The Genetics of Pituitary Adenomas



Abstract
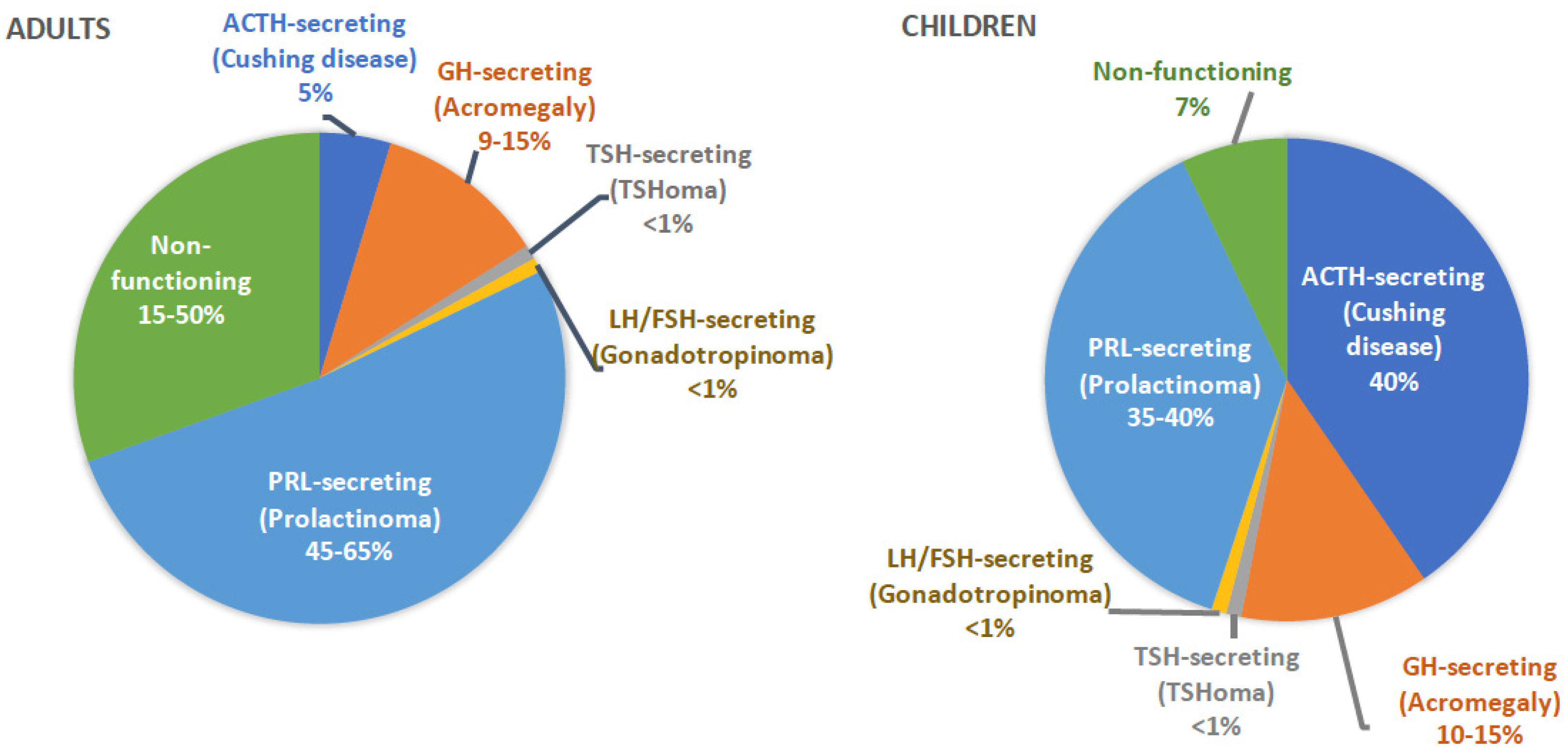
1. Introduction
2. Germline Defect and Associated Syndromes
2.1. Familial Isolated Pituitary Adenomas (FIPA)
2.2. Multiple Endocrine Neoplasia Syndromes
2.3. Carney Complex
2.4. McCune-Albright Syndrome (MAS)
2.5. X-Linked Acrogigantism (X-LAG)
2.6. 3 P Association (3PAs)
2.7. DICER1
2.8. Tuberous Sclerosis (TSC)
2.9. Less Common Germline Genetic Defects Potentially Associated with PAs
3. Somatic Changes
3.1. Ubiquitin Specific Peptidase 8 (USP8)
3.2. USP48 and BRAF
3.3. GNAS
3.4. The Phosphoinositide 3-Kinase (PI3K)/AKT Pathway
3.5. The p53 Tumor Suppressor Gene
3.6. Copy Number Variations (CNVS) at the Chromosomal Level
4. Other Genomic and/or Molecular Events Associated with Various Features of PAs
4.1. Epigenetic Modifications of Pituitary Adenomas
4.2. miRNAs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lim, C.T.; Korbonits, M. Update on the Clinicopathology of Pituitary Adenomas. Endocr. Pract. 2018, 24, 473–488. [Google Scholar] [CrossRef]
- Ezzat, S.; Asa, S.L.; Couldwell, W.T.; Barr, C.E.; Dodge, W.E.; Vance, M.L.; McCutcheon, I.E. The prevalence of pituitary adenomas: A systematic review. Cancer 2004, 101, 613–619. [Google Scholar] [CrossRef]
- Souteiro, P.; Maia, R.; Santos-Silva, R.; Figueiredo, R.; Costa, C.; Belo, S.; Castro-Correia, C.; Carvalho, D.; Fontoura, M. Pituitary incidentalomas in paediatric age are different from those described in adulthood. Pituitary 2019, 22, 124–128. [Google Scholar] [CrossRef]
- Freda, P.U.; Beckers, A.M.; Katznelson, L.; Molitch, M.E.; Montori, V.M.; Post, K.D.; Vance, M.L.; Endocrine, S. Pituitary incidentaloma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 894–904. [Google Scholar] [CrossRef]
- Perry, A.; Graffeo, C.S.; Marcellino, C.; Pollock, B.E.; Wetjen, N.M.; Meyer, F.B. Pediatric Pituitary Adenoma: Case Series, Review of the Literature, and a Skull Base Treatment Paradigm. J. Neurol. Surg. B Skull Base 2018, 79, 91–114. [Google Scholar] [CrossRef]
- Daly, A.F.; Rixhon, M.; Adam, C.; Dempegioti, A.; Tichomirowa, M.A.; Beckers, A. High prevalence of pituitary adenomas: A cross-sectional study in the province of Liege, Belgium. J. Clin. Endocrinol. Metab. 2006, 91, 4769–4775. [Google Scholar] [CrossRef]
- Stiles, C.E.; Korbonits, M. Familial Isolated Pituitary Adenoma. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; South Dartmouth: Dartmouth, MA, USA, 2000. [Google Scholar]
