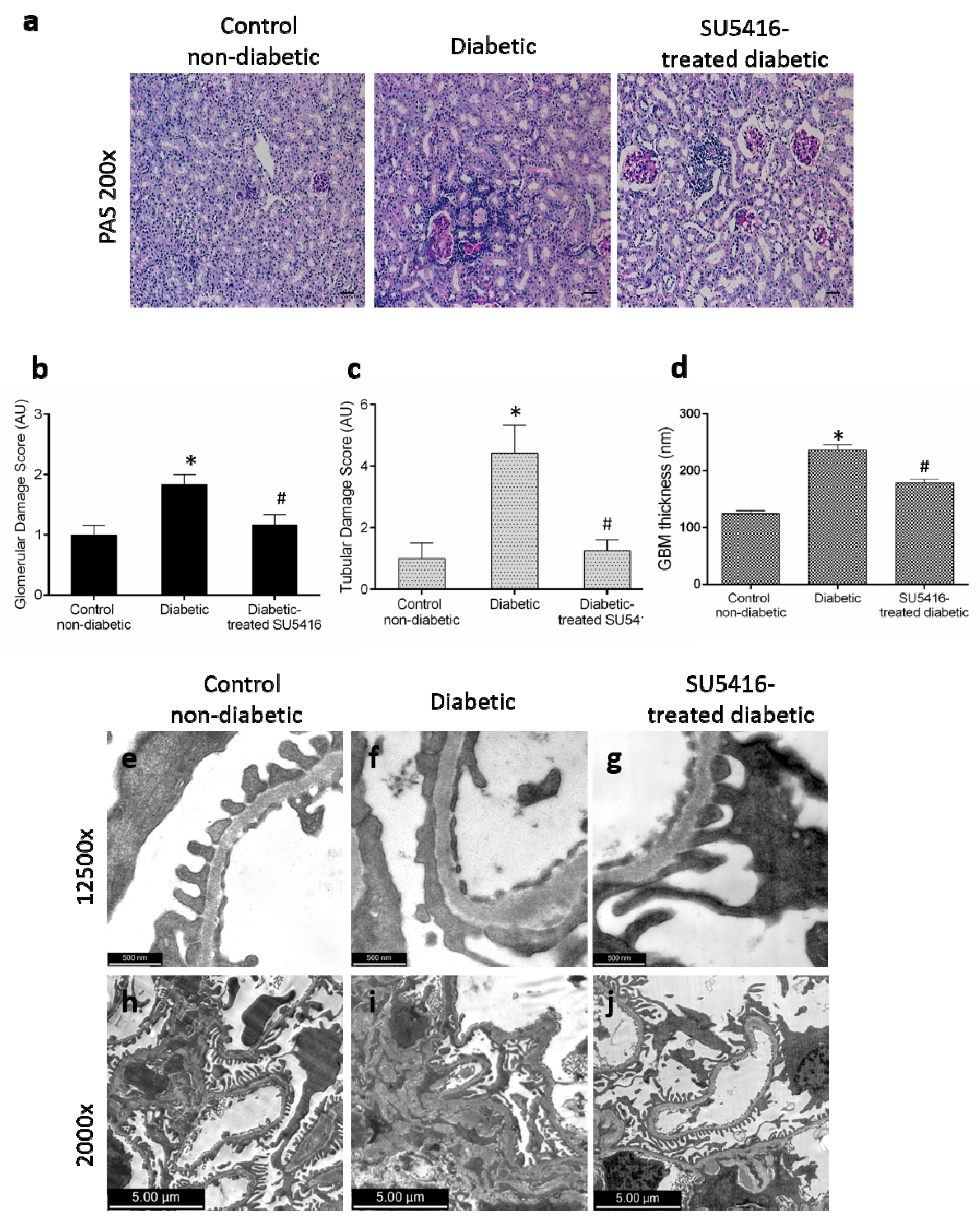
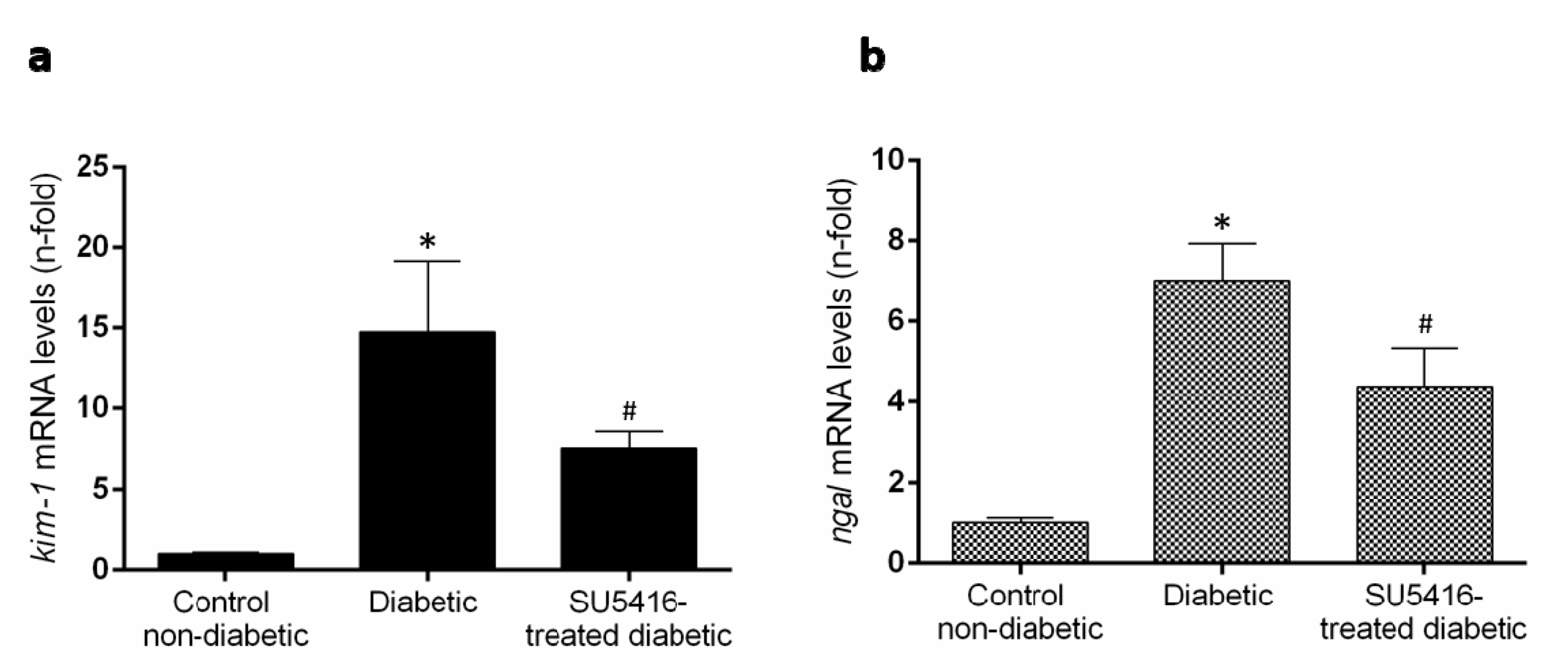
3.2. VEGFR2 Kinase Inhibition Ameliorates Renal Damage in Experimental Diabetic Nephropathy
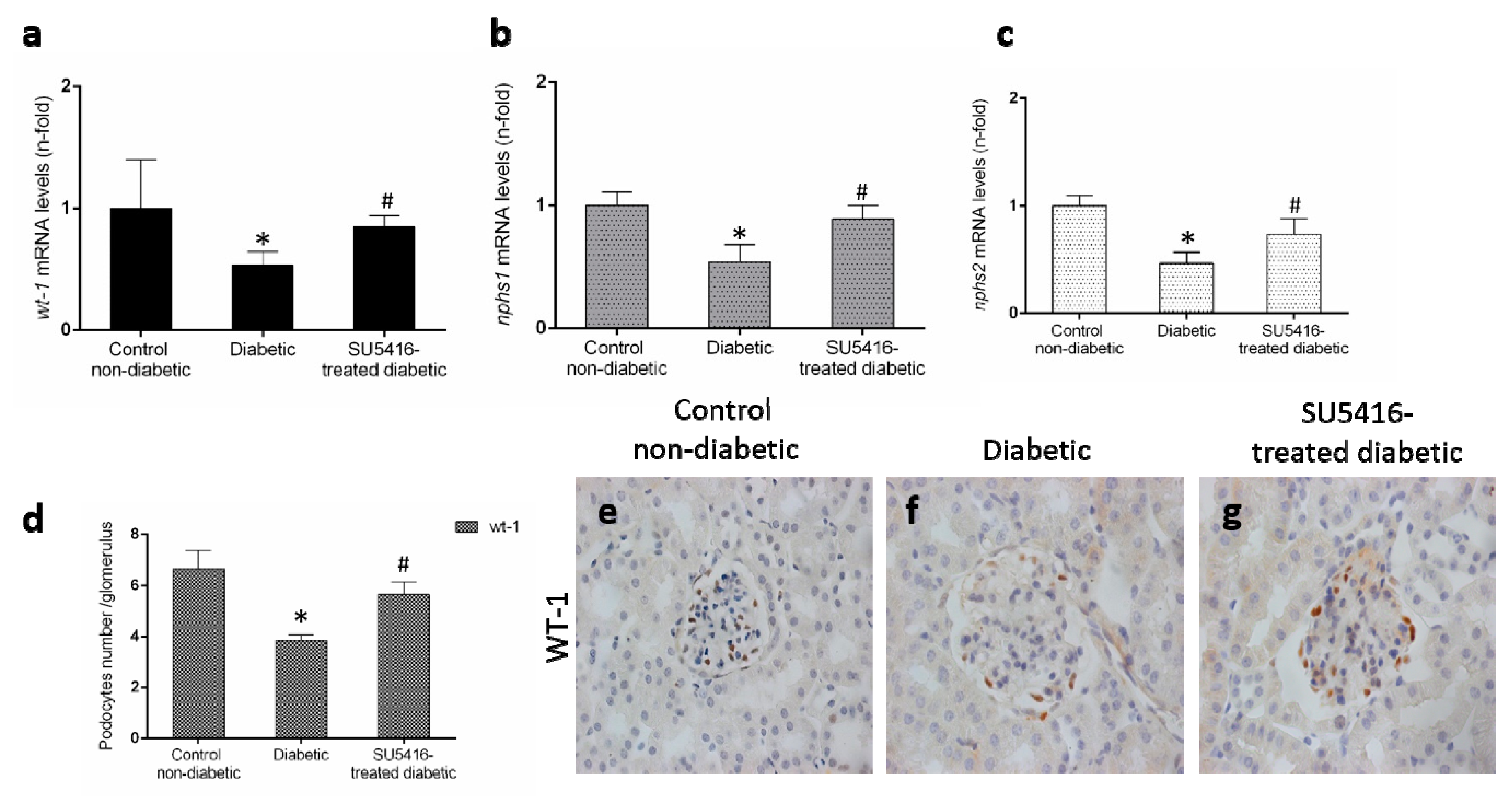
3.3. VEGFR2 Kinase Inhibition Diminishes Podocyte Damage in Experimental Diabetic Nephropathy
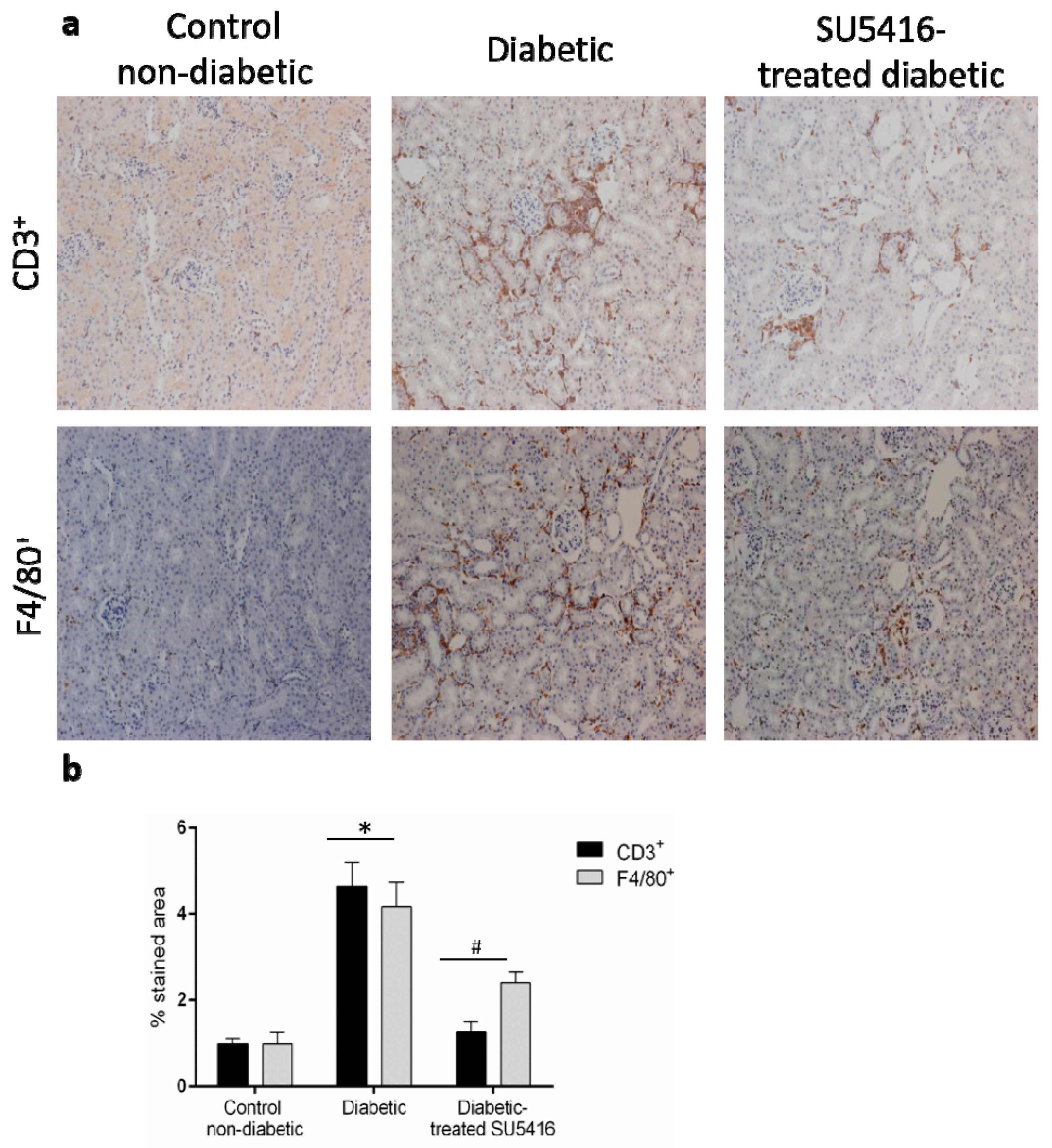
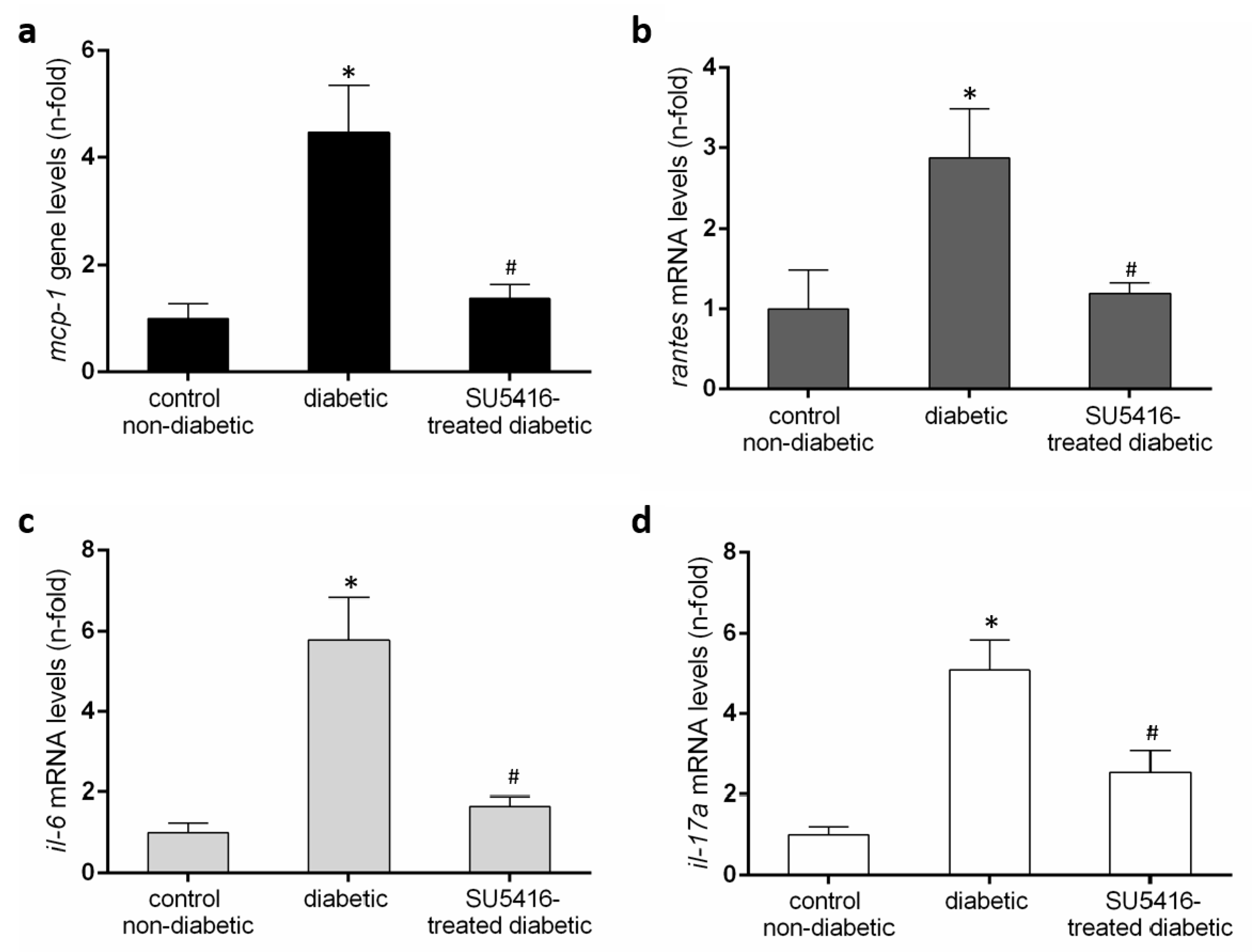
3.4. VEGFR2 Kinase Inhibition Decreases Renal Inflammation in Experimental Diabetic Nephropathy
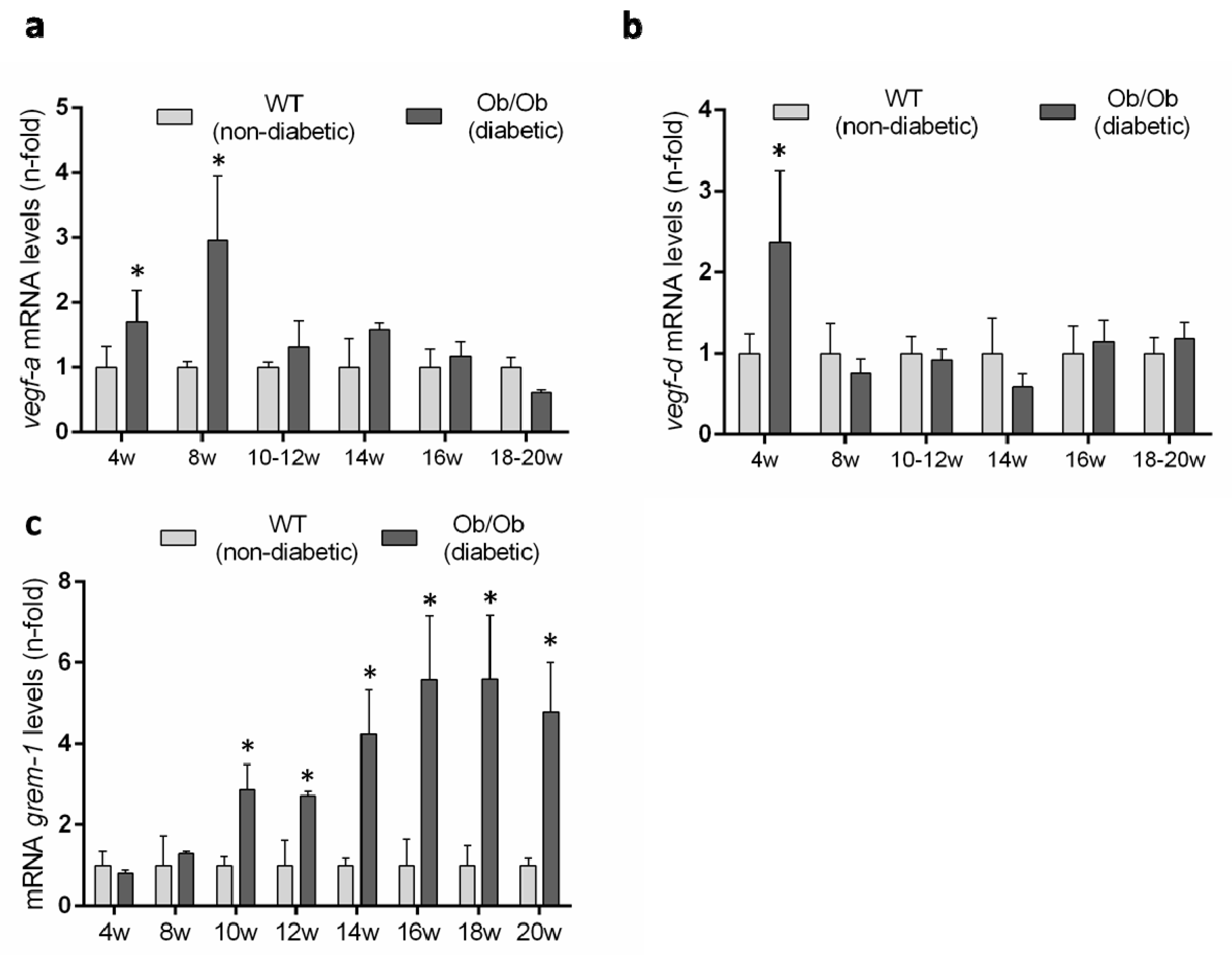
3.5. Evaluation of the Renal VEGFR2 Ligands Expression in Experimental Diabetic Nephropathy
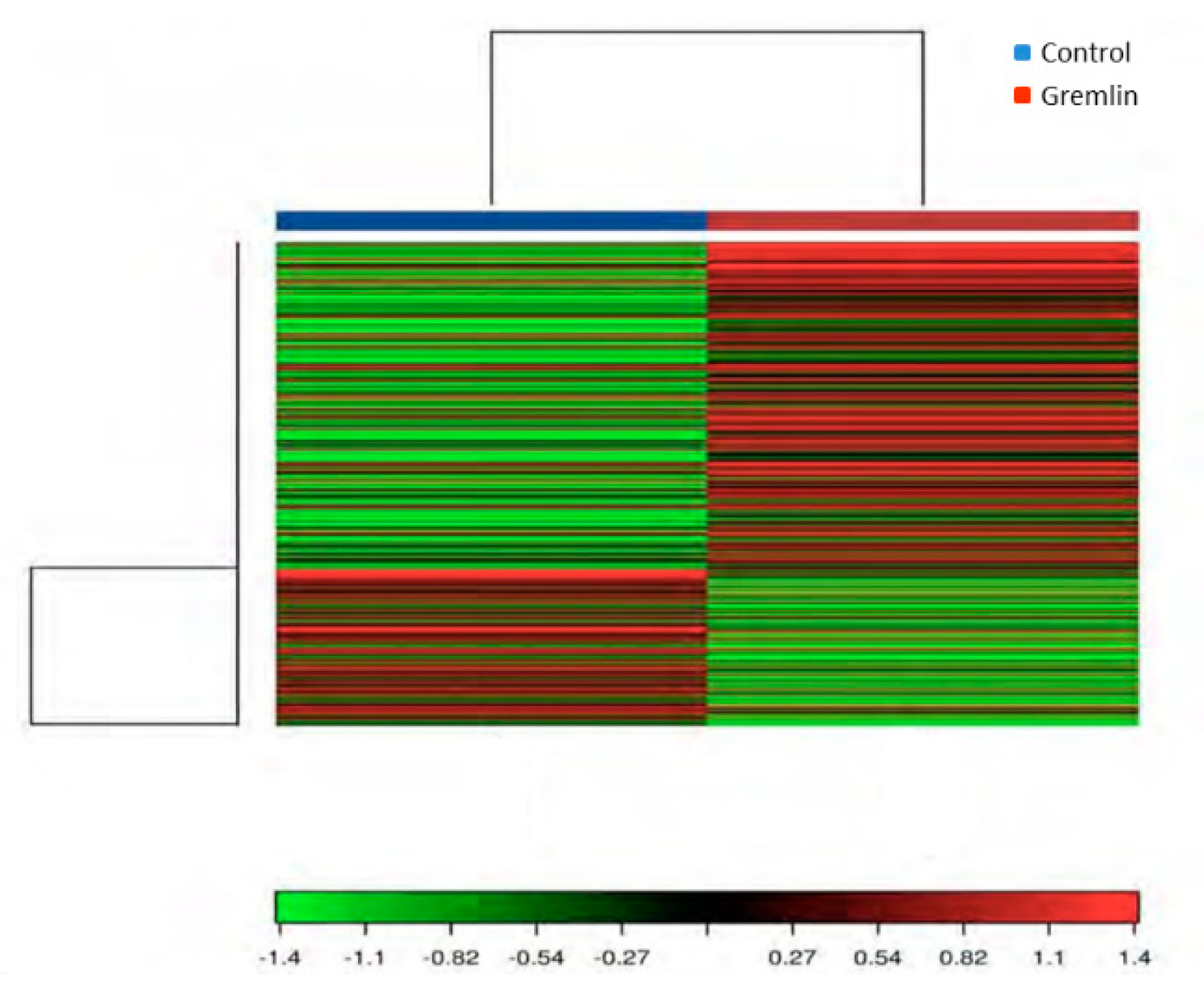
3.6. Gene Expression Levels Regulated by Gremlin in the Kidney, and Their Related Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analysis
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- National Kidney Foundation. KDOQI clinical practice guideline for diabetes and CKD: 2012 update notice section I: Use of the clinical practice guideline. Am. J. Kidney Dis. 2012, 60, 850–886. [Google Scholar] [CrossRef]
- Neumiller, J.J.; Alicic, R.Z.; Tuttle, K.R. Therapeutic Considerations for Antihyperglycemic Agents in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2017, 28, 2263–2274. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Gomez-Guerrero, C.; Mas, S.; Sanz, A.B.; Lorenzo, O.; Ruiz-Ortega, M.; Opazo, L.; Mezzano, S.; Egido, J. Targeting inflammation in diabetic nephropathy: A tale of hope. Expert Opin. Investig. Drugs 2018, 27, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Dutton, L.R.; O’Neill, C.L.; Medina, R.J.; Brazil, D.P. No evidence of Gremlin1-mediated activation of VEGFR2 signaling in endothelial cells. J. Biol. Chem. 2019, 294, 18041–18045. [Google Scholar] [CrossRef] [PubMed]
- Sison, K.; Eremina, V.; Baelde, H.; Min, W.; Hirashima, M.; Fantus, I.G.; Quaggin, S.E. Glomerular Structure and Function Require Paracrine, Not Autocrine, VEGF–VEGFR-2 Signaling. J. Am. Soc. Nephrol. 2010, 21, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Estrada, C.C.; Maldonado, A.; Mallipattu, S.K. Therapeutic Inhibition of VEGF Signaling and Associated Nephrotoxicities. J. Am. Soc. Nephrol. 2019, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Lavoz, C.; Alique, M.; Rodrigues-Diez, R.; Pato, J.; Keri, G.; Mezzano, S.; Egido, J.; Ruiz-Ortega, M. Gremlin regulates renal inflammation via the vascular endothelial growth factor receptor 2 pathway. J. Pathol. 2015, 236, 407–420. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Vascular endothelial growth factor (VEGF) and VEGF receptor inhibitors in the treatment of renal cell carcinomas. Pharmacol. Res. 2017, 120, 116–132. [Google Scholar]
