Hereditary Hemorrhagic Telangiectasia (HHT) and Survival: The Importance of Systematic Screening and Treatment in HHT Centers of Excellence

 ,
,
Abstract
1. Introduction
2. Materials and Methods
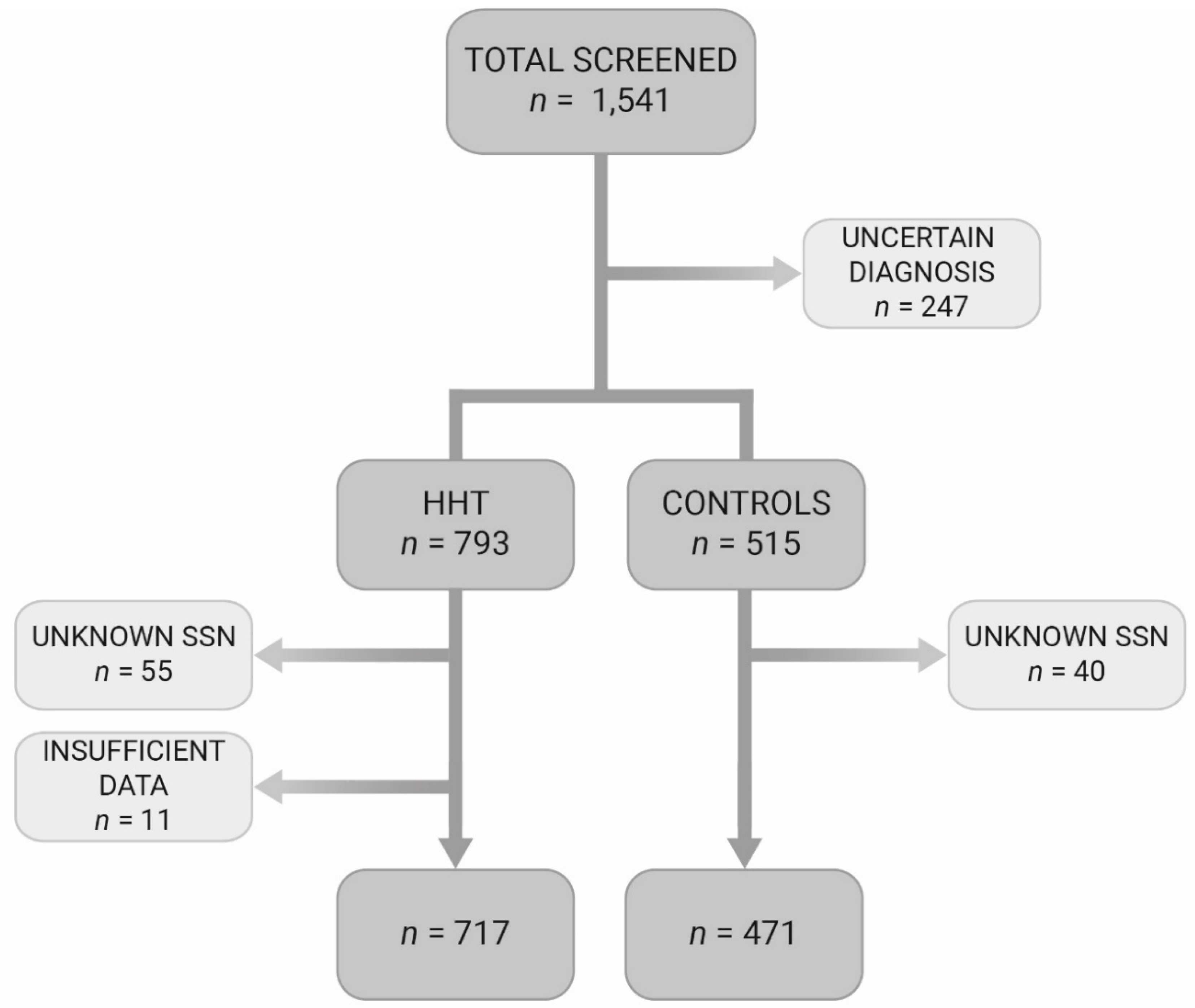
2.1. Study Design and Patient Selection
2.2. Patient Selection
2.3. Screening Protocol for HHT
2.4. Statistical Methods and Ethics
3. Results
3.1. Patient Selection and Baseline Characteristics
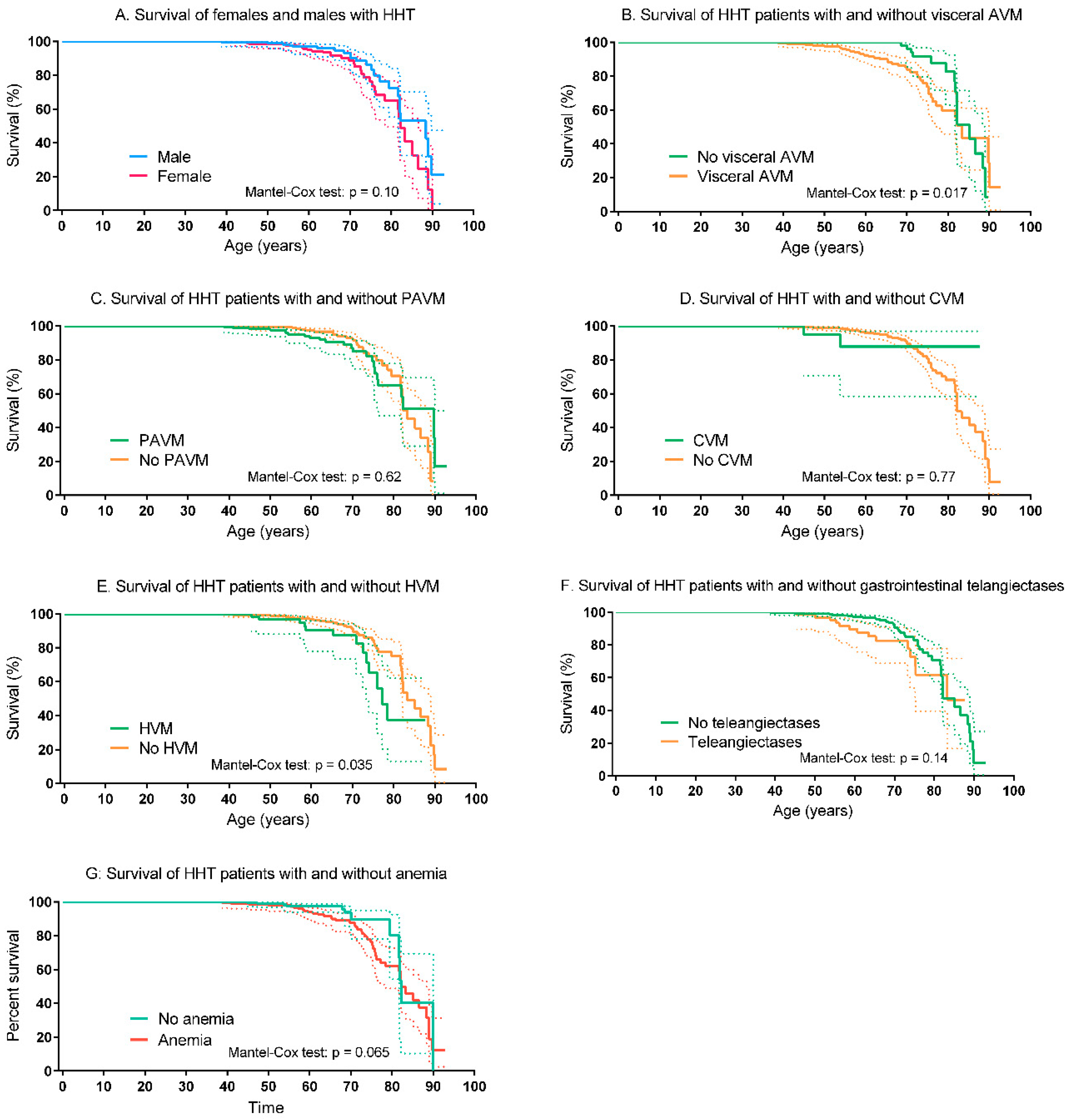
3.2. Visceral AVMs, Comorbidities and Disease Complications
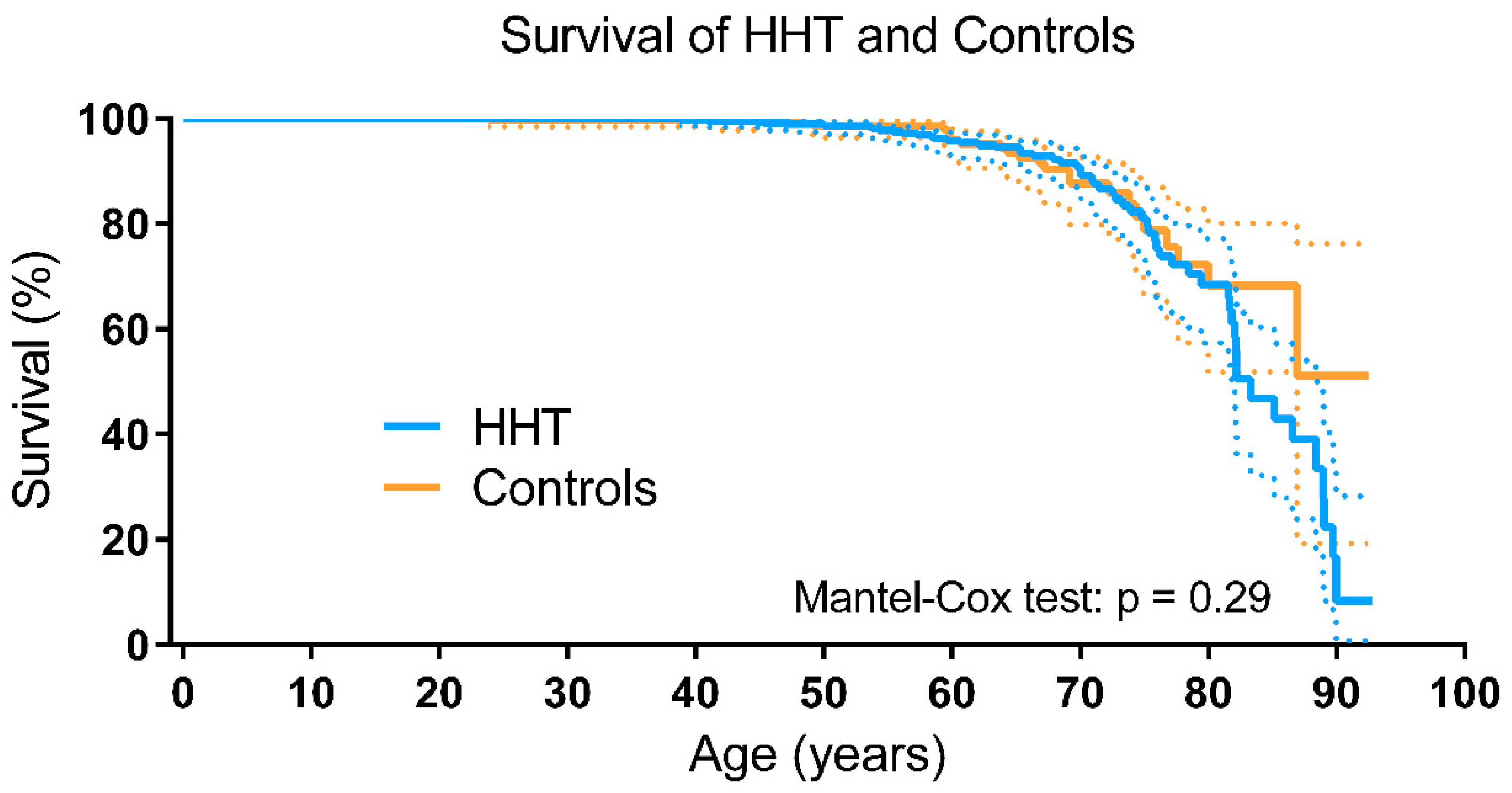
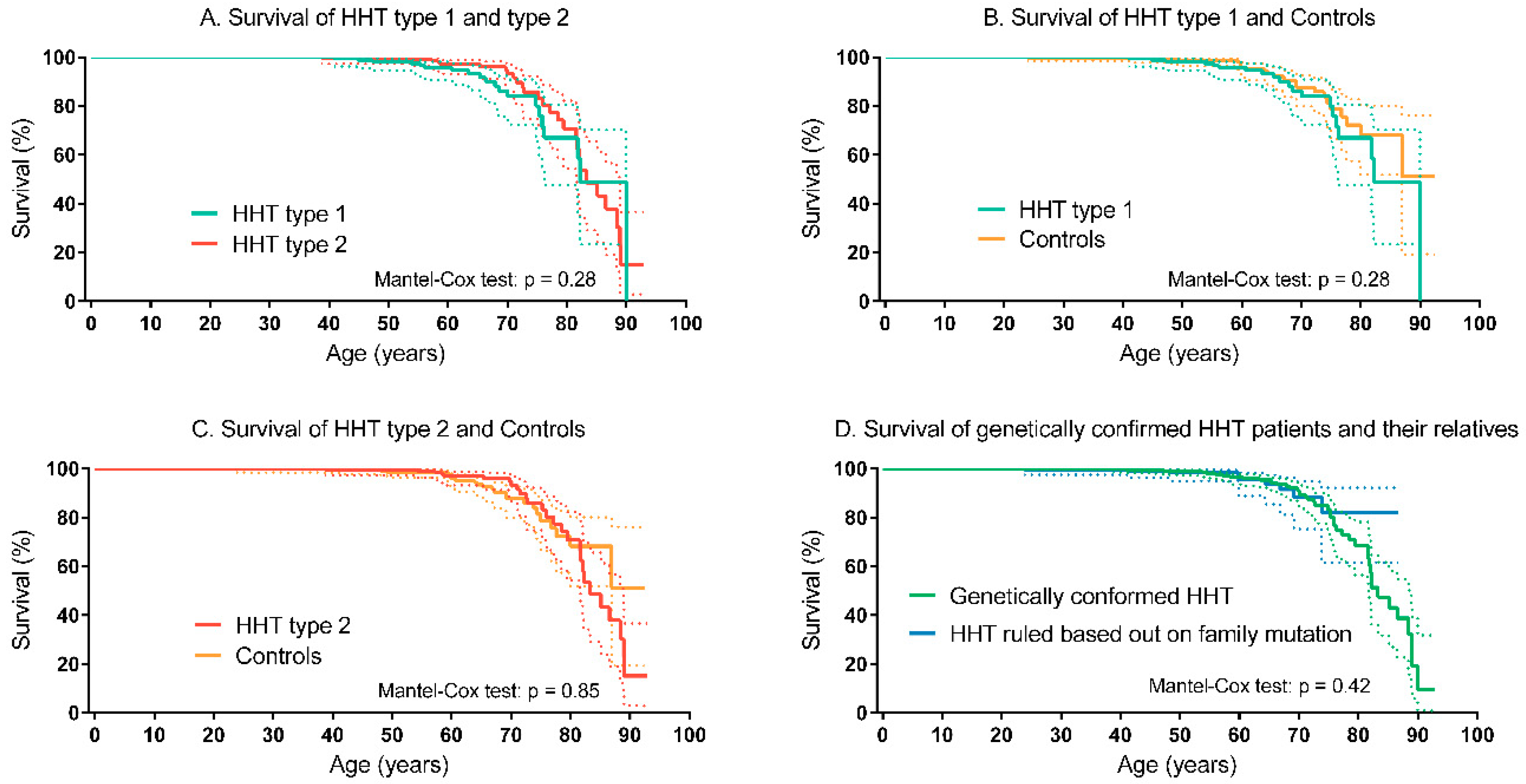
3.3. Survival of HHT Patients and Controls
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Govani, F.S.; Shovlin, C.L. Hereditary haemorrhagic telangiectasia: A clinical and scientific review. Eur. J. Hum. Genet. 2009, 17, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L.; Buscarini, E.; Kjeldsen, A.D.; Mager, H.J.; Sabba, C.; Droege, F.; Geisthoff, U.; Ugolini, S.; Dupuis-Girod, S. European Reference Network for Rare Vascular Diseases (VASCERN) Outcome Measures for Hereditary Haemorrhagic Telangiectasia (HHT). Orphanet J. Rare Dis. 2018, 13, 1–5. [Google Scholar] [CrossRef] [PubMed]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrell, J. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W.; Berg, J.N.; Baldwin, M.A.; Gallione, C.J.; Marondel, I.; Yoon, S.