Predictors of Mortality in Patients with Interstitial Lung Disease-Associated Pulmonary Hypertension


Abstract
1. Introduction
2. Methods
Statistical Analysis
3. Results
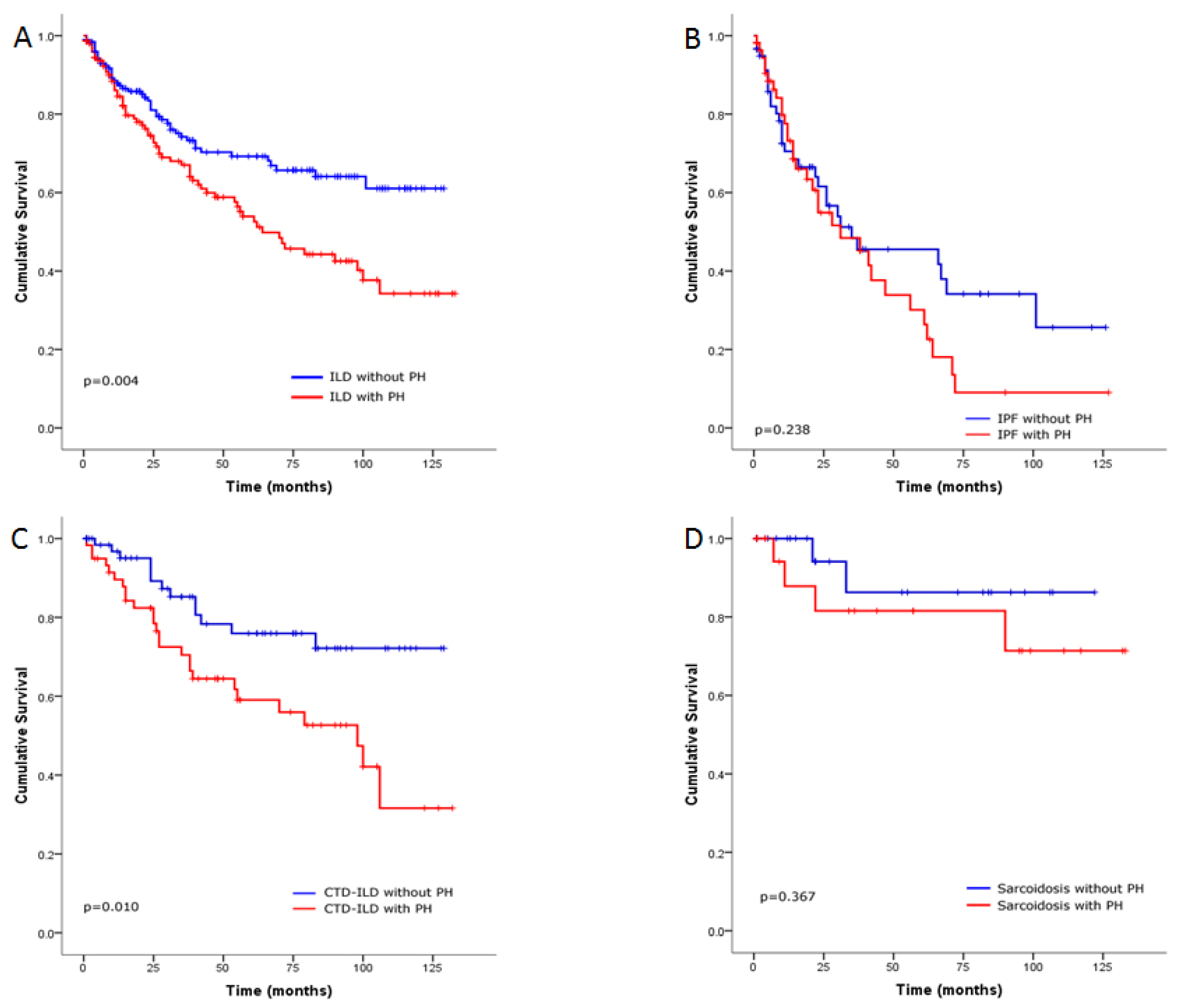
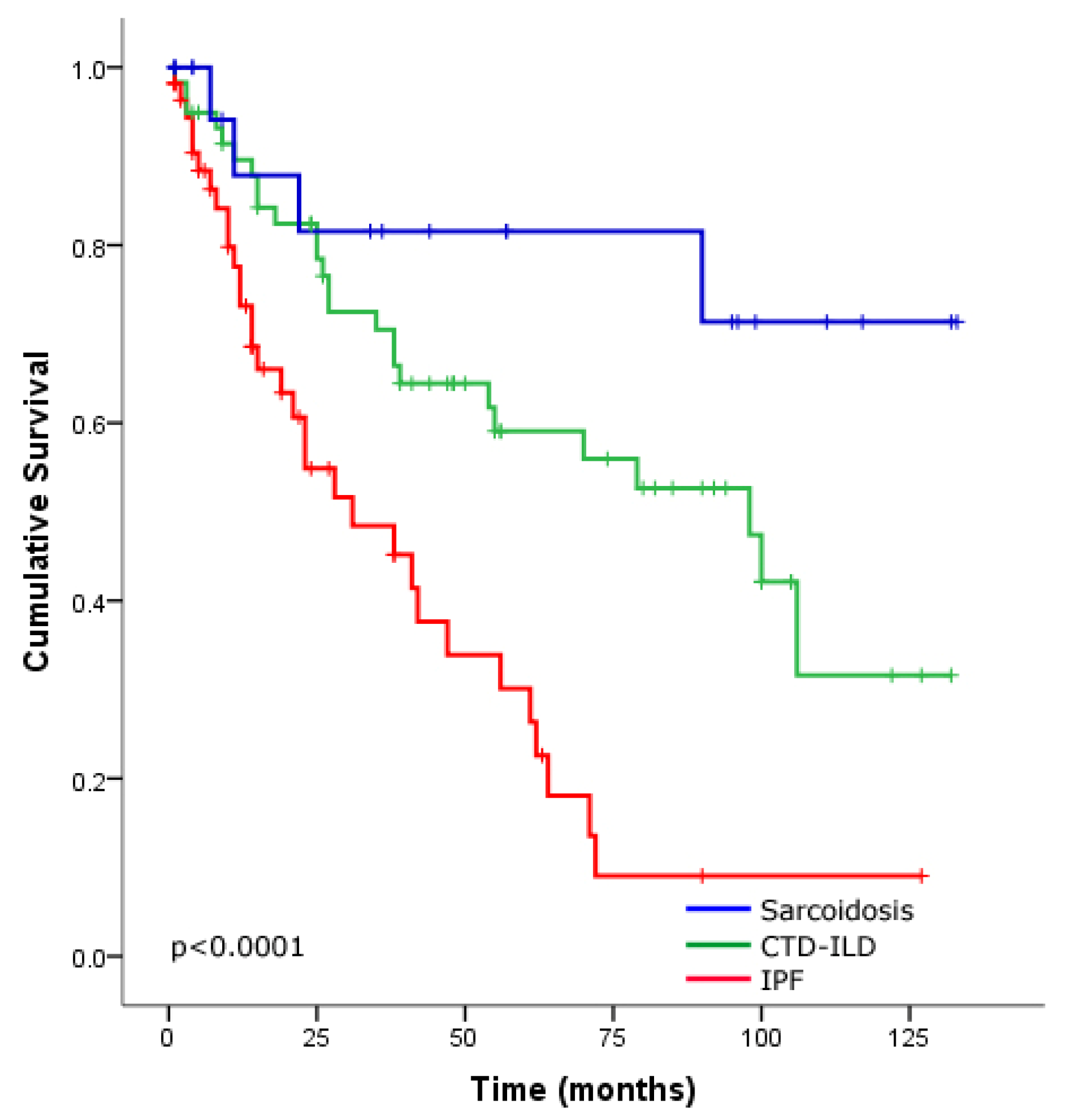
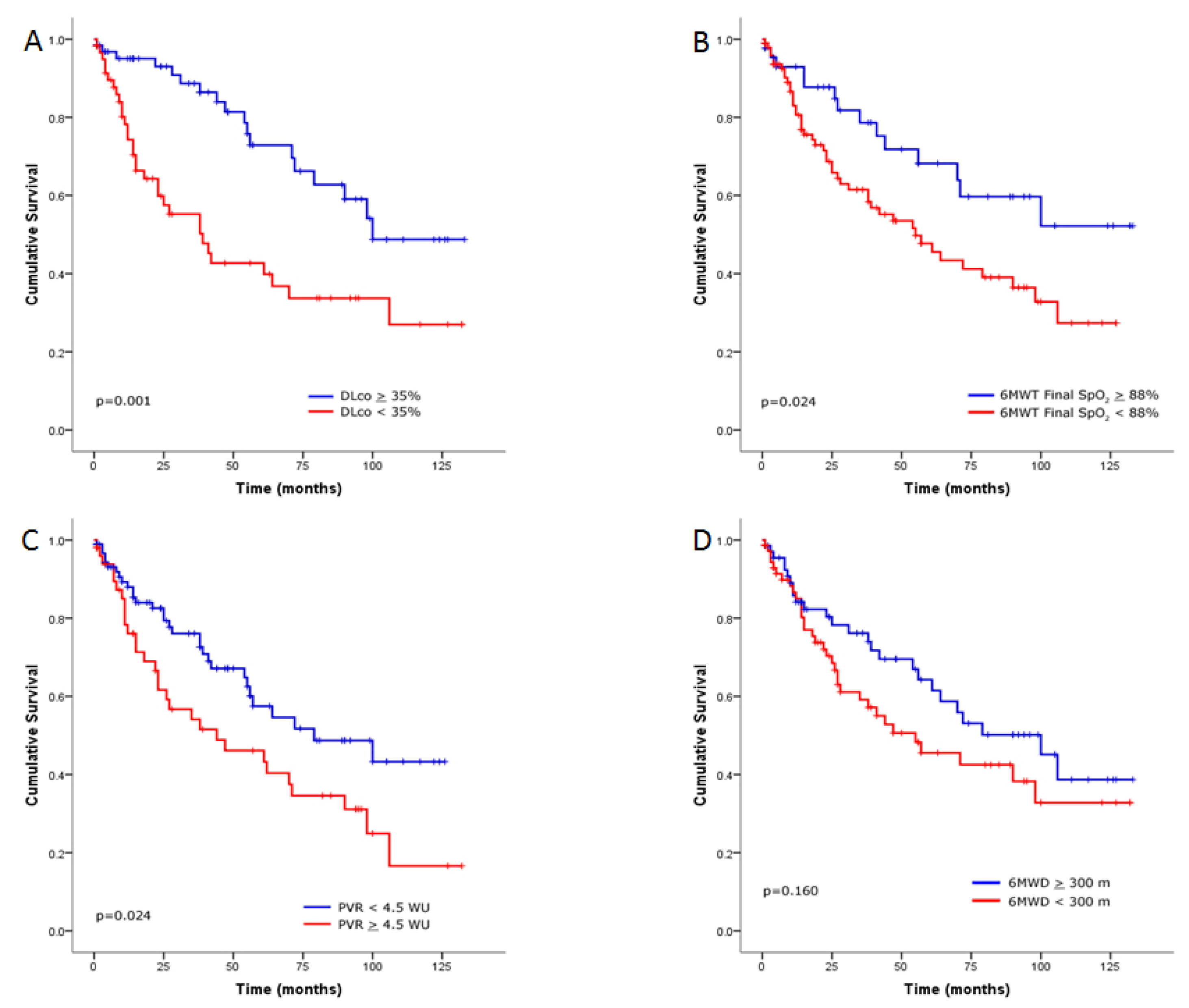
Survival Analysis of the ILD Cohort
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chebib, N.; Mornex, J.F.; Traclet, J.; Philit, F.; Khouatra, C.; Zeghmar, S.; Turquier, S.; Cottin, V. Pulmonary hypertension in chronic lung diseases: Comparison to other pulmonary hypertension groups. Pulm. Circ. 2018, 8, 2045894018775056. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Behr, J.; Held, M.; Grunig, E.; Vizza, C.D.; Vonk-Noordegraaf, A.; Lange, T.J.; Claussen, M.; Grohe, C.; Klose, H.; et al. Pulmonary Hypertension in Patients with Chronic Fibrosing Idiopathic Interstitial Pneumonias. PLoS ONE 2015, 10, e0141911. [Google Scholar] [CrossRef] [PubMed]
- Gall, H.; Felix, J.F.; Schneck, F.K.; Milger, K.; Sommer, N.; Voswinckel, R.; Franco, O.H.; Hofman, A.; Schermuly, R.T.; Weissmann, N.; et al. The Giessen Pulmonary Hypertension Registry: Survival in pulmonary hypertension subgroups. J. Heart Lung Transplant. 2017, 36, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Lettieri, C.J.; Nathan, S.D.; Barnett, S.D.; Ahmad, S.; Shorr, A.F. Prevalence and outcomes of pulmonary arterial ypertension in advanced idiopathic pulmonary fibrosis. Chest 2006, 129, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Shlobin, O.A.; Kouranos, V.; Barnett, S.D.; Alhamad, E.H.; Culver, D.A.; Barney, J.; Cordova, F.C.; Carmona, E.M.; Scholand, M.B.; Wijsenbeek, M.; et al. Physiological predictors of survival in patients with sarcoidosis-associated pulmonary hypertension: Results from an international registry. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef] [PubMed]
- King, C.S.; Shlobin, O.A. The Trouble with Group 3 Pulmonary Hypertension in Interstitial Lung Disease: Dilemmas in Diagnosis and the Conundrum of Treatment. Chest 2020. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Barbera, J.A.; Gaine, S.P.; Harari, S.; Martinez, F.J.; Olschewski, H.; Olsson, K.M.; Peacock, A.J.; Pepke-Zaba, J.; Provencher, S.; et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Macintyre, N.; Crapo, R.O.; Viegi, G.; Johnson, D.C.; van der Grinten, C.P.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Enright, P.; et al. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur. Respir. J. 2005, 26, 720–735. [Google Scholar] [CrossRef]