- Logue, O.C.; McGowan, J.W.D.; George, E.M.; Bidwell, G.L. Therapeutic angiogenesis by vascular endothelial growth factor supplementation for treatment of renal disease. Curr. Opin. Nephrol. Hypertens. 2016, 25, 404–409. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. Pathophysiological consequences of VEGF-induced vascular permeability. Nature 2005, 437, 497–504. [Google Scholar] [CrossRef]
- Hale, L.J.; Hurcombe, J.; Lay, A.; Santamaría, B.; Valverde, A.M.; Saleem, M.A.; Mathieson, P.W.; Welsh, G.I.; Coward, R.J. Insulin directly stimulates VEGF-A production in the glomerular podocyte. Am. J. Physiol.-Ren. Physiol. 2013, 305, F182–F188. [Google Scholar] [CrossRef] [PubMed]
- Coward, R.J.M.; Welsh, G.I.; Koziell, A.; Hussain, S.; Lennon, R.; Ni, L.; Tavare, J.M.; Mathieson, P.W.; Saleem, M.A. Nephrin Is Critical for the Action of Insulin on Human Glomerular Podocytes. Diabetes 2007, 56, 1127–1135. [Google Scholar] [CrossRef]
- Salvatore, S.P.; Reddi, A.S.; Chandran, C.B.; Chevalier, J.M.; Okechukwu, C.N.; Seshan, S. V Collapsing glomerulopathy superimposed on diabetic nephropathy: Insights into etiology of an under-recognized, severe pattern of glomerular injury. Nephrol. Dial. Transplant. 2014, 29, 392–399. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mitola, S.; Ravelli, C.; Moroni, E.; Salvi, V.; Leali, D.; Ballmer-Hofer, K.; Zammataro, L.; Presta, M. Gremlin is a novel agonist of the major proangiogenic receptor VEGFR2. Blood 2010, 116, 3677–3680. [Google Scholar] [CrossRef] [PubMed]
- Mezzano, S.; Droguett, A.; Lavoz, C.; Krall, P.; Egido, J.; Ruiz-Ortega, M. Gremlin and renal diseases: Ready to jump the fence to clinical utility? Nephrol. Dial. Transplant. 2018, 33, 735–741. [Google Scholar] [CrossRef]
- McMahon, R.; Murphy, M.; Clarkson, M.; Taal, M.; Mackenzie, H.S.; Godson, C.; Martin, F.; Brady, H.R. IHG-2, a Mesangial Cell Gene Induced by High Glucose, Is Human gremlin. J. Biol. Chem. 2000, 275, 9901–9904. [Google Scholar] [CrossRef]
- Dolan, V.; Murphy, M.; Sadlier, D.; Lappin, D.; Doran, P.; Godson, C.; Martin, F.; O’Meara, Y.; Schmid, H.; Henger, A.; et al. Expression of gremlin, a bone morphogenetic protein antagonist, in human diabetic nephropathy. Am. J. Kidney Dis. 2005, 45, 1034–1039. [Google Scholar] [CrossRef]
- Afkarian, M.; Hirsch, I.B.; Tuttle, K.R.; Greenbaum, C.; Himmelfarb, J.; De Boer, I.H. Urinary excretion of RAS, BMP, and WNT pathway components in diabetic kidney disease. Physiol. Rep. 2014, 2, e12010. [Google Scholar] [CrossRef]
- Roxburgh, S.A.; Kattla, J.J.; Curran, S.P.; O’Meara, Y.M.; Pollock, C.A.; Goldschmeding, R.; Godson, C.; Martin, F.; Brazil, D.P. Allelic depletion of grem1 attenuates diabetic kidney disease. Diabetes 2009, 58, 1641–1650. [Google Scholar] [CrossRef]
- Zhang, Q.; Shi, Y.; Wada, J.; Malakauskas, S.M.; Liu, M.; Ren, Y.; Du, C.; Duan, H.; Li, Y.; Li, Y.; et al. In Vivo Delivery of Gremlin siRNA Plasmid Reveals Therapeutic Potential against Diabetic Nephropathy by Recovering Bone Morphogenetic Protein-7. PLoS ONE 2010, 5, e11709. [Google Scholar] [CrossRef][Green Version]