- Vierimaa, O.; Georgitsi, M.; Lehtonen, R.; Vahteristo, P.; Kokko, A.; Raitila, A.; Tuppurainen, K.; Ebeling, T.M.; Salmela, P.I.; Paschke, R.; et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 2006, 312, 1228–1230. [Google Scholar] [CrossRef]
- Daly, A.F.; Vanbellinghen, J.F.; Khoo, S.K.; Jaffrain-Rea, M.L.; Naves, L.A.; Guitelman, M.A.; Murat, A.; Emy, P.; Gimenez-Roqueplo, A.P.; Tamburrano, G.; et al. Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: Analysis in 73 families. J. Clin. Endocrinol. Metab. 2007, 92, 1891–1896. [Google Scholar] [CrossRef]
- Iwata, T.; Yamada, S.; Mizusawa, N.; Golam, H.M.; Sano, T.; Yoshimoto, K. The aryl hydrocarbon receptor-interacting protein gene is rarely mutated in sporadic GH-secreting adenomas. Clin. Endocrinol. (Oxf.) 2007, 66, 499–502. [Google Scholar] [CrossRef]
- Hernandez-Ramirez, L.C.; Gabrovska, P.; Denes, J.; Stals, K.; Trivellin, G.; Tilley, D.; Ferrau, F.; Evanson, J.; Ellard, S.; Grossman, A.B.; et al. Landscape of Familial Isolated and Young-Onset Pituitary Adenomas: Prospective Diagnosis in AIP Mutation Carriers. J. Clin. Endocrinol. Metab. 2015, 100, E1242–E1254. [Google Scholar] [CrossRef]
- Schofl, C.; Honegger, J.; Droste, M.; Grussendorf, M.; Finke, R.; Plockinger, U.; Berg, C.; Willenberg, H.S.; Lammert, A.; Klingmuller, D.; et al. Frequency of AIP gene mutations in young patients with acromegaly: A registry-based study. J. Clin. Endocrinol. Metab. 2014, 99, E2789–E2793. [Google Scholar] [CrossRef][Green Version]
- Georgitsi, M.; De Menis, E.; Cannavo, S.; Makinen, M.J.; Tuppurainen, K.; Pauletto, P.; Curto, L.; Weil, R.J.; Paschke, R.; Zielinski, G.; et al. Aryl hydrocarbon receptor interacting protein (AIP) gene mutation analysis in children and adolescents with sporadic pituitary adenomas. Clin. Endocrinol. (Oxf.) 2008, 69, 621–627. [Google Scholar] [CrossRef]
- Daly, A.; Cano, D.A.; Venegas, E.; Petrossians, P.; Dios, E.; Castermans, E.; Flores-Martinez, A.; Bours, V.; Beckers, A.; Soto, A. AIP and MEN1 mutations and AIP immunohistochemistry in pituitary adenomas in a tertiary referral center. Endocr. Connect. 2019, 8, 338–348. [Google Scholar] [CrossRef]
- Tichomirowa, M.A.; Barlier, A.; Daly, A.F.; Jaffrain-Rea, M.L.; Ronchi, C.; Yaneva, M.; Urban, J.D.; Petrossians, P.; Elenkova, A.; Tabarin, A.; et al. High prevalence of AIP gene mutations following focused screening in young patients with sporadic pituitary macroadenomas. Eur. J. Endocrinol. 2011, 165, 509–515. [Google Scholar] [CrossRef]
- Tuominen, I.; Heliovaara, E.; Raitila, A.; Rautiainen, M.R.; Mehine, M.; Katainen, R.; Donner, I.; Aittomaki, V.; Lehtonen, H.J.; Ahlsten, M.; et al. AIP inactivation leads to pituitary tumorigenesis through defective Galphai-cAMP signaling. Oncogene 2015, 34, 1174–1184. [Google Scholar] [CrossRef]