-J.; Stenzel, T.T.; Speer, M.; Pericak-Vance, M.A.; Diamond, A.; et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 1996, 13, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Kroon, S.; Snijder, R.J.; Faughnan, M.E.; Mager, H.J. Systematic screening in hereditary hemorrhagic telangiectasia: A review. Curr. Opin. Pulm. Med. 2018, 24, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Khurshid, I.; Downie, G.H. Pulmonary arteriovenous malformation. Postgrad. Med. J. 2002, 78, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Faughnan, M.E.; Palda, V.A.; Garcia-Tsao, G.; Geisthoff, U.W.; McDonald, J.; Proctor, D.D.; Spears, J.; Brown, D.H.; Chesnutt, M.S.; Cottin, V.; et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J. Med. Genet. 2011, 48, 73–87. [Google Scholar] [CrossRef]
- Boother, E.J.; Brownlow, S.; Tighe, H.C.; Bamford, K.B.; Jackson, J.E.; Shovlin, C.L. Cerebral Abscess Associated with Odontogenic Bacteremias, Hypoxemia, and Iron Loading in Immunocompetent Patients with Right-to-Left Shunting Through Pulmonary Arteriovenous Malformations. Clin. Infect. Dis. 2017, 65, 595–603. [Google Scholar] [CrossRef]
- Donaldson, J.W.; McKeever, T.M.; Hall, I.P.; Hubbard, R.B.; Fogarty, A.W. Complications and mortality in hereditary hemorrhagic telangiectasia. Neurology 2015, 84, 1886–1893. [Google Scholar] [CrossRef]
- de Gussem, E.M.; Edwards, C.P.; Hosman, A.E.; Westermann, C.J.J.; Snijder, R.J.; Faughnan, M.E.; Mager, J.J. Life expectancy of parents with Hereditary Haemorrhagic Telangiectasia. Orphanet J. Rare Dis. 2016, 11, 46. [Google Scholar] [CrossRef]
