Disorders of Sex Development: Classification, Review, and Impact on Fertility


Abstract

1. Introduction
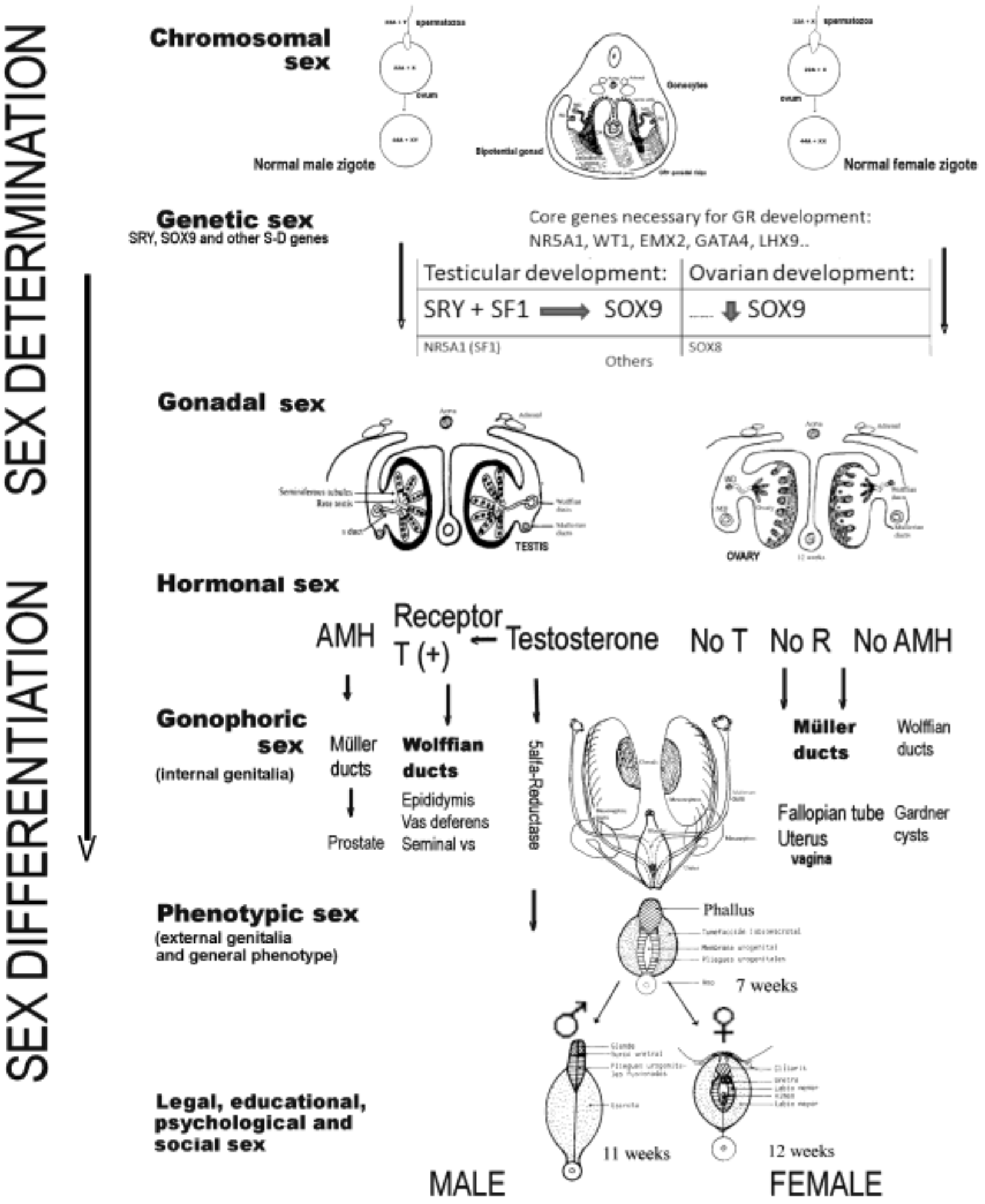
2. Determination and Differentiation of the Human Sex
3. Disorders of Sex Development: Classification
- 1876: Klebs made the first attempt at a classification, noting (1) true sexual ambiguity or true hermaphroditism (TH) and (2) pseudohermaphroditism (Ps): (a) male, if there were testicles, and (b) female, if there were ovaries [22].
- 1931: Goldschmidt introduced the term intersex states and dropped homosexuality from his theory of intersexuality.
- 1949: Baar discovered sex chromatin [23].
- 1956: The karyotype was discovered and the chromosomes were typed.
- 1960: Foss [24] published an article in Brit Med J on intersex states and made a controversial classification of them.
- 1973–1975: The Y-linked histocompatibility (H-Y) antigen [25] was described and, later, the testis-determining factor (TDF) gene and the sex-determining region Y (SRY).
- 2005–2006: The Chicago Consensus Meeting took place, in which the terms used on the subject were discussed, delegates considering them confusing or pejorative, and it was recommended to use the term “disorders of sex development” and a new classification proposal [3]. Dreger et al. [4] had already analyzed changing the nomenclature/taxonomy for intersex, calling for the abandonment of all terms based on the root “hermaphrodite.” Another expert consensus document was proposed in 2018, recommending the term “differences of sex development” and following a classification similar to that of Chicago, also based on the karyotype [6].
- Due to anomalies in the determination of the chromosomes and/or formation of the gonad; and,
- Due to anomalies in the differentiation of the genitalia and phenotype, which will be due to anomalies in hormonal secretions, or their action on the target organs, without anomalies in the chromosomes or in the gonads (at least from the point of view of the histological architecture) [26].
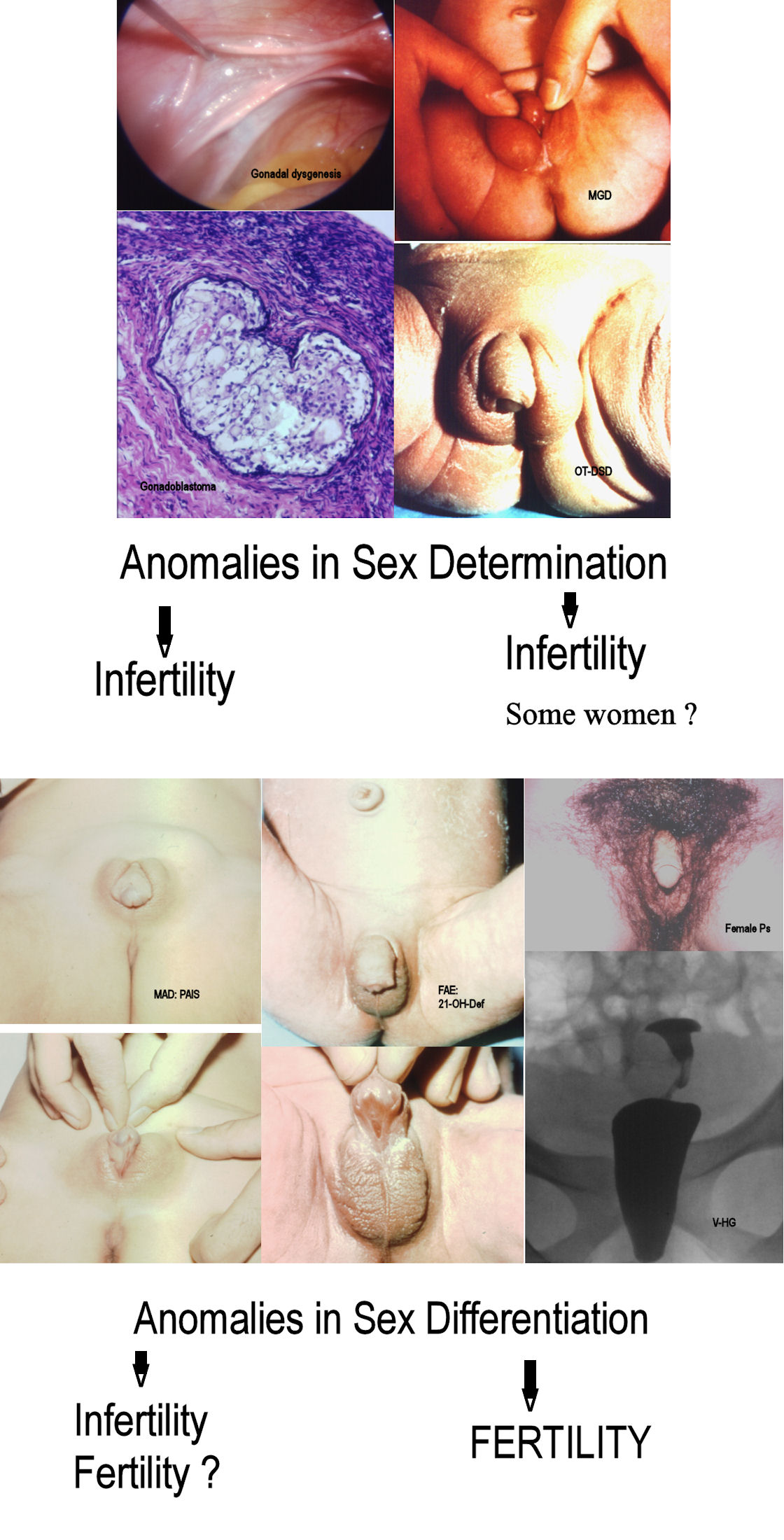
4. DSDs due to Anomalies in Sex Determination (Chromosomes and/or Gonads)
4.1. Anomalies in Sex Determination without Sex Ambiguity (usually)
4.1.1. Gonadal Dysgenesis
4.1.2. Variants of Gonadal Dysgenesis
4.1.3. Polysomies
4.1.4. Sex Reversal or Male XX
4.1.5. Klinefelter Syndrome (KS)
4.1.6. Dysgenetic Infertility
4.2. Anomalies in Sex Determination with Sex Ambiguity (Usually)
4.2.1. Testicular Dysgenesis
4.2.2. Ovotesticular Disorders or TH (OT-ASD or OT-DSD)
4.2.3. Mutations in the NR5A1 Gene
5. DSDs due to Anomalies in Sex Differentiation (Hormones and Enzymes)
5.1. MALE Ps or Male (XY) with Androgen Deficiency (MAD) (by Abnormal Fetal Endocrinology, without Gonadal Abnormality, or Non-Dysgenetic)
- Fetal gonadotropic deficiency: Luteinizing hormone (LH) (or human chorionic gonadotropin -hCG-) that stimulates Leydig cells (Leydig cell hypoplasia), although this is rare. Park et al. [83] described “A Case of Male Pseudohermaphroditism Associated With Elevated LH, Normal FSH, and Low Testosterone Possibly Due to the Secretion of an Abnormal LH,” as a primary defect of the CNS with secretion of an abnormal LH and producing male Ps. In general, if LH is absent, androgens are decreased, the testes do not descend (cryptorchidism), and there is microphalo; however, more frequently, this may lead to gonadotrophin-resistant testes [84].