- Miller, M.R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Crapo, R.; Enright, P.; van der Grinten, C.P.; Gustafsson, P.; et al. Standardisation of spirometry. Eur. Respir. J. 2005, 26, 319–338. [Google Scholar] [CrossRef]
- Wanger, J.; Clausen, J.L.; Coates, A.; Pedersen, O.F.; Brusasco, V.; Burgos, F.; Casaburi, R.; Crapo, R.; Enright, P.; van der Grinten, C.P.; et al. Standardisation of the measurement of lung volumes. Eur. Respir. J. 2005, 26, 511–522. [Google Scholar] [CrossRef]
- ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: Guidelines for the six-minute walk test. Am. J Respir. Crit. Care Med. 2002, 166, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E.; Lynch, D.A., Jr.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef]
- Shiboski, S.C.; Shiboski, C.H.; Criswell, L.; Baer, A.; Challacombe, S.; Lanfranchi, H.; Schiodt, M.; Umehara, H.; Vivino, F.; Zhao, Y.; et al. American College of Rheumatology classification criteria for Sjogren’s syndrome: A data-driven, expert consensus approach in the Sjogren’s International Collaborative Clinical Alliance cohort. Arthritis Care Res. (Hoboken) 2012, 64, 475–487. [Google Scholar] [CrossRef]
- Shiboski, C.H.; Shiboski, S.C.; Seror, R.; Criswell, L.A.; Labetoulle, M.; Lietman, T.M.; Rasmussen, A.; Scofield, H.; Vitali, C.; Bowman, S.J.; et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjogren’s syndrome: A consensus and data-driven methodology involving three international patient cohorts. Ann. Rheum. Dis. 2017, 76, 9–16. [Google Scholar] [CrossRef]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O.; Birnbaum, N.S., 3rd; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef]
- Aringer, M.; Costenbader, K.; Daikh, D.; Brinks, R.; Mosca, M.; Ramsey-Goldman, R.; Smolen, J.S.; Wofsy, D.; Boumpas, D.T.; Kamen, D.L.; et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 1400–1412. [Google Scholar] [CrossRef]
- Van den Hoogen, F.; Khanna, D.; Fransen, J.; Johnson, S.R.; Baron, M.; Tyndall, A.; Matucci-Cerinic, M.; Naden, R.P.; Medsger, T.A., Jr.; Carreira, P.E.; et al. 2013 classification criteria for systemic sclerosis: An American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013, 65, 2737–2747. [Google Scholar] [CrossRef]