- Daly, A.F.; Tichomirowa, M.A.; Petrossians, P.; Heliovaara, E.; Jaffrain-Rea, M.L.; Barlier, A.; Naves, L.A.; Ebeling, T.; Karhu, A.; Raappana, A.; et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: An international collaborative study. J. Clin. Endocrinol. Metab. 2010, 95, E373–E383. [Google Scholar] [CrossRef]
- Caimari, F.; Hernandez-Ramirez, L.C.; Dang, M.N.; Gabrovska, P.; Iacovazzo, D.; Stals, K.; Ellard, S.; Korbonits, M.; International, F.C. Risk category system to identify pituitary adenoma patients with AIP mutations. J. Med. Genet. 2018, 55, 254–260. [Google Scholar] [CrossRef]
- Naves, L.A.; Jaffrain-Rea, M.L.; Vencio, S.A.; Jacomini, C.Z.; Casulari, L.A.; Daly, A.F.; Beckers, A. Aggressive prolactinoma in a child related to germline mutation in the ARYL hydrocarbon receptor interacting protein (AIP) gene. Arq. Bras. Endocrinol. Metabol. 2010, 54, 761–767. [Google Scholar] [CrossRef]
- Joshi, K.; Daly, A.F.; Beckers, A.; Zacharin, M. Resistant Paediatric Somatotropinomas due to AIP Mutations: Role of Pegvisomant. Horm. Res. Paediatr. 2018, 90, 196–202. [Google Scholar] [CrossRef]
- Personnier, C.; Cazabat, L.; Bertherat, J.; Gaillard, S.; Souberbielle, J.C.; Habrand, J.L.; Dufour, C.; Clauser, E.; SainteRose, C.; Polak, M. Clinical features and treatment of pediatric somatotropinoma: Case study of an aggressive tumor due to a new AIP mutation and extensive literature review. Horm. Res. Paediatr. 2011, 75, 392–402. [Google Scholar] [CrossRef]
- Dutta, P.; Reddy, K.S.; Rai, A.; Madugundu, A.K.; Solanki, H.S.; Bhansali, A.; Radotra, B.D.; Kumar, N.; Collier, D.; Iacovazzo, D.; et al. Surgery, Octreotide, Temozolomide, Bevacizumab, Radiotherapy, and Pegvisomant Treatment of an AIP MutationPositive Child. J. Clin. Endocrinol. Metab. 2019, 104, 3539–3544. [Google Scholar] [CrossRef]
- Falchetti, A.; Marini, F.; Luzi, E.; Tonelli, F.; Brandi, M.L. Multiple endocrine neoplasms. Best Pract. Res. Clin. Rheumatol. 2008, 22, 149–163. [Google Scholar] [CrossRef]
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Collins, F.S.; Emmert-Buck, M.R.; Debelenko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A.; et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276, 404–407. [Google Scholar] [CrossRef]
- De Laat, J.M.; Dekkers, O.M.; Pieterman, C.R.; Kluijfhout, W.P.; Hermus, A.R.; Pereira, A.M.; van der Horst-Schrivers, A.N.; Drent, M.L.; Bisschop, P.H.; Havekes, B.; et al. Long-Term Natural Course of Pituitary Tumors in Patients With MEN1: Results From the DutchMEN1 Study Group (DMSG). J. Clin. Endocrinol. Metab. 2015, 100, 3288–3296. [Google Scholar] [CrossRef]