- Deficiency of the testicle itself and its secretions; therefore, there may be male Ps by:
- (a)
- Embryonic testicular regression, described by Edman et al. in 1977 [85] and later by Coulam in 1979 [86], as testicular regression syndrome (TRS). TRS is attributed to an early regression of the embryonic testicle, and therefore, there are no Müllerian derivatives, unlike in Swyer’s syndrome, in which there is a uterus, but it may depend on the moment of such embryonic regression.
- (b)
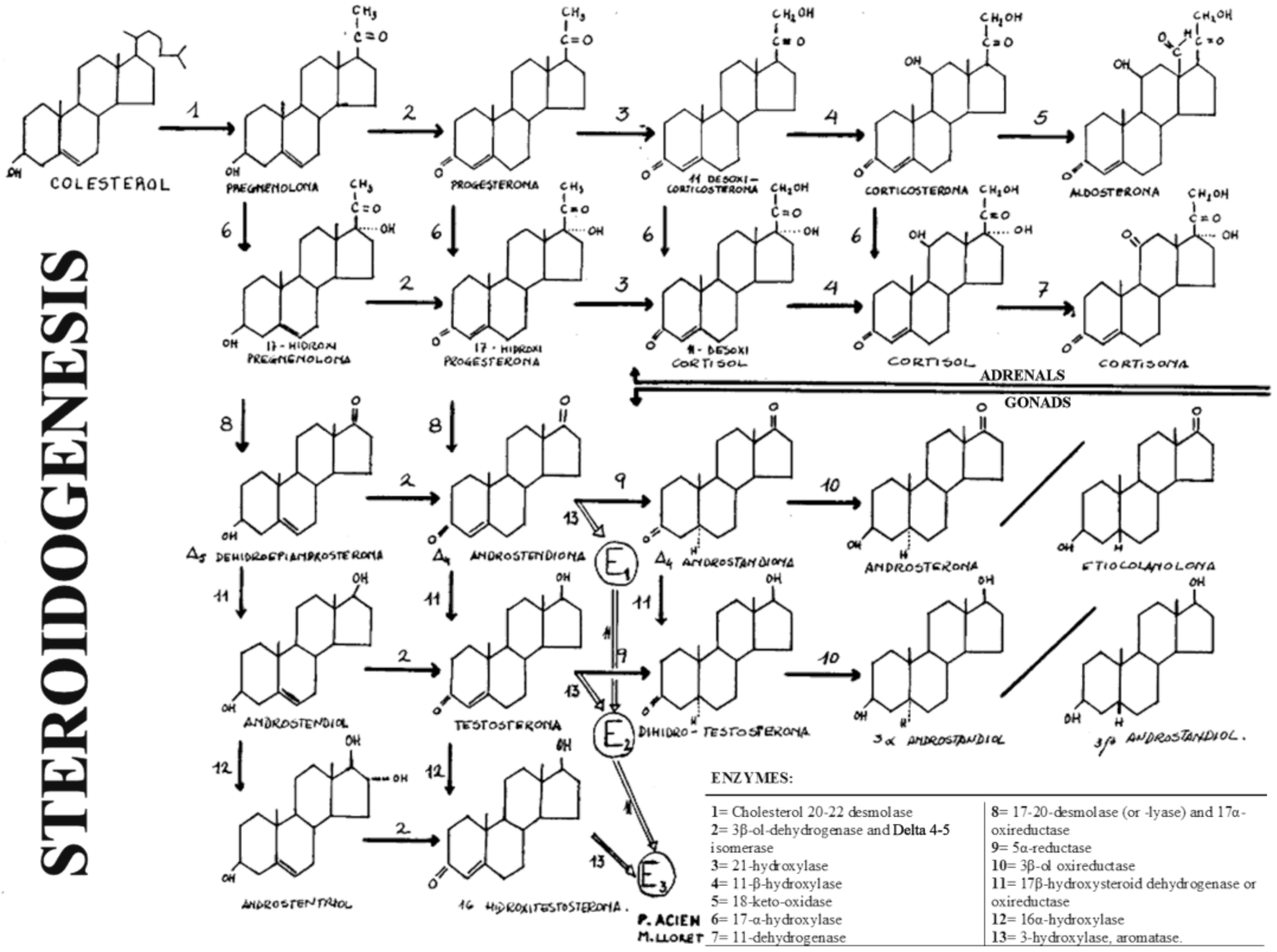
- Enzyme block in steroidogenesis, and consequently, deficient androgen formation.
- (c)
- Poor response to androgens, or defects in androgenic action at the target organ level, which would include complete and incomplete testicular feminization syndromes and 5α-reductase deficiency.
- (d)
- Other mild forms of male Ps, or unambiguous sex, including cryptorchidism, hypospadias, and defects in the formation or action of AMH.
5.1.1. Gonadotrophin-Resistant Testes and Fetal Gonadotropic Deficiency
5.1.2. Embryonic Testicular Regression Syndrome (TRS; Anorchia)
5.1.3. Disorders of Androgen Production (Male Ps or MAD due to Blockage in Steroidogenesis)
5.1.4. Male Ps or MAD Due to Defects in Androgenic Action
5.1.5. Other Mild or Unambiguous Sex Forms of MAD.
5.2. FEMALE Ps or Female (XX) with Androgen Excess (FAE)
- If the noxa acts before 12 weeks, the androgenic stimulus will produce severe ambiguity, with a penile urethra and the external appearance of a male, except that there are no testes, but the internal genitalia and upper vagina are normal.
- In less severe cases, there is scrotal hypospadias, with the sinus and vagina communicating with the urethral opening.
- If androgen acts after 12 weeks, then the external genitalia are normal and there is only an enlargement of the clitoris as a sign of virilization.