- Marchant, V.; Droguett, A.; Valderrama, G.; Burgos, M.E.; Carpio, D.; Kerr, B.; Ruiz-Ortega, M.; Egido, J.; Mezzano, S. Tubular overexpression of Gremlin in transgenic mice aggravates renal damage in diabetic nephropathy. Am. J. Physiology. Ren. Physiol. 2015, 309, F559–F568. [Google Scholar] [CrossRef]
- Brosius, F.C.; Alpers, C.E. New targets for treatment of diabetic nephropathy: What we have learned from animal models. Curr. Opin. Nephrol. Hypertens. 2013, 22, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Betz, B.; Conway, B.R. An Update on the Use of Animal Models in Diabetic Nephropathy Research. Curr. Diabetes Rep. 2016, 16, 18. [Google Scholar] [CrossRef]
- Clee, S.M.; Nadler, S.T.; Attie, A.D. Genetic and Genomic Studies of the BTBR ob/ob Mouse Model of Type 2 Diabetes. Am. J. Ther. 2005, 12, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Hudkins, K.L.; Pichaiwong, W.; Wietecha, T.; Kowalewska, J.; Banas, M.C.; Spencer, M.W.; Mühlfeld, A.; Koelling, M.; Pippin, J.W.; Shankland, S.J.; et al. BTBR Ob/Ob Mutant Mice Model Progressive Diabetic Nephropathy. J. Am. Soc. Nephrol. 2010, 21, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Corna, D.; Camozzi, D.; Cattaneo, D.; Rottoli, D.; Batani, C.; Zanchi, C.; Abbate, M.; Remuzzi, G. How to fully protect the kidney in a severe model of progressive nephropathy: A multidrug approach. J. Am. Soc. Nephrol. 2002, 13, 2898–2908. [Google Scholar] [CrossRef] [PubMed]
- Lavoz, C.; Matus, Y.S.; Orejudo, M.; Carpio, J.D.; Droguett, A.; Egido, J.; Mezzano, S.; Ruiz-Ortega, M. Interleukin-17A blockade reduces albuminuria and kidney injury in an accelerated model of diabetic nephropathy. Kidney Int. 2019, 95, 1418–1432. [Google Scholar] [CrossRef]
- Rajkumar, A.P.; Qvist, P.; Lazarus, R.; Lescai, F.; Ju, J.; Nyegaard, M.; Mors, O.; Børglum, A.D.; Li, Q.; Christensen, J.H. Experimental validation of methods for differential gene expression analysis and sample pooling in RNA-seq. BMC Genom. 2015, 16, 548. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Majumder, S.; Advani, A. VEGF and the diabetic kidney: More than too much of a good thing. J. Diabetes Complicat. 2017, 31, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.J.; Hunsicker, L.G.; Bain, R.P.; Rohde, R.D. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N. Engl. J. Med. 1993, 329, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I.; et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Leehey, D.J.; Singh, A.K.; Alavi, N.; Singh, R. Role of angiotensin II in diabetic nephropathy. Kidney Int. Suppl. 2000, 77, S93–S98. [Google Scholar] [CrossRef]
- Fernández Fernández, B.; Elewa, U.; Sánchez-Niño, M.D.; Rojas-Rivera, J.E.; Martin-Cleary, C.; Egido, J.; Ortiz, A. 2012 update on diabetic kidney disease: The expanding spectrum, novel pathogenic insights and recent clinical trials. Minerva Med. 2012, 103, 219–234. [Google Scholar]