- Droege, F.; Thangavelu, K.; Stuck, B.A.; Stang, A.; Lang, S.; Geisthoff, U. Life expectancy and comorbidities in patients with hereditary hemorrhagic telangiectasia. Vasc. Med. 2018, 23, 377–383. [Google Scholar] [CrossRef]
- Sabbà, C.; Pasculli, G.; Suppressa, P.; D’Ovidio, F.; Lenato, G.M.; Resta, F.; Assennato, G.; Guanti, G. Life expectancy in patients with hereditary haemorrhagic telangiectasia. QJM Mon. J. Assoc. Physicians 2006, 99, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L.; Guttmacher, A.E.; Buscarini, E.; Faughnan, M.E.; Hyland, R.H.; Westermann, C.J.; Kjeldsen, A.D.; Plauchu, H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am. J. Med. Genet. 2000, 91, 66–67. [Google Scholar] [CrossRef]
- Gallione, C.J.; Richards, J.A.; Letteboer, T.G.W.; Rushlow, D.; Prigoda, N.L.; Leedom, T.P.; Ganguly, A.; Castells, A.; van Amstel, J.K.P.; Westermann, C.J.J.; et al. SMAD4 mutations found in unselected HHT patients. J. Med. Genet. 2006, 43, 793–797. [Google Scholar] [CrossRef]
- Pahl, K.S.; Choudhury, A.; Wusik, K.; Hammill, A.; White, A.; Henderson, K.; Pollak, J.; Kasthuri, T.S. Applicability of the Curaçao Criteria for the Diagnosis of Hereditary Hemorrhagic Telangiectasia in the Pediatric Population. J. Pediatr. 2018, 197, 207–213. [Google Scholar] [CrossRef] [PubMed]