5.2.1. Congenital Adrenal Hyperplasia in Female/Adrenogenital Syndrome (AGS)
5.2.2. Hormone Therapy (Iatrogenic FAE)
5.2.3. Maternal Virilizing Tumor
5.2.4. Aromatase Deficiency (P450arom)
5.3. Congenital Hypogonadotropic Hypogonadism (Kallmann Syndrome)
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, P.A.; Houk, C.P.; Ahmed, S.F.; Hughes, I.A. Consensus Statement on Management of Intersex Disorders: In collaboration with the participants in the International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. Pediatrics 2006, 118, e488–e500. [Google Scholar] [CrossRef] [PubMed]
- Houk, C.P.; Hughes, I.A.; Ahmed, S.F.; Lee, P.A. Summary of Consensus Statement on Intersex Disorders and Their Management: Writing Committee for the International Intersex Consensus Conference Participants. Pediatrics 2006, 118, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Hughes, I.A.; Houk, C.P.; Ahmed, S.F.; Lee, P.A.; LWPES1/ESPE2 Consensus Group. Consensus statement on management of intersex disorders. Arch. Dis. Child. 2006, 91, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Dreger, A.D.; Chase, C.; Sousa, A.; Gruppuso, P.A.; Frader, J. Changing the nomenclature/taxonomy for Intersex: A Scientific and Clinical Rationale. J. Pediatr. Endocrinol. Metab. 2005, 18, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Conn, J.; Gillam, L.; Conway, G. Revealing the diagnosis of androgen insensitivity syndrome in adulthood. BMJ 2005, 331, 628e30. [Google Scholar] [CrossRef] [PubMed]
- Cools, M.; Nordenström, A.; Robeva, R.; Hall, J.; Westerveld, P.; Flück, C.; Köhler, B.; Berra, M.; Springer, A.; Schweizer, K.; et al. Caring for Individuals with a Difference of Sex Development (DSD): A Consensus Statement. Nat. Rev. Endocrinol. 2018, 14, 415–429. [Google Scholar] [CrossRef]
- Camats, N.; Flück, C.E.; Audí, L. Oligogenic Origin of Differences of Sex Development in Humans. Int. J. Mol. Sci. 2020, 21, 1809. [Google Scholar] [CrossRef]
- Acién, P. Disorders of Sex Development. Open Acc. J. Repro. Sex. Disord. 2020, 2. [Google Scholar] [CrossRef]
- Acién, P. Classification of the human sex development anomalies. Curr. Pathophysiol. Bases 2020. submitted, under review. [Google Scholar]
- Eid, W.; Biason-Lauber, A. Why boys will be boys and girls will be girls: Human sex development and its defects. Embryo Today 2016, 108, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Biason-Lauber, A. The Battle of the Sexes: Human Sex Development and Its Disorders. Results Probl. Cell Differ. 2016, 58, 337–382. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Gonadal steroid influences on brain development and sexual differentiation. Int. Rev. Physiol. 1983, 27, 99–145. [Google Scholar] [PubMed]
- Peper, J.S.; Koolschijn, P.C. Sex steroids and the organization of the human brain. J. Neurosci. 2012, 32, 6745–6746. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, M.V.; Ashwin, E.; Auyeung, B.; Chakrabarti, B.; Taylor, K.; Hackett, G.; Bullmore, E.T.; Baron-Cohen, S. Fetal testosterone influences sexually dimorphic gray matter in the human brain. Version 2. J. Neurosci. 2012, 32, 674–680. [Google Scholar] [CrossRef]
- Negri-Cesi, P.; Colciago, A.; Celotti, F.; Motta, M. Sexual differentiation of the brain: Role of testosterone and its active metabolites. J. Endocrinol. Invest. 2004, 27 (Suppl. to no. 6), 120–127. [Google Scholar]
- Kiesow, H.; Dunbar, R.I.M.; Kable, J.W.; Kalenscher, T.; Vogeley, K.; Schilbach, L.; Marquand, A.F.; Wiecki, T.V.; Bzdok, D. 10,000 social brains: Sex differentiation in human brain anatomy. Sci. Adv. 2020, 6, eaaz1170. [Google Scholar] [CrossRef]
- Ristori, J.; Cocchetti, C.; Romani, A.; Mazzoli, F.; Vignozzi, L.; Maggi, M.; Fisher, A.D. Brain Sex Differences Related to Gender Identity Development: Genes or Hormones? Int. J. Mol. Sci. 2020, 21, 2123. [Google Scholar] [CrossRef]
- Cisternas, C.D.; Cortes, L.R.; Golynker, I.; Castillo-Ruiz, A.; Forger, N.G. Neonatal Inhibition of DNA Methylation Disrupts Testosterone-Dependent Masculinization of Neurochemical Phenotype. Endocrinology 2020, 161, bqz022. [Google Scholar] [CrossRef]
- Joel, D.; Garcia-Falgueras, A.; Swaab, D. The Complex Relationships between Sex and the Brain. Neuroscientist 2020, 26, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.P. Sexual differentiation of brain and other tissues: Five questions for the next 50 years. Horm. Behav. 2020, 120, 104691. [Google Scholar] [CrossRef]
- Biason-Lauber, A. Human Sex Development: From Basic Science to Clinical Practice and Back. Pediatr. Endocrinol. Rev. 2017, 15, 8–20. [Google Scholar] [CrossRef]
- Acien, P. Tratado de Obstetricia y Ginecología: Ginecología; Ed Molloy: Alicante, Spain, 2004; Volume 2, pp. 371–428. ISBN 84-609-0564-0. [Google Scholar]
- Barr, M.L.; Bertram, E.G. A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. Nature 1949, 163, 676–677. [Google Scholar] [CrossRef] [PubMed]
- Foss, G.L. Intersex States. Br. Med. J. 1960, 2, 1907–1909. [Google Scholar] [CrossRef]
- Wachtel, S.S.; Ohno, S.; Koo, G.C.; Boyse, E.A. Possible role for H-Y antigen in the primary determination of sex. Nature 1975, 257, 235–236. [Google Scholar] [CrossRef] [PubMed]
- Lepais, L.; Morel, Y.; Mouriquand, P.; Gorduza, D.; Plotton, I.; Collardeau-Frachon, S.; Dijoud, F. A novel morphological approach to gonads in disorders of sex development. Mod. Pathol. 2016, 29, 1399–1414. [Google Scholar] [CrossRef]
- Acien, P. Tratado de Obstetricia y Ginecología: Obstetricia; Ed Molloy: Alicante, Spain, 1998; Volume 1, p. 28. ISBN 84-605-8341-4. [Google Scholar]
- Acién, P.; Acién, M.I. The history of female genital tract malformation classifications and proposal of an updated system. Hum. Reprod. Update 2011, 17, 693–705. [Google Scholar] [CrossRef]
- Acién, M.; Acién, P. Classification of the Female Genital Tract Malformations and its Embryological Origin. Diagnostic and Therapeutical Considerations. Curr. Womens Health Rev. 2013, 9, 1–29. [Google Scholar] [CrossRef]
- Acién, P.; Acién, M.I. Embryological considerations and update classification of female genital malformations. G. Ital. Ostet. Ginecol. 2013, XXXV/4, 507–511. [Google Scholar]
- Acién, P.; Acién, M.I.; Sánchez-Ferrer, M. Complex malformations of the female genital tract. New types and revision of classification. Hum. Reprod. 2004, 19, 2377–2384. [Google Scholar] [CrossRef]
- Acién, P.; Acién, M. The presentation and management of complex female genital malformations. Hum. Reprod. Update 2016, 22, 48–69. [Google Scholar] [CrossRef]
- Wilkins, L.; Fleischmann, W. Ovarian Agenesis: Pathology, Associated Clinical Symptoms and the Bearing on the Theories of Sex Differentiation. J. Clin. Endocrinol. 1944, 4, 357–375. [Google Scholar] [CrossRef]
- Linden, H.; Overzier, C. Echter Agonadismus (Anorchismus) bei Geschwistern. Gynaecologia 1956, 142, 215–233. [Google Scholar]
- Abir, R.; Fisch, B.; Nahum, R.; Orvieto, R.; Nitke, S.; Ben Rafael, Z. Turner’s syndrome and fertility: Current status and possible putative prospects. Hum. Reprod. Update 2001, 7, 603–610. [Google Scholar] [CrossRef]