- Tani, C.; Carli, L.; Vagnani, S.; Talarico, R.; Baldini, C.; Mosca, M.; Bombardieri, S. The diagnosis and classification of mixed connective tissue disease. J. Autoimmun. 2014, 48–49, 46–49. [Google Scholar] [CrossRef]
- Doria, A.; Mosca, M.; Gambari, P.F.; Bombardieri, S. Defining unclassifiable connective tissue diseases: Incomplete, undifferentiated, or both? J. Rheumatol. 2005, 32, 213–215. [Google Scholar] [PubMed]
- Judson, M.A.; Costabel, U.; Drent, M.; Wells, A.; Maier, L.; Koth, L.; Shigemitsu, H.; Culver, D.A.; Gelfand, J.; Valeyre, D.; et al. The WASOG Sarcoidosis Organ Assessment Instrument: An update of a previous clinical tool. Sarcoidosis Vasc. Diffuse Lung Dis. 2014, 31, 19–27. [Google Scholar] [PubMed]
- Hamada, K.; Nagai, S.; Tanaka, S.; Handa, T.; Shigematsu, M.; Nagao, T.; Mishima, M.; Kitaichi, M.; Izumi, T. Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest 2007, 131, 650–656. [Google Scholar] [CrossRef]
- Kimura, M.; Taniguchi, H.; Kondoh, Y.; Kimura, T.; Kataoka, K.; Nishiyama, O.; Aso, H.; Sakamoto, K.; Hasegawa, Y. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration 2013, 85, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Amatto, V.C.; Behr, J.; Stowasser, S. Comorbidities in idiopathic pulmonary fibrosis patients: A systematic literature review. Eur. Respir. J. 2015, 46, 1113–1130. [Google Scholar] [CrossRef] [PubMed]
- Shorr, A.F.; Wainright, J.L.; Cors, C.S.; Lettieri, C.J.; Nathan, S.D. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur. Respir. J. 2007, 30, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Le Pavec, J.; Prevot, G.; Mal, H.; Humbert, M.; Simonneau, G.; Cordier, J.F. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur. Respir. J. 2010, 35, 105–111. [Google Scholar] [CrossRef]
- Hurdman, J.; Condliffe, R.; Elliot, C.A.; Davies, C.; Hill, C.; Wild, J.M.; Capener, D.; Sephton, P.; Hamilton, N.; Armstrong, I.J.; et al. ASPIRE registry: Assessing the Spectrum of Pulmonary hypertension Identified at a REferral centre. Eur. Respir. J. 2012, 39, 945–955. [Google Scholar] [CrossRef]
- Alhamad, E.H.; Cal, J.G. Predictors of mortality in interstitial lung disease patients without pulmonary hypertension. Ann. Thorac. Med. 2020, 15, 238–243. [Google Scholar] [CrossRef]
- D’Andrea, A.; Stanziola, A.; Di Palma, E.; Martino, M.; D’Alto, M.; Dellegrottaglie, S.; Cocchia, R.; Riegler, L.; Betancourt Cordido, M.V.; Lanza, M. Right ventricular structure and function in idiopathic pulmonary fibrosis with or without pulmonary hypertension. Echocardiography 2016, 33, 57–65. [Google Scholar] [CrossRef]