- Verges, B.; Boureille, F.; Goudet, P.; Murat, A.; Beckers, A.; Sassolas, G.; Cougard, P.; Chambe, B.; Montvernay, C.; Calender, A. Pituitary disease in MEN type 1 (MEN1): Data from the France-Belgium MEN1 multicenter study. J. Clin. Endocrinol. Metab. 2002, 87, 457–465. [Google Scholar] [CrossRef]
- Trouillas, J.; Labat-Moleur, F.; Sturm, N.; Kujas, M.; Heymann, M.F.; Figarella-Branger, D.; Patey, M.; Mazucca, M.; Decullier, E.; Verges, B.; et al. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): A case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am. J. Surg. Pathol. 2008, 32, 534–543. [Google Scholar] [CrossRef]
- Giusti, F.; Cianferotti, L.; Boaretto, F.; Cetani, F.; Cioppi, F.; Colao, A.; Davi, M.V.; Faggiano, A.; Fanciulli, G.; Ferolla, P.; et al. Multiple endocrine neoplasia syndrome type 1: Institution, management, and data analysis of a nationwide multicenter patient database. Endocrine 2017, 58, 349–359. [Google Scholar] [CrossRef]
- Rix, M.; Hertel, N.T.; Nielsen, F.C.; Jacobsen, B.B.; Hoejberg, A.S.; Brixen, K.; Hangaard, J.; Kroustrup, J.P. Cushing’s disease in childhood as the first manifestation of multiple endocrine neoplasia syndrome type 1. Eur. J. Endocrinol. 2004, 151, 709–715. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Schussheim, D.H.; Freedman, S.M.; Keil, M.F.; Pack, S.D.; Agarwal, S.K.; Skarulis, M.C.; Weil, R.J.; Lubensky, I.A.; Zhuang, Z.; et al. Pituitary macroadenoma in a 5-year-old: An early expression of multiple endocrine neoplasia type 1. J. Clin. Endocrinol. Metab. 2000, 85, 4776–4780. [Google Scholar] [CrossRef]
- Makri, A.; Bonella, M.B.; Keil, M.F.; Hernandez-Ramirez, L.; Paluch, G.; Tirosh, A.; Saldarriaga, C.; Chittiboina, P.; Marx, S.J.; Stratakis, C.A.; et al. Children with MEN1 gene mutations may present first (and at a young age) with Cushing disease. Clin. Endocrinol. (Oxf.) 2018, 89, 437–443. [Google Scholar] [CrossRef]
- Salenave, S.; Ancelle, D.; Bahougne, T.; Raverot, G.; Kamenicky, P.; Bouligand, J.; Guiochon-Mantel, A.; Linglart, A.; Souchon, P.F.; Nicolino, M.; et al. Macroprolactinomas in children and adolescents: Factors associated with the response to treatment in 77 patients. J. Clin. Endocrinol. Metab. 2015, 100, 1177–1186. [Google Scholar] [CrossRef]
- Wenbin, C.; Asai, A.; Teramoto, A.; Sanno, N.; Kirino, T. Mutations of the MEN1 tumor suppressor gene in sporadic pituitary tumors. Cancer Lett. 1999, 142, 43–47. [Google Scholar] [CrossRef]