- Folsom, L.J.; Fuqua, J.S. Reproductive Issues in Women with Turner Syndrome. Endocrinol. Metab. Clin. N. Am. 2015, 44, 723–737. [Google Scholar] [CrossRef]
- Bernard, V.; Donadille, B.; Zenaty, D.; Courtillot, C.; Salenave, S.; Brac de la Perriere, A.; Albarel, F.; Fèvre, A.; Kerlan, V.; Brue, T.; et al. Spontaneous fertility and pregnancy outcomes amongst 480 women with Turner syndrome. Hum. Reprod. 2016, 31, 782–788. [Google Scholar] [CrossRef]
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef]
- Kalra, R.; Cameron, M.; Stern, C. Female fertility preservation in DSD. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101289. [Google Scholar] [CrossRef]
- Mamsen, L.S.; Charkiewicz, K.; Anderson, R.A.; Telfer, E.E.; McLaughlin, M.; Kelsey, T.W.; Kristensen, S.G.; Gook, D.A.; Ernst, E.; Andersen, C.Y. Characterization of follicles in girls and young women with Turner syndrome who underwent ovarian tissue cryopreservation. Fertil. Steril. 2019, 111, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.L.; Chetty, T.; Jorgensen, A.; Mitchell, R. Disorders of Sex Development—Novel Regulators, Impacts on Fertility, and Options for Fertility Preservation. Int. J. Mol. Sci. 2020, 21, 2282. [Google Scholar] [CrossRef]
- Greenblatt, R.B.; Byrd, J.R.; McDonough, P.G.; Mahesh, V.B. The spectrum of gonadal dysgenesis. A clinical, cytogenetic, and pathologic study. Am. J. Obstet. Gynecol. 1967, 98, 151–172. [Google Scholar] [CrossRef]
- Portnoi, M.F.; Dumargne, M.C.; Rojo, S.; Witchel, S.F.; Duncan, A.J.; Eozenou, C.; Bignon-Topalovic, J.; Yatsenko, S.A.; Rajkovic, A.; Reyes-Mugica, M.; et al. Mutations involving the SRY-related gene SOX8 are associated with a spectrum of human reproductive anomalies. Hum. Mol. Genet. 2018, 27, 1228–1240. [Google Scholar] [CrossRef]
- Lim, H.H.; Kil, H.R.; Koo, S.H. Incidence, puberty, and fertility in 45,X/47,XXX mosaicism: Report of a patient and a literature review. Am. J. Med. Genet. A 2017, 173, 1961–1964. [Google Scholar] [CrossRef]
- Martin, R.J.; Smith, G.; Hughes, J.; Morrison, P.J. Letter: Incidence, puberty, and fertility in 45,X/47,XXX mosaicism: Report of a patient and a literature review. Am. J. Med. Genet. A. 2018, 176, 1029. [Google Scholar] [CrossRef]
- De Leon, F.D.; Hersh, J.H.; Sanfilippo, J.S.; Schikler, K.N.; Yen, F.F. Gonadal and Müllerian duct agenesis in a girl with 46, X, i(Xq). Obstet. Gynecol. 1984, 63(3), 81S–83S. [Google Scholar]
- Kim, J.W.; Park, S.Y.; Ryu, H.M.; Lee, D.E.; Lee, B.Y.; Kim, S.Y.; Park, Y.S.; Lee, H.S.; Seo, J.T. Molecular and Clinical Characteristics of 26 Cases with Structural Y Chromosome Aberrations. Cytogenet. Genome Res. 2012, 136, 270–277. [Google Scholar] [CrossRef]
- Stankiewicz, P.; Hélias-Rodzewicz, Z.; Jakubów-Durska, K.; Bocian, E.; Obersztyn, E.; Rappold, G.A.; Mazurczak, T. Cytogenetic and Molecular Characterization of Two Isodicentric Y Chromosomes. Am. J. Med. Genet. 2001, 101, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.R.F.; Pereira, A.C.N.; Souza, M.T.V.L.; Ramos, M.R.B.P. FISH, PCR and Cytogenetic Characterization in a Girl with Ambiguous Genitalia and Karyotype mos46,X,iso(Y)(qter→p11.3::p11.3→qter)[80]/45,X[17]/46,X,+mar[3]. Genet. Mol. Res. 2008, 7, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Bergendi, E.; Plöchl, E.; Vlasak, I.; Rittinger, O.; Muss, W. A Turner-like Phenotype in a Girl with an Isodicentric Fluorescent Y Chromosome Mosaicism. Klin. Pediatr. 1997, 209, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Bouayed Abdelmoula, N.; Amouri, A. Dicentric Y Chromosome. Ann. Biol. Clin. (Paris) 2005, 63, 363–375. (In French) [Google Scholar]
- Pan, S.; Guo, S.; Liu, L.; Yang, X.; Liang, H. Functional study of a novel c.630delG (p.Y211Tfs*85) mutation in NR5A1 gene in a Chinese boy with 46,XY disorders of sex development. J. Assist. Reprod. Genet. 2020, 37, 477–486. [Google Scholar] [CrossRef]
- Pertusa, S.; Palacios, A. 46 XX pure gonadal dysgenesis: An infrequent cause of primary amenorrhoea. BMJ Case Rep. 2009, 2009, bcr0720080485. [Google Scholar] [CrossRef]
- Schokman, F.C.M.; Correy, J.F. A Patient with Pure Gonadal Dysgenesis, Gonadal Tumour and Virilisation. Aust. N. Z. J. Obs. Gynaecol. 1979, 19, 241–244. [Google Scholar] [CrossRef]
- Maeyama, M.; Kagami, T.; Miyakawa, I.; Tooya, T.; Kawasaki, N.; Iwamasa, T. Case report of dysgerminoma in a patient with 46,XX pure gonadal dysgenesis. Gynecol. Oncol. 1983, 16, 405–413. [Google Scholar] [CrossRef]
- Swyer, G.I.M. Male Pseudohermaphroditism: A Hitherto Undescribed Form. Br. Med. J. 1955, 2, 709–712. [Google Scholar] [CrossRef]
- Gupta, A.; Bajaj, R.; Jindal, U.N. A Rare Case of Swyer Syndrome in Two Sisters with Successful Pregnancy Outcome in Both. J. Hum. Reprod. Sci. 2019, 12, 267–269. [Google Scholar] [CrossRef]
- Tartaglia, N.R.; Howell, S.; Sutherland, A.; Wilson, R.; Wilson, L. A review of trisomy X (47,XXX). Orphanet J. Rare Dis. 2010, 5, 8. [Google Scholar] [CrossRef]
- DelaChapelle, A.; Hortling, H.; Niemi, M.; Wennstroem, J. XX Sex Chromosomes in a Human Male. First case. Acta Med. Scand. 1964, 175 (Suppl. 412), 25–28. [Google Scholar] [CrossRef]
- Page, D.C.; de la Chapelle, A.; Weissenbach, J. Chromosome Y-specific DNA in Related Human XX Males. Nature 1985, 315, 224–226. [Google Scholar] [CrossRef]
- Vorona, E.; Zitzmann, M.; Gromoll, J.; Schuring, A.N.; Nieschlag, E. Clinical, Endocrinological, and Epigenetic Features of the 46,XX Male Syndrome, Compared with 47,XXY Klinefelter Patients. J. Clin. Endocrinol. Metab. 2007, 92, 3458–3465. [Google Scholar] [CrossRef]
- Van Batavia, J.P.; Kolon, T.F. Fertility in disorders of sex development: A review. J. Pediatr. Urol. 2016, 12, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Klinefelter, H.F.; Reifenstein, E.C.; Albright, F. Syndrome characterized by gynecomastia, aspermatogenesis without Leydigism, increased excretion of follicle-stimulating hormone. J. Clin. Endocrinol. 1942, 2, 615–627. [Google Scholar] [CrossRef]
- Jacobs, P.A.; Baikie, A.G.; Court Brown, W.M.; Forrest, H.; Roy, J.R.; Stewart, J.S.S.; Lennox, B. Chromosomal sex in the syndrome of testicular feminization. Lancet 1959, 2, 591–592. [Google Scholar] [CrossRef]
- Schwartz, I.; Root, A. The Klinefelter Syndrome of Testicular Dysgenesis. Endocrinol. Metab. Clin. N. Am. 1991, 20, 153–162. [Google Scholar] [CrossRef]
- Hanna, E.S.; Cheetham, T.; Fearon, K.; Herbrand, C.; Hudson, N.; McEleny, K.; Quinton, R.; Stevenson, E.; Wilkes, S. The Lived Experience of Klinefelter Syndrome: A Narrative Review of the Literature. Front. Endocrinol. 2019, 10, 825. [Google Scholar] [CrossRef] [PubMed]
- Zitzmann, M.; Aksglaede, L.; Corona, G.; Isidori, A.M.; Juul, A.; T’Sjoen, G.; Kliesch, S.; D’Hauwers, K.; Toppari, J.; Słowikowska-Hilczer, J.; et al. European academy of andrology guidelines on Klinefelter Syndrome: Endorsing Organization: European Society of Endocrinology. Andrology 2020, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Corona, G.; Pizzocaro, A.; Lanfranco, F.; Garolla, A.; Pelliccione, F.; Vignozzi, L.; Ferlin, A.; Foresta, C.; Jannini, E.A.; Maggi, M.; et al. Sperm recovery and ICSI outcomes in Klinefelter syndrome: A systematic review and meta-analysis. Hum. Reprod. Update 2017, 23, 265–275. [Google Scholar] [CrossRef]