- Lin, J.S.; Susztak, K. Podocytes: The Weakest Link in Diabetic Kidney Disease? Curr. Diabetes Rep. 2016, 16, 45. [Google Scholar] [CrossRef]
- Zhu, J.; Sun, N.; Aoudjit, L.; Li, H.; Kawachi, H.; Lemay, S.; Takano, T. Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int. 2008, 73, 556–566. [Google Scholar] [CrossRef]
- Doublier, S.; Salvidio, G.; Lupia, E.; Ruotsalainen, V.; Verzola, D.; Deferrari, G.; Camussi, G. Nephrin expression is reduced in human diabetic nephropathy: Evidence for a distinct role for glycated albumin and angiotensin II. Diabetes 2003, 52, 1023–1030. [Google Scholar] [CrossRef]
- Veron, D.; Reidy, K.J.; Bertuccio, C.; Teichman, J.; Villegas, G.; Jimenez, J.; Shen, W.; Kopp, J.B.; Thomas, D.B.; Tufro, A. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int. 2010, 77, 989–999. [Google Scholar] [CrossRef]
- Wiggins, R.C. The spectrum of podocytopathies: A unifying view of glomerular diseases. Kidney Int. 2007, 71, 1205–1214. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Sanz, A.B.; Sanchez-Lopez, E.; Ruiz-Ortega, M.; Benito-Martin, A.; Saleem, M.A.; Mathieson, P.W.; Mezzano, S.; Egido, J.; Ortiz, A. HSP27/HSPB1 as an adaptive podocyte antiapoptotic protein activated by high glucose and angiotensin II. Lab. Investig. A J. Tech. Methods Pathol. 2012, 92, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Sivaskandarajah, G.A.; Jeansson, M.; Maezawa, Y.; Eremina, V.; Baelde, H.J.; Quaggin, S.E. Vegfa protects the glomerular microvasculature in diabetes. Diabetes 2012, 61, 2958–2966. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-B.; Zhu, H.; Song, W.; Su, J.-H. Gremlin Regulates Podocyte Apoptosis via Transforming Growth Factor-β (TGF-β) Pathway in Diabetic Nephropathy. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 183–189. [Google Scholar] [CrossRef]
- Li, G.; Li, Y.; Liu, S.; Shi, Y.; Chi, Y.; Liu, G.; Shan, T. Gremlin aggravates hyperglycemia-induced podocyte injury by a TGFβ/smad dependent signaling pathway. J. Cell. Biochem. 2013, 114, 2101–2113. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Huang, H.; Li, Y.; Liu, M.; Shi, Y.; Chi, Y.; Zhang, T. Gremlin induces cell proliferation and extra cellular matrix accumulation in mouse mesangial cells exposed to high glucose via the ERK1/2 pathway. BMC Nephrol. 2013, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V. Can we target tubular damage to prevent renal function decline in diabetes? Semin. Nephrol. 2012, 32, 452–462. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Díez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, in press. [Google Scholar] [CrossRef]
- Rodrigues-Diez, R.; Lavoz, C.; Carvajal, G.; Rayego-Mateos, S.; Rodrigues Diez, R.R.; Ortiz, A.; Egido, J.; Mezzano, S.; Ruiz-Ortega, M. Gremlin Is a Downstream Profibrotic Mediator of Transforming Growth Factor-Beta in Cultured Renal Cells. Nephron Exp. Nephrol. 2012, 122, 62–74. [Google Scholar] [CrossRef]