- van Gent, M.W.F.; Post, M.C.; Luermans, J.G.L.M.; Snijder, R.J.; Westermann, C.J.J.; Plokker, H.W.M.; Overtoom, T.T.; Mager, J.J. Screening for pulmonary arteriovenous malformations using transthoracic contrast echocardiography: A prospective study. Eur. Respir. J. 2009, 33, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Hosman, A.E.; de Gussem, E.M.; Balemans, W.A.F.; Gauthier, A.; Westermann, C.J.J.; Snijder, R.J.; Post, M.C.; Mager, J.J. Screening children for pulmonary arteriovenous malformations: Evaluation of 18 years of experience. Pediatr. Pulmonol. 2017, 52, 1206–1211. [Google Scholar] [CrossRef]
- Kjeldsen, A.; Aagaard, K.S.; Tørring, P.M.; Möller, S.; Green, A. 20-year follow-up study of Danish HHT patients—Survival and causes of death. Orphanet J. Rare Dis. 2016, 11, 1–8. [Google Scholar] [CrossRef]
- Hosman, A.E.; Devlin, H.L.; Silva, B.M.; Shovlin, C.L. Specific cancer rates may differ in patients with hereditary haemorrhagic telangiectasia compared to controls. Orphanet J. Rare Dis. 2013, 8, 1–15. [Google Scholar] [CrossRef]
- Shovlin, C.L.; Awan, I.; Cahilog, Z.; Abdulla, F.N.; Guttmacher, A.E. Reported cardiac phenotypes in hereditary hemorrhagic telangiectasia emphasize burdens from arrhythmias, anemia and its treatments, but suggest reduced rates of myocardial infarction. Int. J. Cardiol. 2016, 215, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Geirdal, A.Ø.; Dheyauldeen, S.; Bachmann-Harildstad, G.; Heimdal, K. Quality of life in patients with hereditary hemorrhagic telangiectasia in Norway: A population based study. Am. J. Med. Genet. A 2012, 158A, 1269–1278. [Google Scholar] [CrossRef]