- Van Saen, D.; Vloeberghs, V.; Gies, I.; Mateizel, I.; Sermon, K.; De Schepper, J.; Tournaye, H.; Goossens, E. When does germ cell loss and fibrosis occur in patients with Klinefelter syndrome? Hum. Reprod. 2018, 33, 1009–1022. [Google Scholar] [CrossRef]
- Deebel, N.A.; Galdon, G.; Zarandi, N.P.; Stogner-Underwood, K.; Howards, S.; Lovato, J.; Kogan, S.; Atala, A.; Lue, Y.; Sadri-Ardekani, H. Age-related presence of spermatogonia in patients with Klinefelter syndrome: A systematic review and meta-analysis. Hum. Reprod. Update 2020, 26, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Fabbri-Scallet, H.; de Sousa, L.M.; Maciel-Guerra, A.T.; Guerra-Júnior, G.; de Mello, M.P. Mutation update for the NR5A1 gene involved in DSD and infertility. Hum. Mutat. 2020, 41, 58–68. [Google Scholar] [CrossRef]
- De Paula, G.B.; Barros, B.A.; Carpini, S.; Tincani, B.J.; Mazzola, T.N.; Sanches Guaragna, M.; Santos da Cruz Piveta, C.; Candido de Oliveira, L.; Ribeiro Andrade, J.G.; Guaragna-Filho, G.; et al. 408 Cases of Genital Ambiguity Followed by Single Multidisciplinary Team during 23 Years: Etiologic Diagnosis and Sex of Rearing. Int. J. Endocrinol. 2016, 2016, 4963574. [Google Scholar] [CrossRef]
- Andrade, J.G.R.; Fabbri-Scallet, H.; Dos Santos, A.P.; Cools, M.; Werner, R.; Hiort, O.; de Mello, M.P.; Guerra-Júnior, G.; Maciel-Guerra, A.T. Clinical Findings and Follow-Up of 46,XY and 45,X/46,XY Testicular Dysgenesis. Sex. Dev. 2019, 13, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Andrade, J.G.; Guerra-Júnior, G.; Maciel-Guerra, A.T. 46,XY and 45,X/46,XY testicular dysgenesis: Similar gonadal and genital phenotype, different prognosis. Arq. Bras. Endocrinol. Metabol. 2010, 54, 331–334. [Google Scholar] [CrossRef]
- Weidler, E.M.; Pearson, M.; van Leeuwen, K.; Garvey, E. Clinical management in mixed gonadal dysgenesis with chromosomal mosaicism: Considerations in newborns and adolescents. Semin. Pediatr. Surg. 2019, 28, 150841. [Google Scholar] [CrossRef]
- Kim, Y.M.; Oh, A.; Kim, K.S.; Yoo, H.W.; Choi, J.H. Pubertal outcomes and sex of rearing of patients with ovotesticular disorder of sex development and mixed gonadal dysgenesis. Ann. Pediatr. Endocrinol. Metab. 2019, 24, 231–236. [Google Scholar] [CrossRef]
- Gabriel Ribeiro de Andrade, J.; Marques-de-Faria, A.P.; Fabbri, H.C.; de Mello, M.P.; Guerra-Júnior, G.; Maciel-Guerra, A.T. Long-Term Follow-Up of Patients with 46,XY Partial Gonadal Dysgenesis Reared as Males. Int. J. Endocrinol. 2014, 2014, 480724. [Google Scholar] [CrossRef]
- Tiltman, A.J.; Sweerts, M. Multiparity in a covert true hermaphrodite. Obstet. Gynecol. 1982, 60(6), 752–754. [Google Scholar] [PubMed]
- Schultz, B.A.; Roberts, S.; Rodgers, A.; Ataya, K. Pregnancy in true hermaphrodites and all male offspring to date. Obstet. Gynecol. 2009, 113 Pt 2, 534–536. [Google Scholar] [CrossRef]
- Sugawara, N.; Kimura, Y.; Araki, Y. Successful second delivery outcome using refrozen thawed testicular sperm from an infertile male true hermaphrodite with a 46,XX/46,XY karyotype: Case report. Hum. Cell. 2012, 25, 96–99. [Google Scholar] [CrossRef]
- Georgopapadakos, N.; Manoli, A.; Passia, G.; Skandalakis, P.N.; Filippou, D. Uterus Transplantation as a Therapy Method in Mayer-Rokitansky-Küster-Hauser Syndrome. Cureus 2019, 11, e6333. [Google Scholar] [CrossRef]
- Fabbri, H.C.; Ribeiro de Andrade, J.G.; Maciel-Guerra, A.T.; Guerra-Júnior, G.; de Mello, M.P. NR5A1 Loss-of-Function Mutations Lead to 46,XY Partial Gonadal Dysgenesis Phenotype: Report of Three Novel Mutations. Sex. Dev. 2016, 10, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Park, I.J.; Burnett, L.; Jones Jr., H.; Migeon, C.; Blizzard, R.M. A Case of Male Pseudohermaphroditism Associated with Elevated LH, Normal FSH and Low Testosterone Possibly Due to the Secretion of an Abnormal LH Molecule. Eur. J. Endocrinol. 1976, 83, 173–181. [Google Scholar] [CrossRef]
- Latronico, A.C.; Prado Arnhold, I.J. Gonadotropin Resistance. Endocr. Dev. 2013, 24, 25–32. [Google Scholar] [CrossRef]
- Edman, C.D.; Winters, A.J.; Porter, J.C.; Wilson, J.; MacDonald, P.C. Embryonic testicular regression. A clinical spectrum of XY agonadal individuals. Obstet. Gynecol. 1977, 49, 208–217. [Google Scholar]
- Coulam, C.B. Testicular regression syndrome. Obstet. Gynecol. 1979, 53, 44–49. [Google Scholar] [PubMed]
- Berthezène, F.; Forest, M.G.; Grimaud, J.A.; Claustrat, B.; Mornex, R. Leydig-cell agenesis: A cause of male pseudohermaphroditism. N. Engl. J. Med. 1976, 295, 969–972. [Google Scholar] [CrossRef]
- Schwartz, M.; Imperato-McGinley, J.; Peterson, R.E.; Cooper, G.; Morris, P.I.; MacGillivray, M.; Hensle, T. Male pseudohermaphroditism secondary to an abnormality in Leydig cell differentiation. J. Clin. Endocrinol. Metab. 1981, 53, 123–127. [Google Scholar] [CrossRef]
- David, R.; Yoon, D.J.; Landin, L.; Lew, L.; Sklar, C.; Schinella, R.; Golimbu, M. A syndrome of gonadotropin resistance possibly due to a luteinizing hormone receptor defect. J. Clin. Endocrinol. Metab. 1984, 59, 156–160. [Google Scholar] [CrossRef]
- Saldanha, P.H.; Arnhold, I.J.P.; Mendonca, B.B.; Bloise, W.; Toledo, S.P.A. A clinico-genetic investigation of Leydig cell hypoplasia. Am. J. Med. Genet. 1987, 26, 337–344. [Google Scholar] [CrossRef]
- Huhtaniemi, I.; Alevizaki, M. Gonadotrophin Resistance. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 561–576. [Google Scholar] [CrossRef]
- Latronico, A.C.; Anasti, J.; Arnhold, I.J.P.; Rapaport, R.; Mendonca, B.B.; Bloise, W. Testicular and Ovarian Resistance to Luteinizing Hormone Caused by Inactivating Mutations of the Luteinizing Hormone–Receptor Gene. N. Engl. J. Med. 1996, 334, 507–512. [Google Scholar] [CrossRef]
- Arnhold, I.J.; Latronico, A.C.; Batista, M.C.; Izzo, C.R.; Mendonca, B.B. Clinical Features of Women with Resistance to Luteinizing Hormone. Clin. Endocrinol. (Oxf.) 1999, 51, 701–707. [Google Scholar] [CrossRef]
- Simoni, M.; Tüttelmann, F.; Michel, C.; Böckenfeld, Y.; Nieschlag, E.; Gromoll, J. Polymorphisms of the Luteinizing Hormone/Chorionic Gonadotropin Receptor Gene: Association with Maldescended Testes and Male Infertility. Pharmacogenet. Genomics 2008, 18, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Bakircioglu, M.E.; Tulay, P.; Findikli, N.; Erzik, B.; Gultomruk, M.; Bahceci, M. Successful testicular sperm recovery and IVF treatment in a man with Leydig cell hypoplasia. J. Assist. Reprod. Genet. 2014, 31, 817–821. [Google Scholar] [CrossRef][Green Version]
- Heksch, R.A.; Matheson, M.A.; Tishelman, A.C.; Swartz, J.M.; Jayanthi, V.R.; Diamond, D.A.; Harrison, C.J.; Chan, Y.M.; Nahata, L. Testicular Regression Syndrome: Practice Variation in Diagnosis and Management. Endocr. Pract. 2019, 25, 779–786. [Google Scholar] [CrossRef]