- Rodrigues-Diez, R.; Rodrigues-Diez, R.R.; Lavoz, C.; Carvajal, G.; Droguett, A.; Garcia-Redondo, A.B.; Rodriguez, I.; Ortiz, A.; Egido, J.; Mezzano, S.; et al. Gremlin activates the smad pathway linked to epithelial mesenchymal transdifferentiation in cultured tubular epithelial cells. BioMed Res. Int. 2014, 2014, 802841. [Google Scholar] [CrossRef]
- Lavoz, C.; Poveda, J.; Marquez-Exposito, L.; Rayego-Mateos, S.; Rodrigues-Diez, R.R.; Ortiz, A.; Egido, J.; Mezzano, S.; Ruiz-Ortega, M. Gremlin activates the Notch pathway linked to renal inflammation. Clin. Sci. (London, England: 1979) 2018, 132, 1097–1115. [Google Scholar] [CrossRef]
- Marquez-Exposito, L.; Lavoz, C.; Rodrigues-Diez, R.R.; Rayego-Mateos, S.; Orejudo, M.; Cantero-Navarro, E.; Ortiz, A.; Egido, J.; Selgas, R.; Mezzano, S.; et al. Gremlin Regulates Tubular Epithelial to Mesenchymal Transition via VEGFR2: Potential Role in Renal Fibrosis. Front. Pharmacol. 2018, 9, 1195. [Google Scholar] [CrossRef] [PubMed]
- Niranjan, T.; Murea, M.; Susztak, K. The pathogenic role of Notch activation in podocytes. Nephron. Exp. Nephrol. 2009, 111, e73–e79. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.W.; Roxburgh, S.A.; McGettigan, P.; Berthier, C.C.; Higgins, D.G.; Kretzler, M.; Cohen, C.D.; Mezzano, S.; Brazil, D.P.; Martin, F. Co-regulation of Gremlin and Notch signalling in diabetic nephropathy. Biochim. Biophys. Acta 2008, 1782, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Niranjan, T.; Bielesz, B.; Gruenwald, A.; Ponda, M.P.; Kopp, J.B.; Thomas, D.B.; Susztak, K. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat. Med. 2008, 14, 290–298. [Google Scholar] [CrossRef]
- Lin, C.-L.; Wang, F.-S.; Hsu, Y.-C.; Chen, C.-N.; Tseng, M.-J.; Saleem, M.A.; Chang, P.-J.; Wang, J.-Y. Modulation of notch-1 signaling alleviates vascular endothelial growth factor-mediated diabetic nephropathy. Diabetes 2010, 59, 1915–1925. [Google Scholar] [CrossRef]
- Droguett, A.; Valderrama, G.; Burgos, M.E.; Carpio, D.; Saka, C.; Egido, J.; Ruiz-Ortega, M.; Mezzano, S. Gremlin, A Potential Urinary Biomarker of Anca-Associated Crescentic Glomerulonephritis. Sci. Rep. 2019, 9, 6867. [Google Scholar] [CrossRef]
- Guan, Y.; Cheng, W.; Zou, C.; Wang, T.; Cao, Z. Gremlin1 promotes carcinogenesis of glioma in vitro. Clin. Exp. Pharmacol. Physiol. 2017, 44, 244–256. [Google Scholar] [CrossRef]
- McCormack, N.; Molloy, E.L.; O’Dea, S. Bone morphogenetic proteins enhance an epithelial-mesenchymal transition in normal airway epithelial cells during restitution of a disrupted epithelium. Respir. Res. 2013, 14, 36. [Google Scholar] [CrossRef]
- Lee, H.; O’Meara, S.J.; O’Brien, C.; Kane, R. The role of gremlin, a BMP antagonist, and epithelial-to-mesenchymal transition in proliferative vitreoretinopathy. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4291–4299. [Google Scholar] [CrossRef]