- Zarrabeitia, R.; Farinas-Alvarez, C.; Santibanez, M.; Senaris, B.; Fontalba, A.; Botella, L.M.; Parra, J.A. Quality of life in patients with hereditary haemorrhagic telangiectasia (HHT). Health Qual. Life Outcomes 2017, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- The World Bank—Life Expectancy at Birth, Total (Years)—Netherlands. 2020. Available online: https://data.worldbank.org/indicator/SP.DYN.LE00.IN?locations=NL (accessed on 18 October 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HHT Group (n = 717) | Control Group (n = 471) | p-Value | |
|---|---|---|---|
| Gender (%) | 0.28 | ||
| Female | 384 (54) | 268 (57) | |
| Male | 333 (46) | 203 (43) | |
| Genetic mutation (%) | - | - | |
| ENG (HHT type 1) | 319 (45) | ||
| ACVRL1 (HHT type 2) | 325 (45) | ||
| SMAD4 | 29 (4) | ||
| Mutation unknown | 44 (6) | ||
| Mean age at presentation, years (SD) | 40.8 (19.4) | 40.6 (17.4) | 0.86 |
| ENG (n = 319) | 35.7 (19.9) | - | - |
| ACVRL1 (n = 325) | 44.9 (17.8) | - | - |
| SMAD4 (n = 29) | 31.4 (17.2) | - | - |
| Mutation unknown (n = 44) | 54.1 (13.9) | - | - |
| Mean birth year | 1969 | 1969 | - |
| HHT Group (n = 717) | Control Group (n = 471) | |
|---|---|---|
| PAVM (%) | 255 (36) | 32 (7) |
| ENG (n = 319) | 176 | - |
| ACVRL1 (n = 325) | 47 | - |
| SMAD4 (n = 29) | 12 | - |
| Mutation unknown (n = 44) | 20 | - |
| PAVM embolotherapy | 175 | 28 |