- Macdonald, J.; Kilcoyne, K.R.; Sharpe, R.M.; Kavanagh, Á.; Anderson, R.A.; Brown, P.; Smith, L.B.; Jørgensen, A.; Mitchell, R.T. DMRT1 repression using a novel approach to genetic manipulation induces testicular dysgenesis in human fetal gonads. Hum. Reprod. 2018, 33, 2107–2121. [Google Scholar] [CrossRef]
- Fujitani, K.; Otomo, A.; Nagayama, Y.; Tachibana, T.; Kato, R.; Kawashima, Y.; Kodera, Y.; Kato, T.; Takada, S.; Tamura, K.; et al. PACT/PRKRA and p53 regulate transcriptional activity of DMRT1. Genet. Mol. Biol. 2020, 43, e20190017. [Google Scholar] [CrossRef]
- McElreavey, K.; Jorgensen, A.; Eozenou, C.; Merel, T.; Bignon-Topalovic, J.; Tan, D.S.; Houzelstein, D.; Buonocore, F.; Warr, N.; Kay, R.G.G. Pathogenic variants in the DEAH-box RNA helicase DHX37 are a frequent cause of 46,XY gonadal dysgenesis and 46,XY testicular regression syndrome. Genet. Med. 2020, 22, 150–159. [Google Scholar] [CrossRef]
- Nataraja, R.M.; Yeap, E.; Healy, C.J.; Nandhra, I.S.; Murphy, F.L.; Hutson, J.M.; Kimber, C. Presence of viable germ cells in testicular regression syndrome remnants: Is routine excision indicated? A systematic review. Pediatr. Surg. Int. 2018, 34, 353–361. [Google Scholar] [CrossRef]
- Cabrera, M.S.; Vogiatzi, M.G.; New, M.I. Long term outcome in adult males with classic congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2001, 86, 3070–3078. [Google Scholar] [CrossRef]
- Claahsen-van der Grinten, H.L.; Hermus, A.R.M.M.; Otten, B.J. Testicular Adrenal Rest Tumours in Congenital Adrenal Hyperplasia. Int. J. Pediatr. Endocrinol. 2009, 2009, 624823. [Google Scholar] [CrossRef]
- Reisch, N.; Flade, L.; Scherr, M.; Rottenkolber, M.; Pedrosa Gil, F.; Bidlingmaier, M.; Wolff, H.; Schwarz, H.P.; Quinkler, M.; Beuschlein, F. High prevalence of reduced fecundity in men with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2009, 94, 1665–1670. [Google Scholar] [CrossRef] [PubMed]
- Reichman, D.E.; White, P.C.; New, M.I.; Rosenwaks, Z. Fertility in patients with congenital adrenal hyperplasia. Fertil. Steril. 2014, 101, 301–309. [Google Scholar] [CrossRef]
- Claahsen-van der Grinten, H.L.; Otten, B.J.; Takahashi, S.; Meuleman, E.J.; Hulsbergen-van de Kaa, C.; Sweep, F.C.; Hermus, A.R. Testicular adrenal rest tumors in adult males with congenital adrenal hyperplasia: Evaluation of pituitary-gonadal function before and after successful testis-sparing surgery in eight patients. J. Clin. Endocrinol. Metab. 2007, 92, 612–615. [Google Scholar] [CrossRef]
- Bizzarri, C.; Pisaneschi, E.; Mucciolo, M.; Pedicelli, S.; Galeazzi, D.; Novelli, A.; Cappa, M. Lipoid Congenital Adrenal Hyperplasia by Steroidogenic Acute Regulatory Protein (STAR) Gene Mutation in an Italian Infant: An Uncommon Cause of Adrenal Insufficiency. Ital. J. Pediatr. 2017, 43, 57. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Casas, L.; Bose, H.S. Lipoid Congenital Adrenal Hyperplasia Due to STAR Mutations in a Caucasian Patient. Endocrinol. Diabetes Metab. Case Rep. 2016, 2016, 150119. [Google Scholar] [CrossRef]
- Hauffa, B.; Hiort, O. P450 Side-Chain Cleavage Deficiency—A Rare Cause of Congenital Adrenal Hyperplasia. Endocr. Dev. 2011, 20, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Rice, A.; O’Connor, E.; Piya, A.; Buckler, B.; Bose, H.S. Novel SCC Mutation in a Patient of Mexican Descent with Sex Reversal, Salt-Losing Crisis and Adrenal Failure. Endocrinol. Diabetes Metab. Case Rep. 2016, 2016, 16–59. [Google Scholar] [CrossRef]
- Zhao, X.; Su, Z.; Liu, X.; Song, J.; Gan, Y.; Wen, P.; Li, S.; Wang, L.; Pan, L. Long-term Follow-Up in a Chinese Child with Congenital Lipoid Adrenal Hyperplasia Due to a StAR Gene Mutation. BMC Endocr. Disord. 2018, 18, 78. [Google Scholar] [CrossRef]
- Kallali, W.; Gray, E.; Mehdi, M.Z.; Lindsay, R.; Metherell, L.A.; Buonocore, F.; Suntharalingham, J.P.; Achermann, J.C.; Donaldson, M. Long-term outcome of partial P450 side-chain cleavage enzyme deficiency in three brothers: The importance of early diagnosis. Eur. J. Endocrinol. 2020, 182, K15–K24. [Google Scholar] [CrossRef]
- Piya, A.; Kaur, J.; Rice, A.; Bose, H. De novo Disruption of Promoter and Exon 1 of STAR Gene Reveals Essential Role for Gonadal Development. Endocrinol. Diabetes Metab. Case Rep. 2017, 2017, 16–120. [Google Scholar] [CrossRef]
- Simard, J.; Ricketts, M.L.; Gingras, S.; Soucy, P.; Feltus, F.A.; Melner, M. Molecular Biology of the 3beta-hydroxysteroid dehydrogenase/delta5-delta4 Isomerase Gene Family. Endocr. Rev. 2005, 26, 525–582. [Google Scholar] [CrossRef] [PubMed]
- Levy-Shraga, Y.; Pinhas-Hamiel, O. High 17-hydroxyprogesterone Level in Newborn Screening Test for Congenital Adrenal Hyperplasia. BMJ Case Rep. 2016, 2016, bcr2015213939. [Google Scholar] [CrossRef] [PubMed]
- Bahíllo-Curieses, M.P.; Fernández de Trocóniz, L.L.; del Cañizo López, A.; Martínez-Sopena, M.J. Partial 3ß-hydroxysteroid Dehydrogenase Type 2 Deficiency: Diagnosis of a Novel Mutation After Positive Newborn Screening for 21-hydroxylase Deficiency. Med. Clin. (Barc.) 2016, 146, 92–93. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Herrera, F.; Espín, E.; Tito-Álvarez, A.; Beltrán, L.; Gómez-Correa, D.; Burgos, G.; Llamos, A.; Zurita, C.; Rojas, S.; Dueñas-Espín, I.; et al. A Report of Congenital Adrenal Hyperplasia Due to 17α-hydroxylase Deficiency in Two 46,XX Sisters. Gynecol. Endocrinol. 2020, 36, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Kater, C.E.; Biglieri, E.G. Disorders of steroid 17 alpha-hydroxylase deficiency. Endocrinol. Metab. Clin. N. Am. 1994, 23, 341–357. [Google Scholar] [CrossRef]
- Dhir, V.; Reisch, N.; Bleicken, C.M.; Lebl, J.; Kamrath, C.; Schwarz, H.P.; Grötzinger, J.; Sippell, W.G.; Riepe, F.G.; Arlt, W.; et al. Steroid 17alpha-hydroxylase deficiency: Functional characterization of four mutations (A174E, V178D, R440C, L465P) in the CYP17A1 gene. J. Clin. Endocrinol. Metab. 2009, 94, 3058–3064. [Google Scholar] [CrossRef]
- Auchus, R.J. Steroid 17-hydroxylase and 17,20-lyase Deficiencies, Genetic and Pharmacologic. J. Steroid Biochem. Mol. Biol. 2017, 165 Pt A, 71–78. [Google Scholar] [CrossRef]
- Zachmann, M.; Werder, E.A.; Prader, A. Two types of male pseudohermaphroditism due to 17, 20-desmolase deficiency. J. Clin. Endocrinol. Metab. 1982, 55, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, F.R.; Costin, G.; Goebelsmann, U.; Stanczyk, F.Z.; Zachmann, M. Male Pseudohermaphroditism due to 17,20-Desmolase Deficiency. J. Clin. Endocrinol. Metab. 1983, 57, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Zachmann, M.; Kempken, B.; Manella, B.; Navarro, E. Conversion from pure 17,20-desmolase- to combined 17,20-desmolase/17 alpha-hydroxylase deficiency with age. Acta Endocrinol. (Copenh.) 1992, 127, 97–99. [Google Scholar] [CrossRef]
- Zachmann, M. Endocrine findings in male pseudohermaphroditism. Eur. J. Pediatr. 1993, 152, S58–S61. [Google Scholar] [CrossRef]
- Miller, W. The Syndrome of 17,20 Lyase Deficiency. J. Clin. Endocrinol. Metab. 2012, 97, 59–67. [Google Scholar] [CrossRef]