- Rivera-Lebron, B.N.; Forfia, P.R.; Kreider, M.; Lee, J.C.; Holmes, J.H.; Kawut, S.M. Echocardiographic and hemodynamic predictors of mortality in idiopathic pulmonary fibrosis. Chest 2013, 144, 564–570. [Google Scholar] [CrossRef]
- Weill, D.; Benden, C.; Corris, P.A.; Dark, J.H.; Davis, R.D.; Keshavjee, S.; Lederer, D.J.; Mulligan, M.J.; Patterson, G.A.; Singer, L.G.; et al. A consensus document for the selection of lung transplant candidates: 2014—An update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 2015, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.; Liu, J.; Parsons, L.; Hassoun, P.M.; McGoon, M.; Badesch, D.B.; Miller, D.P.; Nicolls, M.R.; Zamanian, R.T. Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL: Identifying systemic sclerosis as a unique phenotype. Chest 2010, 138, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.J.; Jiang, X.; Zhou, W.; Wang, Y.; Gao, L.; Li, G.T.; Hong, T.; Huo, Y.; Jing, Z.C.; Zhang, Z.L. Connective tissue disease-associated pulmonary arterial hypertension in Chinese patients. Eur. Respir. J. 2014, 44, 963–972. [Google Scholar] [CrossRef]
- Zhang, R.; Dai, L.Z.; Xie, W.P.; Yu, Z.X.; Wu, B.X.; Pan, L.; Yuan, P.; Jiang, X.; He, J.; Humbert, M.; et al. Survival of Chinese patients with pulmonary arterial hypertension in the modern treatment era. Chest 2011, 140, 301–309. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, Q.; Liu, Y.; Tian, Z.; Guo, X.; Wang, H.; Lai, J.; Huang, C.; Yang, X.; Li, M.; et al. Clinical characteristics and survival of pulmonary arterial hypertension associated with three major connective tissue diseases: A cohort study in China. Int. J. Cardiol. 2017, 236, 432–437. [Google Scholar] [CrossRef]
- Mathai, S.C.; Hummers, L.K.; Champion, H.C.; Wigley, F.M.; Zaiman, A.; Hassoun, P.M.; Girgis, R.E. Survival in pulmonary hypertension associated with the scleroderma spectrum of diseases: Impact of interstitial lung disease. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2009, 60, 569–577. [Google Scholar] [CrossRef]
- Condliffe, R.; Kiely, D.G.; Peacock, A.J.; Corris, P.A.; Gibbs, J.S.R.; Vrapi, F.; Das, C.; Elliot, C.A.; Johnson, M.; DeSoyza, J. Connective tissue disease–associated pulmonary arterial hypertension in the modern treatment era. Am. J. Respir. Crit. Care Med. 2009, 179, 151–157. [Google Scholar] [CrossRef]