| CVM (%) | ||
| Yes | 28 (4) | 4 (2) |
| No | 404 (56) | 0 |
| Unknown/not screened | 285 (40) | 467 (99) |
| CVM treatment | ||
| No treatment | 14 | 1 |
| Surgery | 5 | 1 |
| Radiotherapy | 3 | 1 |
| Embolotherapy | 2 | 1 |
| Combination | 4 | 0 |
| HVM (%) * | 75 (11) | 3 (<1) |
| Gastrointestinal telangiectases (%) * | 72 (10) | 6 (1) |
| Other AVMs (%) * | 14 (2) | 4 (1) |
| Spinal | 4 | 0 |
| Pancreatic | 3 | 0 |
| Renal | 2 | 1 |
| Urinary bladder | 2 | 1 |
| Splenic | 1 | 0 |
| Muscular | 1 | 2 |
| Ocular | 1 | 0 |
| Anemia (%) | 212 (30) | 28 (6) |
| Comorbidities (%) | ||
| Malignancy | 36 (5) | 21 (5) |
| Atrial fibrillation | 36 (5) | 13 (3) |
| COPD and bronchiectasis | 28 (4) | 16 (3) |
| Acute coronary disease | 26 (4) | 10 (2) |
| Venous thromboembolism | 25 (4) | 8 (2) |
| Autoimmune disease | 21 (3) | 23 (5) |
| Pulmonary hypertension | 19 (3) | 2 (< 1) |
| Diabetes mellitus type 2 | 16 (2) | 19 (4) |
| Peripheral vascular disease | 15 (2) | 7 (2) |
| Disease complications (%) | ||
| Cerebrovascular accident | 49 (7) | 22 (5) |
| High-output heart failure | 14 (2) | 0 (0) |
| Brain abscess | 11 (2) | 3 (1) |
| HHT Group (n = 717) | Control Group (n = 471) | p-Value | |
|---|---|---|---|
| Deceased (%) | 57 (8) | 24 (5) | 0.06 |
| Mean age death, years (SD) | 69.7 (13.3) | 64.3 (13.7) | 0.1 |
| Cause of death | - | - | |
| Infection | 15 | ||
| Heart failure | 9 | ||
| Malignancy | 8 | ||
| Severe anemia | 4 | ||
| Thromboembolism | 2 | ||