- Hiort, O.; Marshall, L.; Birnbaum, W.; Wünsch, L.; Holterhus, P.M.; Döhnert, U.; Werner, R. Pubertal Development in17Beta-Hydroxysteroid Dehydrogenase Type 3 Deficiency. Horm. Res. Paediatr. 2017, 87, 354–358. [Google Scholar] [CrossRef]
- Sagsak, E.; Aycan, Z.; Savas-Erdeve, S.; Keskin, M.; Cetinkayaand, S.; Karaer, K. 17βHSD-3 enzyme deficiency due to novel mutations in the HSD17B3 gene diagnosed in a neonate. J. Ped. Endocrinol. Metab. 2015, 28, 7–8. [Google Scholar] [CrossRef]
- George, M.M.; New, M.I.; Ten, S.; Sultan, C.; Bhangoo, A. The Clinical and Molecular Heterogeneity of 17βHSD-3 Enzyme Deficiency. Horm. Res. Paediatr. 2010, 74, 229–240. [Google Scholar] [CrossRef]
- Goebelsmann, U.; Horton, R.; Mestman, J.H.; Arce, J.J.; Nagata, Y.; Nakamura, R.M.; Thorneycroft, I.H.; Mishell, D.R., Jr. Male pseudohermaphroditism due to testicular 17β-hydroxysteroid dehydrogenase deficiency. J. Clin. Endocrinol. Metab. 1973, 36, 867–879. [Google Scholar] [CrossRef]
- Imperato-McGinley, J.; Guerrero, L.; Gautier, T.; Peterson, R.E. Steroid 5α-Reductase Deficiency in Man: An Inherited Form of Male Pseudohermaphroditism. Science 1974, 186/4170, 1213–1215. [Google Scholar] [CrossRef]
- Imperato-McGinley, J.; Peterson, R.E.; Gautier, T.; Sturla, E. Male Pseudohermaphroditism Secondary to 5 Alpha-Reductase Deficiency—A Model for the Role of Androgens in Both the Development of the Male Phenotype and the Evolution of a Male Gender Identity. J. Steroid Biochem. 1979, 11/1 Pt 2, 637–645. [Google Scholar] [CrossRef]
- Walsh, P.C.; Madden, J.D.; Harrod, M.J.; Goldstein, J.L.; MacDonald, P.C.; Wilson, J.D. Familial incomplete male pseudohermaphroditism, type 2. Decreased dihydrotestosterone formation in pseudovaginal perineoscrotal hypospadias. N. Engl. J. Med. 1974, 291, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Misgar, R.A.; Bhat, M.H.; Masoodi, S.R.; Bashir, M.I.; Wani, A.I.; Baba, A.A.; Mufti, G.N.; Bhat, N.A. Disorders of Sex Development: A 10 Years Experience with 73 Cases from the Kashmir Valley. Indian J. Endocrinol. Metab. 2019, 23, 575–579. [Google Scholar] [CrossRef]
- Kumar, G.; Barboza-Meca, J.J. 5 Alpha Reductase Deficiency; StatPearls Publishing: Treasure Island, San Francisco, FL, USA, 2020. [Google Scholar]
- Sasaki, G.; Ishii, T.; Hori, N.; Amano, N.; Homma, K.; Sato, S.; Hasegawa, T. Effects of pre- and post-pubertal dihydrotestosterone treatment on penile length in 5α-reductase type 2 deficiency. Endocr. J. 2019, 66, 837–842. [Google Scholar] [CrossRef]
- Morris, J.M. The syndrome of testicular feminization in male pseudohermaphrodites. Am. J. Obstet. Gynecol. 1953, 65, 1192–1211. [Google Scholar] [CrossRef]
- Reifenstein, E.C., Jr. Hereditary familiar hypogonadism. Proc. Am. Fed. Clin. Res. 1947, 3, 86–89. [Google Scholar]
- Gilbert-Dreyfus, S.; Sebaoum, C.I.A.; Belaisch, J. Etude d’un cas familial d’androgynoïdisme avec hypospadias grave, gynécomastie et hyperoestrogénie (Case of familial androgynism with severe hypospadias, gynecomastia and excessive estrogen production). Ann. Endocrinol. (Paris) 1957, 18, 93–101. [Google Scholar]
- Rosewater, S.; Gwinup, G.; Hamwi, G. Familial Gynecomastia. Ann. Int. Med. 1965. [Google Scholar] [CrossRef]
- Amrhein, J.A.; Meyer, W.J., 3rd; Jones, H.W., Jr.; Migeon, C.J. Androgen insensitivity in man: Evidence for genetic heterogeneity. Proc. Natl. Acad. Sci. USA 1976, 73, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Rodprasert, W.; Virtanen, H.E.; Mäkelä, J.A.; Toppari, J. Hypogonadism and Cryptorchidism. Front. Endocrinol. (Lausanne) 2020, 10, 906. [Google Scholar] [CrossRef]
- Liu, Q.; Yin, X.; Li, P. Clinical, hormonal and genetic characteristics of androgen insensitivity syndrome in 39 Chinese patients. Reprod. Biol. Endocrinol. 2020, 18, 34. [Google Scholar] [CrossRef]
- Hutson, J.M.; Feingold, K.R.; Anawalt, B.; Boyce, A.; Chrousos, G.; Dungan, K. (Eds.) Cryptorchidism and Hypospadias; Endotext (Internet); MDText.com, Inc.: Dartmouth, MA, USA, 2018; NBK279106. [Google Scholar] [PubMed]
- Li, L.; Su, C.; Fan, L.; Gao, F.; Liang, X.; Gong, C. Clinical and molecular spectrum of 46,XY disorders of sex development that harbour MAMLD1 variations: Case series and review of literature. Orphanet J. Rare Dis. 2020, 15, 188. [Google Scholar] [CrossRef]
- Donaire, A.E.; Mendez, M.D. Hypospadias; StatPearls Publishing: Treasure Island, San Francisco, FL, USA, 2020; NBK482122. [Google Scholar] [PubMed]
- Unal, E.; Yıldırım, R.; Tekin, S.; Demir, V.; Onay, H.; Haspolat, Y.K. A Novel Mutation of AMHR2 in Two Siblings with Persistent Müllerian Duct Syndrome. J. Clin. Res. Pediatr. Endocrinol. 2018, 10, 387–390. [Google Scholar] [CrossRef]
- Marsh, C.A.; Auchus, R.J. Fertility in Patients with Genetic Deficiencies of Cytochrome P450c17 (CYP17A1): Combined 17-hydroxylase/17,20-lyase Deficiency and Isolated 17,20-lyase Deficiency. Fertil. Steril. 2014, 101, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Yatsuga, S.; Amano, N.; Nakamura-Utsunomiya, A.; Kobayashi, H.; Takasawa, K.; Nagasaki, K.; Nakamura, A.; Nishigaki, S.; Numakura, C.; Fujiwara, I.; et al. Clinical Characteristics of Cytochrome P450 Oxidoreductase Deficiency: A Nationwide Survey in Japan. Endocr. J. 2020. [Google Scholar] [CrossRef]
- Papadakis, G.E.; Dumont, A.; Bouligand, J.; Chasseloup, F.; Raggi, A.; Catteau-Jonard, S.; Boute-Benejean, O.; Pitteloud, N.; Young, J.; Dewailly, D.; et al. Non-classic cytochrome P450 oxidoreductase deficiency strongly linked with menstrual cycle disorders and female infertility as primary manifestations. Hum. Reprod. 2020, 35, 939–949. [Google Scholar] [CrossRef]
- Parween, S.; Fernández-Cancio, M.; Benito-Sanz, S.; Camats, N.; Rojas Velazquez, M.N.; López-Siguero, J.P.; Udhane, S.S.; Kagawa, N.; Flück, C.E.; Audí, L.; et al. Molecular Basis of CYP19A1 Deficiency in a 46,XX Patient with R550W Mutation in POR: Expanding the PORD Phenotype. J. Clin. Endocrinol. Metab. 2020, 105, dgaa076. [Google Scholar] [CrossRef]
- Hagenfeldt, K.; Janson, P.O.; Holmdahl, G.; Falhammar, H.; Filipsson, H.; Frisen, L.; Thorén, M.; Nordenskjöld, A. Fertility and pregnancy outcome in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum. Reprod. 2008, 23, 1607–1613. [Google Scholar] [CrossRef]
- Claahsen-van der Grinten, H.L.; Stikkelbroeck, N.M.; Sweep, C.G.; Hermus, A.R.; Otten, B.J. Fertility in patients with congenital adrenal hyperplasia. J. Pediatr. Endocrinol. Metab. 2006, 19, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Simm, P.J.; Zacharin, M.R. Successful pregnancy in a patient with severe 11-beta-hydroxylase deficiency and novel mutations in CYP11B1 gene. Horm. Res. 2007, 68, 294–297. [Google Scholar] [CrossRef]