- Silvério de Barros, R.; Dias, G.S.; Paula do Rosario, A.; Paladino, F.V.; Lopes, G.H.; Campos, A.H. Gremlin-1 potentiates the dedifferentiation of VSMC in early stages of atherosclerosis. Differ. Res. Biol. Divers. 2019, 109, 28–33. [Google Scholar] [CrossRef]
- Yin, M.; Tissari, M.; Tamminen, J.; Ylivinkka, I.; Rönty, M.; von Nandelstadh, P.; Lehti, K.; Hyytiäinen, M.; Myllärniemi, M.; Koli, K. Gremlin-1 is a key regulator of the invasive cell phenotype in mesothelioma. Oncotarget 2017, 8, 98280–98297. [Google Scholar] [CrossRef] [PubMed]
- Staloch, D.; Gao, X.; Liu, K.; Xu, M.; Feng, X.; Aronson, J.F.; Falzon, M.; Greeley, G.H.; Rastellini, C.; Chao, C.; et al. Gremlin is a key pro-fibrogenic factor in chronic pancreatitis. J. Mol. Med. (Berlin Germany) 2015, 93, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Z.; Cheng, H.; Chen, J.; Qian, J. Gremlin promotes retinal pigmentation epithelial (RPE) cell proliferation, migration and VEGF production via activating VEGFR2-Akt-mTORC2 signaling. Oncotarget 2017, 8, 979–987. [Google Scholar] [CrossRef]
- Lien, E.C.; Lyssiotis, C.A.; Cantley, L.C. Metabolic Reprogramming by the PI3K-Akt-mTOR Pathway in Cancer. Recent Results Cancer Res. 2016, 207, 39–72. [Google Scholar]
- Marquard, F.E.; Jücker, M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem. Pharmacol. 2019, 172, 113729. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, F.; Ziaeemehr, A.; Shahidsales, S.; Gharib, M.; Khazaei, M.; Ferns, G.A.; Ryzhikov, M.; Avan, A.; Hassanian, S.M. Role of regulatory miRNAs of the PI3K/AKT/mTOR signaling in the pathogenesis of hepatocellular carcinoma. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Roncolato, F.; Lindemann, K.; Willson, M.L.; Martyn, J.; Mileshkin, L. PI3K/AKT/mTOR inhibitors for advanced or recurrent endometrial cancer. Cochrane Database Syst. Rev. 2019, 10, CD012160. [Google Scholar] [CrossRef] [PubMed]
- Szrejder, M.; Piwkowska, A. AMPK signalling: Implications for podocyte biology in diabetic nephropathy. Biol. Cell 2019, 111, 109–120. [Google Scholar] [CrossRef]
- Eisenreich, A.; Leppert, U. Update on the Protective Renal Effects of Metformin in Diabetic Nephropathy. Curr. Med. Chem. 2017, 24, 3397–3412. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| kim-1 | 5-′TGTCGAGTGGAGATTCCTGGATGGT-3′ | 5-′GGTCTTCCTGTAGCTGTGGGCC-3′ |
| ngal | 5′-GCCCTGAGTGTCATGTGTCT-3′ | 5′-GAACTGATCGCTCCGGAAGT-3´ |
| wt-1 | 5′-CAGCGAAAGTTTTCCCGGTC-3´ | 5′-TGTTGTGATGGCGGACCAAT-3′ |
| nphs1 | 5´-AGGGTCGGAGGATCGAA-3′ | 5′-GGGAAGCTGGGGACTGAAGT-3′ |
| nphs2 | 5′-CCAAAGTGCGGGTGATTGC-3′ | 5′-TGATGCTCCCTTGTGCTCTG-3′ |
| mcp-1 | 5′- AGCTCTCTCTTCCTCCACCA-3′ | 5′ -GGCGTTAACTGCATCTGGCT-3 |
| rantes | 5′-AGAGGACTCTGAGACAGCACA-3′ | 5′- CGAGCCATATGGTGAGGCAG-3′ |
| il-6 | 5′CCCCAATTTCCAATGCTCTCC-3´ | 5′-CGCACTAGGTTTGCCGAGTA-3′ |
| il-17a | 5′-TCTCCACCGCAATGAAGACC-3′ | 5′-GACCAGGATCTCTTGCTGGA-3′ |