- Launay, D.; Hachulla, E.; Hatron, P.Y.; Jais, X.; Simonneau, G.; Humbert, M. Pulmonary arterial hypertension: A rare complication of primary Sjogren syndrome: Report of 9 new cases and review of the literature. Medicine 2007, 86, 299–315. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, X.; Tian, Z.; Qian, J.; Wang, Q.; Zhao, J.; Huang, C.; Liu, Y.; Guo, X.; Wang, H.; et al. The prognosis of pulmonary arterial hypertension associated with primary Sjogren’s syndrome: A cohort study. Lupus 2018, 27, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Mosca, M.; Tani, C.; Talarico, R.; Bombardieri, S. Undifferentiated connective tissue diseases (UCTD): Simplified systemic autoimmune diseases. Autoimmun. Rev. 2011, 10, 256–258. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Antoniou, K.M.; Brown, K.K.; Cadranel, J.; Corte, T.J.; du Bois, R.M.; Lee, J.S.; Leslie, K.O.; Lynch, D.A.; Matteson, E.L.; et al. An official European Respiratory Society/American Thoracic Society research statement: Interstitial pneumonia with autoimmune features. Eur. Respir. J. 2015, 46, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Boucly, A.; Cottin, V.; Nunes, H.; Jaïs, X.; Tazi, A.; Prévôt, G.; Reynaud-Gaubert, M.; Dromer, C.; Viacroze, C.; Horeau-Langlard, D. Management and long-term outcomes of sarcoidosis-associated pulmonary hypertension. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef]
- Rose, L.; Prins, K.W.; Archer, S.L.; Pritzker, M.; Weir, E.K.; Misialek, J.R.; Thenappan, T. Survival in pulmonary hypertension due to chronic lung disease: Influence of low diffusion capacity of the lungs for carbon monoxide. J. Heart Lung Transplant. 2019, 38, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Brewis, M.J.; Church, A.C.; Johnson, M.K.; Peacock, A.J. Severe pulmonary hypertension in lung disease: Phenotypes and response to treatment. Eur. Respir. J. 2015, 46, 1378–1389. [Google Scholar] [CrossRef]
- Stadler, S.; Mergenthaler, N.; Lange, T.J. The prognostic value of DLCO and pulmonary blood flow in patients with pulmonary hypertension. Pulm. Circ. 2019, 9, 2045894019894531. [Google Scholar] [CrossRef]
- Lewis, R.A.; Thompson, A.R.; Billings, C.G.; Charalampopoulos, A.; Elliot, C.A.; Hamilton, N.; Hill, C.; Hurdman, J.; Rajaram, S.; Sabroe, I. Mild parenchymal lung disease and/or low diffusion capacity impacts survival and treatment response in patients diagnosed with idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Variable | Without PH | With PH | Severe PH | p-Value |