- Zhuang, Z.; Ezzat, S.Z.; Vortmeyer, A.O.; Weil, R.; Oldfield, E.H.; Park, W.S.; Pack, S.; Huang, S.; Agarwal, S.K.; Guru, S.C.; et al. Mutations of the MEN1 tumor suppressor gene in pituitary tumors. Cancer Res. 1997, 57, 5446–5451. [Google Scholar]
- Mulligan, L.M.; Kwok, J.B.; Healey, C.S.; Elsdon, M.J.; Eng, C.; Gardner, E.; Love, D.R.; Mole, S.E.; Moore, J.K.; Papi, L.; et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993, 363, 458–460. [Google Scholar] [CrossRef]
- Traugott, A.L.; Moley, J.F. The RET Protooncogene. Cancer Treat Res. 2010, 153, 303–319. [Google Scholar]
- Saito, T.; Miura, D.; Taguchi, M.; Takeshita, A.; Miyakawa, M.; Takeuchi, Y. Coincidence of multiple endocrine neoplasia type 2A with acromegaly. Am. J. Med. Sci. 2010, 340, 329–331. [Google Scholar] [CrossRef]
- Heinlen, J.E.; Buethe, D.D.; Culkin, D.J.; Slobodov, G. Multiple endocrine neoplasia 2a presenting with pheochromocytoma and pituitary macroadenoma. ISRN Oncol. 2011, 2011, 732452. [Google Scholar] [CrossRef][Green Version]
- Ezzat, T.; Paramesawaran, R.; Phillips, B.; Sadler, G. MEN 2 syndrome masquerading as MEN 1. Ann. R Coll. Surg. Engl. 2012, 94, e206–e207. [Google Scholar] [CrossRef]
- Heliovaara, E.; Tuupanen, S.; Ahlsten, M.; Hodgson, S.; de Menis, E.; Kuismin, O.; Izatt, L.; McKinlay Gardner, R.J.; Gundogdu, S.; Lucassen, A.; et al. No evidence of RET germline mutations in familial pituitary adenoma. J. Mol. Endocrinol. 2011, 46, 1–8. [Google Scholar] [CrossRef]
- Komminoth, P.; Roth, J.; Muletta-Feurer, S.; Saremaslani, P.; Seelentag, W.K.; Heitz, P.U. RET proto-oncogene point mutations in sporadic neuroendocrine tumors. J. Clin. Endocrinol. Metab. 1996, 81, 2041–2046. [Google Scholar]
- Yoshimoto, K.; Tanaka, C.; Moritani, M.; Shimizu, E.; Yamaoka, T.; Yamada, S.; Sano, T.; Itakura, M. Infrequent detectable somatic mutations of the RET and glial cell line-derived neurotrophic factor (GDNF) genes in human pituitary adenomas. Endocr. J. 1999, 46, 199–207. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Mateo, C.M.; Marx, S.J. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J. Clin. Endocrinol. Metab. 2009, 94, 1826–1834. [Google Scholar] [CrossRef]
- Georgitsi, M.; Raitila, A.; Karhu, A.; van der Luijt, R.B.; Aalfs, C.M.; Sane, T.; Vierimaa, O.; Makinen, M.J.; Tuppurainen, K.; Paschke, R.; et al. Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. J. Clin. Endocrinol. Metab. 2007, 92, 3321–3325. [Google Scholar] [CrossRef]