| grem-1 | 5′-CCCGGGGAGGAGGTGCTGGAGT-3′ | 5′-CCGGATGTGCCTGGGGATGTAGAA-3′ |
| vegf-a | 5′-TATTCAGCGGACTCACCAGC-3′ | 5′-AACCAACCTCCTCAAACCGT-3′ |
| vegf-d | 5′-TGTGACGCAGTGAGGGAGT-3′ | 5′- AAGCAGTTGTTTCCAGGCAG-3′ |
| cyc | 5′-GGCAATGCTGGACCAAACACAA-3′ | 5′-GTAAAATGCCCGCAAGTCAAAAG-3′ |
| Gene | Control FPKM | Gremlin FPKM | Log2 Fold Change | q Value |
|---|---|---|---|---|
| Havcr1 | 1.83792 | 65.521 | 5.15581 | 0.0636633 |
| AC131780.3 | 1.40074 | 49.7095 | 5.14926 | 0.0636633 |
| Gm10800 | 10.8964 | 212.665 | 4.28666 | 0.0636633 |
| Serpine1 | 0.400392 | 5.56466 | 3.79681 | 0.0636633 |
| Lcn2 | 7.64414 | 98.5245 | 3.68806 | 0.0636633 |
| Aldh1a3 | 1.16126 | 12.0796 | 3.37881 | 0.0636633 |
| Serpina10 | 2.62027 | 22.3256 | 3.09091 | 0.0636633 |
| Col1a1 | 1.29665 | 10.9816 | 3.08223 | 0.0636633 |
| Ugt2b37 | 1.12231 | 7.54508 | 2.74907 | 0.0636633 |
| Col3a1 | 4.35433 | 27.7459 | 2.67175 | 0.0636633 |
| Mki67 | 0.606635 | 2.70504 | 2.15675 | 0.0636633 |
| C3 | 2.35343 | 10.4619 | 2.1523 | 0.0636633 |
| Cdk1 | 1.80221 | 7.87254 | 2.12706 | 0.0636633 |
| Fgg | 6.5632 | 27.6495 | 2.07478 | 0.0636633 |
| Igfbp2 | 3.86836 | 14.4286 | 1.89914 | 0.0636633 |
| Serpina1a | 2.53818 | 9.26732 | 1.86836 | 0.0636633 |
| Tnc | 0.777548 | 2.79985 | 1.84834 | 0.0636633 |
| Spp1 | 2049.39 | 5757.69 | 1.4903 | 0.0636633 |
| Vcam1 | 2.53223 | 7.00542 | 1.46806 | 0.0636633 |
| Mgp | 134.793 | 326.969 | 1.27841 | 0.0636633 |
| Cfi | 6.66434 | 16.1511 | 1.2771 | 0.0636633 |
| Lyz2 | 27.1578 | 60.4045 | 1.15329 | 0.0636633 |
| Fabp4 | 377.135 | 161.948 | −1.21955 | 0.0636633 |
| Car3 | 78.7394 | 11.605 | −2.76233 | 0.0636633 |
| Thrsp | 35.0519 | 3.21214 | −3.44789 | 0.0636633 |
| Retn | 22.4473 | 1.61617 | −3.79589 | 0.0636633 |
| Adipoq | 16.8479 | 0.961797 | −4.13069 | 0.0636633 |
| Cfd | 148.864 | 7.47942 | −4.31493 | 0.0636633 |
| Category | Term | Count | p-Value |
| GOTERM_BP_DIRECT | Response to ethanol | 5 | 1.60E−05 |
| GOTERM_BP_DIRECT | Response to mechanical stimulus | 4 | 7.70E−05 |
| GOTERM_BP_DIRECT | Wound healing | 4 | 2.50E−04 |
| GOTERM_BP_DIRECT | Cellular response to tumor necrosis factor | 4 | 4.00E−04 |
| GOTERM_BP_DIRECT | Aging | 4 | 1.50E−03 |
| GOTERM_BP_DIRECT | Regulation of triglyceride biosynthetic process | 2 | 4.00E−03 |
| GOTERM_BP_DIRECT | Response to nutrient | 3 | 4.20E−03 |
| GOTERM_BP_DIRECT | Cell-matrix adhesion | 3 | 4.80E−03 |
| GOTERM_BP_DIRECT | Blood coagulation | 3 | 5.50E−03 |
| GOTERM_BP_DIRECT | Osteoblast differentiation | 3 | 9.80E−03 |
| GOTERM_BP_DIRECT | Response to drug | 4 | 9.90E−03 |
| GOTERM_BP_DIRECT | Immune system process | 4 | 1.40E−02 |
| GOTERM_BP_DIRECT | Positive regulation of gene expression | 4 | 1.50E−02 |
| GOTERM_BP_DIRECT | Innate immune response | 4 | 1.50E−02 |
| GOTERM_BP_DIRECT | Negative regulation of smooth muscle cell migration | 2 | 2.10E−02 |
| GOTERM_BP_DIRECT | Protein heterotrimerization | 2 | 2.50E−02 |