|---|---|---|---|---|
| (n = 188) | (n = 96) | (n = 56) | ||
| Age | 55.8 ± 15.6 | 60.2 ± 14.3 | 59.3 ± 14.3 | 0.042 |
| Male sex | 93 (49.5) | 36 (37.5) | 25 (44.6) | 0.158 |
| Ever smoker | 46 (24.4) | 17 (17.7) | 17 (30.3) | 0.187 |
| Follow-up duration, months | 42.7 ± 38.5 | 40.8 ± 37.8 | 41.7 ± 37.5 | 0.921 |
| BMI, kg/m2 | 28.9 ± 6.3 | 30.1 ± 6.6 | 30.4 ± 8.4 | 0.202 |
| Underlying disease | ||||
| IPF | 59 (31.3) | 34 (35.4) | 22 (39.2) | 0.508 |
| CTD-ILD | 70 (37.2) | 39 (40.6) | 20 (35.7) | 0.798 |
| Rheumatoid arthritis | 13 (6.9) | 4 (4.1) | 1 (1.7) | 0.272 |
| SLE | 5 (2.6) | 3 (3.1) | 1 (1.7) | 0.884 |
| Systemic sclerosis | 8 (4.2) | 9 (9.3) | 0 | 0.030 |
| Primary Sjogren’s syndrome | 10 (5.3) | 13 (13.5) | 7 (12.5) | 0.039 |
| Polymyositis | 3 (1.5) | 0 | 1 (1.7) | 0.448 |
| MCTD | 11 (5.8) | 2 (2.0) | 2 (3.5) | 0.324 |
| UCTD | 20 (10.6) | 8 (8.3) | 8 (14.2) | 0.516 |
| Sarcoidosis | 26 (13.8) | 11 (11.4) | 11 (19.6) | 0.371 |
| Chronic Hypersensitivity Pneumonitis | 14 (7.4) | 5 (5.2) | 1 (1.7) | 0.272 |
| Idiopathic NSIP | 4 (2.1) | 4 (4.1) | 2 (3.5) | 0.601 |
| Others | ||||
| Organizing Pneumonia | 4 (2.1) | 2 (2.0) | 0 | 0.547 |
| Unclassifiable fibrosis | 6 (3.1) | 0 | 0 | 0.085 |
| RBILD | 3 (1.5) | 1 (1.0) | 0 | 0.617 |
| DIP | 2 (1.0) | 0 | 0 | 0.443 |
| Variable | Without PH | With PH | Severe PH | p-Value |
|---|---|---|---|---|
| (n = 188) | (n = 96) | (n = 56) | ||
| Pulmonary function test | ||||
| FVC, % predicted | 62.3 ± 19.1 | 54.7 ± 18.1 ǂ | 52.9 ± 17.7 | <0.0001 |
| FEV1, % predicted | 67.9 ± 19.3 | 60.7 ± 19.1 ǂ | 59.3 ± 19.1 | 0.001 |
| FEV1/FVC, ratio | 88.0 ± 9.3 | 89.4 ± 8.2 ǂ | 88.9 ± 8.4 | 0.449 |
| DLCO, % predicted | 45.3 ± 20.7 γ | 37.9 ± 17.1 κ | 33.3 ± 19.4 λ | <0.0001 |
| Six-minute walk test | n = 184 | n = 91 | n = 54 | |
| Initial Borg score | 1.0 ± 1.4 | 0.9 ± 1.4 | 0.9 ± 0.9 | 0.984 |
| Final Borg score | 3.4 ± 2.3 | 3.9 ± 2.4 | 3.9 ± 2.2 | 0.153 |
| Initial SpO2, % | 95.7 ± 2.7 | 94.8 ± 2.7 | 94.2 ± 3.8 | 0.001 |
| Final SpO2, % | 86.7 ± 7.0 | 82.9 ± 7.6 | 82.3 ± 8.0 | <0.0001 |
| Distance, meters | 343.6 ± 113.6 | 307.8 ± 103.4 | 251.0 ± 122.0 | <0.0001 |
| Treatment | ||||
| PDE-5i | - | 39 (40.6) | 20 (35.7) | 0.549 |
| ERA | - | 3 (3.1) | 3 (5.3) | 0.670 |
| Prostanoids | - | 0 | 1 (1.7) | 0.368 |