- Levran, D.; Ben-Shlomo, I.; Pariente, C.; Dor, J.; Mashiach, S.; Weissman, A. Familial partial 17,20-desmolase and 17alpha-hydroxylase deficiency presenting as infertility. J. Assist. Reprod. Genet. 2003, 20, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, P.H.; Gouveia, G.R.; Costa, E.M.; Domenice, S.; Martin, R.M.; de Carvalho, L.C.; Pelaes, T.; Inacio, M.; Rocha Codarin, R.; Sator de Faria, M.B.; et al. Successful Live Birth in a Woman With 17α-Hydroxylase Deficiency Through IVF Frozen-Thawed Embryo Transfer. J. Clin. Endocrinol. Metab. 2016, 101, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Ben-Nun, I.; Siegal, A.; Shulman, A.; Ghetler, Y.; Kaneti, H.; Lunenfeld, B.; Beyth, Y.; Fejgin, M. Induction of artificial endometrial cycles with oestradiol implants and injectable progesterone: Establishment of a viable pregnancy in a woman with 17-alpha-hydroxylase deficiency. Hum. Reprod. 1995, 10, 2456–2458. [Google Scholar] [CrossRef] [PubMed]
- Albarel, F.; Perrin, J.; Jegaden, M.; Roucher-Boulez, F.; Reynaud, R.; Brue, T.; Courbiere, B. Successful IVF pregnancy despite inadequate ovarian steroidogenesis due to congenital lipoid adrenal hyperplasia (CLAH): A case report. Hum. Reprod. 2016, 31, 2609–2612. [Google Scholar] [CrossRef]
- Shozu, M.; Akasofu, K.; Harada, T.; Kubota, Y. A new cause of female pseudohermaphroditism: Placental aromatase deficiency. J. Clin. Endocrinol. Metab. 1991, 72, 560–566. [Google Scholar] [CrossRef]
- Alsaleem, M.; Miller, D.E.; Saadeh, L.; Majumdar, I. Aromatase deficiency: A rare cause of maternal virilisation and ambiguous genitalia in neonates. BMJ Case Rep. 2019, 12, e231267. [Google Scholar] [CrossRef] [PubMed]
- Praveen, V.P.; Ladjouze, A.; Sauter, K.S.; Pulickal, A.; Katharopoulos, E.; Trippel, M.; Perren, A.; Pandey, A.V.; Flück, C.E. Novel CYP19A1 Mutations Extend the Genotype-Phenotype Correlation and Reveal the Impact on Ovarian Function. J. Endocr. Soc. 2020, 4, bvaa030. [Google Scholar] [CrossRef]
- Sonne, J.; Lopez-Ojeda, W. Kallmann Syndrome; StatPearls Publishing: Treasure Island, San Francisco, FL, USA, 2020. [Google Scholar]
- Kallmann, F.; Schoenfeld, W.A.; Barrera, S.E. The genetic aspects of primary eunuchoidism. Am. J. Ment. Defic. 1944, 48, 203–236. [Google Scholar]
- Maestre de San Juan, A. Teratologia: Falta total de los nervios oplfatorios con anosmia en un individuo en quien existia una atrofia congenita de los testículos y miembro viril. El Siglo Medico 1856, 3, 211–221. [Google Scholar]
- Meczekalski, B.; Podfigurna-Stopa, A.; Smolarczyk, R.; Katulski, K.; Genazzani, A.R. Kallmann Syndrome in Women: From Genes to Diagnosis and Treatment. Gynecol. Endocrinol. 2013, 29, 296–300. [Google Scholar] [CrossRef]
- Laitinen, E.M.; Vaaralahti, K.; Tommiska, J.; Eklund, E.; Tervaniemi, M.; Valanne, L.; Raivio, T. Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland. Orphanet J. Rare Dis. 2011, 6, 41. [Google Scholar] [CrossRef]
- Rawson, N.E.; Brand, J.G.; Cowart, B.J.; Lowry, L.D.; Pribitkin, E.A.; Rao, V.M.; Restrepo, D. Functionally Mature Olfactory Neurons from Two Anosmic Patients With Kallmann Syndrome. Brain Res. 1995, 681, 58–64. [Google Scholar] [CrossRef]
- Boehm, U.; Bouloux, P.M.; Dattani, M.T.; de Roux, N.; Dodé, C.; Dunkel, L.; Dwyer, A.A.; Giacobini, P.; Hardelin, J.P.; Juul, A.; et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism—Pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2015, 11, 547–564. [Google Scholar] [CrossRef]
- Kim, S.H. Congenital Hypogonadotropic Hypogonadism and Kallmann Syndrome: Past, Present, and Future. Endocrinol. Metab. (Seoul) 2015, 30, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Lima Amato, L.G.; Latronico, A.C.; Gontijo Silveira, L.F. Molecular and Genetic Aspects of Congenital Isolated Hypogonadotropic Hypogonadism. Endocrinol. Metab. Clin. N. Am. 2017, 46, 283–303. [Google Scholar] [CrossRef]
- Stamou, M.I.; Plummer, L.; Galli-Tsinopoulou, A.; Stergidou, D.; Koika, V.; Georgopoulos, N.A. Unilateral Renal Agenesis as an early marker for genetic screening in Kallmann Syndrome. Hormones (Athens) 2019, 18, 103–105. [Google Scholar] [CrossRef]
- Iolascon, G.; Frizzi, L.; Bianco, M.; Gimigliano, F.; Palumbo, V.; Sinisi, A.M.; Sinisi, A.A. Bone involvement in males with Kallmann disease. Aging Clin. Exp. Res. 2015, 27, 31–36. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| ANOMALIES IN SEX DETERMINATION (Chromosomes and Gonads) (Sex chromosome, 46,XY, or 46,XX DSDs in the Chicago Classification) | ANOMALIES IN SEX DIFFERENTIATION (Hormones and Enzymes) (Always 46,XX or 46,XY DSDs in the Chicago Classification) | ||
|---|---|---|---|
| A. Without sex ambiguity (usually) | B. With sex ambiguity (usually) | A. Male Ps (non-dysgenetic Ps) or male (XY) with androgen deficiency (MAD) (if 46,XY and with testes) | B. Female Ps or female (XX) with androgen excess (FAE) (if 46XX and with ovaries) |
| 1. Gonadal dysgenesis: (a) Ovarian agenesis (gonadal agenesis) (b) Gonadal dysgenesis with chromosomal and/or phenotypic alteration: Turner syndrome (c) Ovarian dysgenesis or hypoplasia | 1. Testicular dysgenesis (XY): (a) Dysgenetic male Ps or partial gonadal dysgenesis. (b) Mixed gonadal dysgenesis | 1. Gonadotropin-resistant testes and Fetal gonadotropic deficiency (Leydig cell hypoplasia) | 1. Congenital adrenal hyperplasia in female/adrenogenital syndrome: (1) 17-α-hydroxilase deficiency (CYP17A1) (2) Cytochrome P450-oxidoreductase deficiency (PORD) (3) 21-hydroxylase deficiency (P450c21) (4) 11-β-hydroxylase deficiency (P450c11) |
| 2. Variants of gonadal dysgenesis: (a) Mosaic variants(b) Structural abnormalities of the second sex chromosome(c) Pure gonadal dysgenesis. Swyer syndrome | 2. True hermaphroditism or Ovotesticular disorders, OT-DSD | 2. Deficiencies in the testicle itself or its secretions, including: (1) Embryonic testicular regression or testicular regression syndrome (anorquia) (2) Disorders of androgen production, i.e., male Ps or MAD due to blockage in steroidogenesis by enzyme deficiencies | 2. Hormone therapy, iatrogenic |
| 3. Triple X constitution and other polysomies | 3. Mutations in the NR5A1 gene/SF-1 | 3. Defects in androgenic action or androgenic insensitivity syndromes (AIS) due to 5α-reductase deficiency or disorders in androgen receptor function | 3. Maternal virilizing tumor |
| 4. Sex reversal, males XX | 4. Other mild forms of male Ps, or unambiguous (infertile male, cryptorchidism, hypospadias, persistent Müllerian duct syndrome). | 4. Aromatase deficiency (P450arom) | |
| 5. Klinefelter syndrome, males | |||
| 6. Dysgenetic infertility | C. Without Ps: Congenital hypogonadotropic hypogonadism (CHH) (Kallmann syndrome, males and females) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acién, P.; Acién, M. Disorders of Sex Development: Classification, Review, and Impact on Fertility. J. Clin. Med. 2020, 9, 3555. https://doi.org/10.3390/jcm9113555
Acién P, Acién M. Disorders of Sex Development: Classification, Review, and Impact on Fertility. Journal of Clinical Medicine. 2020; 9(11):3555. https://doi.org/10.3390/jcm9113555
Chicago/Turabian StyleAcién, Pedro, and Maribel Acién. 2020. "Disorders of Sex Development: Classification, Review, and Impact on Fertility" Journal of Clinical Medicine 9, no. 11: 3555. https://doi.org/10.3390/jcm9113555
APA StyleAcién, P., & Acién, M. (2020). Disorders of Sex Development: Classification, Review, and Impact on Fertility. Journal of Clinical Medicine, 9(11), 3555. https://doi.org/10.3390/jcm9113555