| GOTERM_BP_DIRECT | Negative regulation of extrinsic apoptotic signaling pathway via death domain receptors | 2 | 3.30E−02 |
| GOTERM_BP_DIRECT | Positive regulation of interleukin-8 production | 2 | 3.30E−02 |
| GOTERM_BP_DIRECT | Negative regulation of endothelial cell apoptotic process | 2 | 3.70E−02 |
| GOTERM_BP_DIRECT | Brown fat cell differentiation | 2 | 4.40E−02 |
| Category | Term | Count | p-Value |
| GOTERM_CC_DIRECT | Extracellular space | 17 | 5.20E-13 |
| GOTERM_CC_DIRECT | Extracellular region | 16 | 1.00E-10 |
| GOTERM_CC_DIRECT | Extracellular exosome | 17 | 3.30E−09 |
| GOTERM_CC_DIRECT | Extracellular matrix | 5 | 4.10E−04 |
| GOTERM_CC_DIRECT | Collagen trimer | 3 | 4.60E−03 |
| GOTERM_CC_DIRECT | Endoplasmic reticulum | 6 | 2.00E−02 |
| GOTERM_CC_DIRECT | Cell surface | 4 | 4.00E−02 |
| Category | Term | Count | p-Value |
| GOTERM_MF_DIRECT | Extracellular matrix structural constituent | 3 | 1.40E−03 |
| GOTERM_MF_DIRECT | Protease binding | 3 | 1.10E−02 |
| GOTERM_MF_DIRECT | Serine-type endopeptidase inhibitor activity | 3 | 1.20E−02 |
| GOTERM_MF_DIRECT | Platelet-derived growth factor binding | 2 | 1.60E−02 |
| GOTERM_MF_DIRECT | Protein homodimerization activity | 5 | 2.20E−02 |
| GOTERM_MF_DIRECT | Endopeptidase inhibitor activity | 2 | 4.20E−02 |
| Category | Term | Count | p-Value |
| KEGG_PATHWAY | Complement and coagulation cascades | 5 | 1.50E−05 |
| KEGG_PATHWAY | ECM-receptor interaction | 4 | 7.30E−04 |
| KEGG_PATHWAY | Staphylococcus aureus infection | 3 | 4.70E−03 |
| KEGG_PATHWAY | Focal adhesion | 4 | 8.30E−03 |
| KEGG_PATHWAY | Platelet activation | 3 | 3.00E−02 |
| KEGG_PATHWAY | PI3K-Akt signaling pathway | 4 | 3.40E−02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lavoz, C.; Rodrigues-Diez, R.R.; Plaza, A.; Carpio, D.; Egido, J.; Ruiz-Ortega, M.; Mezzano, S. VEGFR2 Blockade Improves Renal Damage in an Experimental Model of Type 2 Diabetic Nephropathy. J. Clin. Med. 2020, 9, 302. https://doi.org/10.3390/jcm9020302
Lavoz C, Rodrigues-Diez RR, Plaza A, Carpio D, Egido J, Ruiz-Ortega M, Mezzano S. VEGFR2 Blockade Improves Renal Damage in an Experimental Model of Type 2 Diabetic Nephropathy. Journal of Clinical Medicine. 2020; 9(2):302. https://doi.org/10.3390/jcm9020302
Chicago/Turabian StyleLavoz, Carolina, Raul R. Rodrigues-Diez, Anita Plaza, Daniel Carpio, Jesús Egido, Marta Ruiz-Ortega, and Sergio Mezzano. 2020. "VEGFR2 Blockade Improves Renal Damage in an Experimental Model of Type 2 Diabetic Nephropathy" Journal of Clinical Medicine 9, no. 2: 302. https://doi.org/10.3390/jcm9020302
APA StyleLavoz, C., Rodrigues-Diez, R. R., Plaza, A., Carpio, D., Egido, J., Ruiz-Ortega, M., & Mezzano, S. (2020). VEGFR2 Blockade Improves Renal Damage in an Experimental Model of Type 2 Diabetic Nephropathy. Journal of Clinical Medicine, 9(2), 302. https://doi.org/10.3390/jcm9020302