| Combination therapy | ||||
| PDE-5i + ERA | - | 10 (10.4) | 7 (12.5) | 0.694 |
| PDE-5i + Prostanoids | - | 2 (2.0) | 1 (1.7) | 1.000 |
| PDE-5i + ERA + Prostanoids | - | 3 (3.1) | 2 (3.5) | 1.000 |
| Antifibrotic therapy | 31 (16.4) | 16 (16.6) | 8 (14.2) | 0.909 |
| Immuomodulator therapy | 44 (23.4) | 31 (32.2) | 18 (32.1) | 0.245 |
| Oxygen supplementation | 106 (56.3) | 75 (78.1) | 44 (78.5) | <0.0001 |
| Variable | Without PH | With PH | Severe PH | p-Value |
|---|---|---|---|---|
| (n = 188) | (n = 96) | (n = 56) | ||
| RAP, mmHg | 4.5 ± 3.0 | 5.7 ± 3.2 | 8.5 ± 4.2 | <0.0001 |
| sPAP, mmHg | 29.3 ± 6.0 | 41.1 ± 5.7 | 60.7 ± 14.9 | <0.0001 |
| dPAP, mmHg | 11.2 ± 3.9 | 17.2 ± 5.1 | 25.6 ± 6.6 | <0.0001 |
| mPAP, mmHg | 18.6 ± 3.5 | 27.0 ± 3.2 | 40.0 ± 7.5 | <0.0001 |
| PCWP, mmHg | 8.5 ± 3.5 | 9.9 ± 3.5 | 14.4 ± 7.7 | <0.0001 |
| PVR, Wood units | 2.1 ± 0.8 | 3.6 ± 1.2 | 6.0 ± 3.4 | <0.0001 |
| CO, L/min | 5.0 ± 1.3 | 4.8 ± 1.0 | 4.7 ± 1.8 | 0.193 |
| CI, L/min/m2 | 2.9 ± 0.6 | 2.8 ± 0.5 | 2.7 ± 0.9 | 0.260 |
| Variable | Univariate Predictors | Multivariable Predictors | ||
|---|---|---|---|---|
| HR (95% CI) | p-Value | HR (95% CI) | p-Value | |
| Age | 1.027 (1.008–1.046) | 0.006 | ||
| Male sex | 2.426 (1.465–4.016) | 0.001 | 1.931 (0.934–3.992) | 0.076 |
| Ever smoker | 2.235 (1.291–3.870) | 0.004 | ||
| BMI, kg/m2 | 0.956 (0.919–0.995) | 0.026 | ||
| IPF diagnosis | 3.292 (1.982–5.468) | <0.0001 | 2.579 (1.293–5.147) | 0.007 |
| CTD-ILD diagnosis | 0.751 (0.455–1.240) | 0.263 | ||
| Sarcoidosis diagnosis | 0.320 (0.116–0.885) | 0.028 | ||
| FVC, % predicted | 0.971 (0.955–0.988) | 0.001 | 0.972 (0.949–0.995) | 0.016 |
| DLCO, % predicted | 0.970 (0.954–0.986) | <0.0001 | 0.978 (0.961–0.995) | 0.013 |
| 6MWD < 300 m | 1.439 (0.862–2.403) | 0.164 | ||
| 6MWT final SpO2 < 88% | 1.969 (1.081–3.589) | 0.027 | ||
| mPAP, mmHg | 1.023 (0.993–1.054) | 0.134 | ||
| RAP, mmHg | 1.042 (0.977–1.112) | 0.210 | ||
| sPAP, mmHg | 1.017 (1.000–1.035) | 0.048 | 1.023 (1.002–1.045) | 0.034 |
| dPAP, mmHg | 1.031 (0.994–1.069) | 0.107 | ||
| PCWP, mmHg | 1.045 (1.002–1.091) | 0.042 | ||
| PVR, Wood units | 1.109 (1.021–1.206) | 0.014 | ||
| CI, L/min/m2 | 0.551 (0.373–0.815) | 0.003 | 0.639 (0.421–0.971) | 0.036 |
| Antifibrotic therapy | 1.764 (0.988–3.150) | 0.055 | ||
| Immunomodulator therapy | 0.574 (0.337–0.979) | 0.041 | ||
| PH-specific therapy | 0.473 (0.286–0.783) | 0.004 | ||