| Postoperative complications | 2 | ||
| Hemorrhagic stroke | 1 | ||
| Unknown | 1 | ||
| 17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Gussem, E.M.; Kroon, S.; Hosman, A.E.; Kelder, J.C.; Post, M.C.; Snijder, R.J.; Mager, J.J. Hereditary Hemorrhagic Telangiectasia (HHT) and Survival: The Importance of Systematic Screening and Treatment in HHT Centers of Excellence. J. Clin. Med. 2020, 9, 3581. https://doi.org/10.3390/jcm9113581
de Gussem EM, Kroon S, Hosman AE, Kelder JC, Post MC, Snijder RJ, Mager JJ. Hereditary Hemorrhagic Telangiectasia (HHT) and Survival: The Importance of Systematic Screening and Treatment in HHT Centers of Excellence. Journal of Clinical Medicine. 2020; 9(11):3581. https://doi.org/10.3390/jcm9113581
Chicago/Turabian Stylede Gussem, Els M., Steven Kroon, Anna E. Hosman, Johannes C. Kelder, Martijn C. Post, Repke J. Snijder, and Johannes J. Mager. 2020. "Hereditary Hemorrhagic Telangiectasia (HHT) and Survival: The Importance of Systematic Screening and Treatment in HHT Centers of Excellence" Journal of Clinical Medicine 9, no. 11: 3581. https://doi.org/10.3390/jcm9113581
APA Stylede Gussem, E. M., Kroon, S., Hosman, A. E., Kelder, J. C., Post, M. C., Snijder, R. J., & Mager, J. J. (2020). Hereditary Hemorrhagic Telangiectasia (HHT) and Survival: The Importance of Systematic Screening and Treatment in HHT Centers of Excellence. Journal of Clinical Medicine, 9(11), 3581. https://doi.org/10.3390/jcm9113581