| Oxygen supplementation | 1.721 (0.898–3.299) | 0.102 | ||
| Variable | Univariate Predictors | Multivariable Predictors | ||
|---|---|---|---|---|
| HR (95% CI) | p-Value | HR (95% CI) | p-Value | |
| Age | 1.020 (0.995–1.046) | 0.110 | ||
| Male sex | 1.787 (0.876–3.646) | 0.110 | ||
| Ever smoker | 1.867 (0.885–3.943) | 0.101 | ||
| BMI, kg/m2 | 0.963 (0.915–1.013) | 0.140 | ||
| IPF diagnosis | 3.479 (1.659–7.294) | 0.001 | 2.544 (1.106–5.853) | 0.028 |
| CTD-ILD diagnosis | 0.689 (0.330–1.442) | 0.323 | ||
| Sarcoidosis diagnosis | 0.235 (0.055–0.993) | 0.049 | ||
| FVC, % predicted | 0.981 (0.958–1.004) | 0.099 | ||
| DLCO, % predicted | 0.949 (0.922–0.977) | <0.0001 | 0.945 (0.914–0.977) | 0.001 |
| 6MWD < 300 m | 1.612 (0.733–3.545) | 0.235 | ||
| 6MWT final SpO2 < 88% | 2.055 (0.867–4.871) | 0.102 | ||
| mPAP, mmHg | 1.006 (0.946–1.069) | 0.850 | ||
| RAP, mmHg | 1.045 (0.961–1.137) | 0.306 | ||
| sPAP, mmHg | 1.005 (0.976–1.034) | 0.757 | ||
| dPAP, mmHg | 1.015 (0.948–1.087) | 0.662 | ||
| PCWP, mmHg | 1.060 (1.009–1.114) | 0.020 | ||
| PVR, Wood units | 1.025 (0.913–1.151) | 0.670 | ||
| CI, L/min/m2 | 0.621 (0.401–0.963) | 0.033 | ||
| Antifibrotic therapy | 1.482 (0.606–3.620) | 0.388 | ||
| Immunomodulator therapy | 0.409 (0.182–0.918) | 0.030 | ||
| PH-specific therapy | 0.375 (0.180–0.784) | 0.009 | ||
| Oxygen supplementation | 1.673 (0.681–4.108) | 0.262 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhamad, E.H.; Cal, J.G.; Alrajhi, N.N.; Alharbi, W.M. Predictors of Mortality in Patients with Interstitial Lung Disease-Associated Pulmonary Hypertension. J. Clin. Med. 2020, 9, 3828. https://doi.org/10.3390/jcm9123828
Alhamad EH, Cal JG, Alrajhi NN, Alharbi WM. Predictors of Mortality in Patients with Interstitial Lung Disease-Associated Pulmonary Hypertension. Journal of Clinical Medicine. 2020; 9(12):3828. https://doi.org/10.3390/jcm9123828
Chicago/Turabian StyleAlhamad, Esam H., Joseph G. Cal, Nuha N. Alrajhi, and Waleed M. Alharbi. 2020. "Predictors of Mortality in Patients with Interstitial Lung Disease-Associated Pulmonary Hypertension" Journal of Clinical Medicine 9, no. 12: 3828. https://doi.org/10.3390/jcm9123828
APA StyleAlhamad, E. H., Cal, J. G., Alrajhi, N. N., & Alharbi, W. M. (2020). Predictors of Mortality in Patients with Interstitial Lung Disease-Associated Pulmonary Hypertension. Journal of Clinical Medicine, 9(12), 3828. https://doi.org/10.3390/jcm9123828