- Occhi, G.; Regazzo, D.; Trivellin, G.; Boaretto, F.; Ciato, D.; Bobisse, S.; Ferasin, S.; Cetani, F.; Pardi, E.; Korbonits, M.; et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS Genet. 2013, 9, e1003350. [Google Scholar] [CrossRef]
- Pellegata, N.S.; Quintanilla-Martinez, L.; Siggelkow, H.; Samson, E.; Bink, K.; Hofler, H.; Fend, F.; Graw, J.; Atkinson, M.J. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc. Natl. Acad. Sci. USA 2006, 103, 15558–15563. [Google Scholar] [CrossRef]
- Molatore, S.; Marinoni, I.; Lee, M.; Pulz, E.; Ambrosio, M.R.; degli Uberti, E.C.; Zatelli, M.C.; Pellegata, N.S. A novel germline CDKN1B mutation causing multiple endocrine tumors: Clinical, genetic and functional characterization. Hum. Mutat. 2010, 31, E1825–E1835. [Google Scholar] [CrossRef]
- Frederiksen, A.; Rossing, M.; Hermann, P.; Ejersted, C.; Thakker, R.V.; Frost, M. Clinical Features of Multiple Endocrine Neoplasia Type 4: Novel Pathogenic Variant and Review of Published Cases. J. Clin. Endocrinol. Metab. 2019, 104, 3637–3646. [Google Scholar] [CrossRef]
- Tichomirowa, M.A.; Lee, M.; Barlier, A.; Daly, A.F.; Marinoni, I.; Jaffrain-Rea, M.L.; Naves, L.A.; Rodien, P.; Rohmer, V.; Faucz, F.R.; et al. Cyclin-dependent kinase inhibitor 1B (CDKN1B) gene variants in AIP mutation-negative familial isolated pituitary adenoma kindreds. Endocr. Relat. Cancer 2012, 19, 233–241. [Google Scholar] [CrossRef]
- Sambugaro, S.; Di Ruvo, M.; Ambrosio, M.R.; Pellegata, N.S.; Bellio, M.; Guerra, A.; Buratto, M.; Foschini, M.P.; Tagliati, F.; degli Uberti, E.; et al. Early onset acromegaly associated with a novel deletion in CDKN1B 5’UTR region. Endocrine 2015, 49, 58–64. [Google Scholar] [CrossRef]
- Stratakis, C.A. Carney complex: A familial lentiginosis predisposing to a variety of tumors. Rev. Endocr. Metab. Disord. 2016, 17, 367–371. [Google Scholar] [CrossRef]
- Salpea, P.; Stratakis, C.A. Carney complex and McCune Albright syndrome: An overview of clinical manifestations and human molecular genetics. Mol. Cell. Endocrinol. 2014, 386, 85–91. [Google Scholar] [CrossRef]
- Kirschner, L.S.; Carney, J.A.; Pack, S.D.; Taymans, S.E.; Giatzakis, C.; Cho, Y.S.; Cho-Chung, Y.S.; Stratakis, C.A. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat. Genet. 2000, 26, 89–92. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Carney, J.A.; Lin, J.P.; Papanicolaou, D.A.; Karl, M.; Kastner, D.L.; Pras, E.; Chrousos, G.P. Carney complex, a familial multiple neoplasia and lentiginosis syndrome. Analysis of 11 kindreds and linkage to the short arm of chromosome 2. J. Clin. Investig. 1996, 97, 699–705. [Google Scholar] [CrossRef]
- Courcoutsakis, N.A.; Tatsi, C.; Patronas, N.J.; Lee, C.C.; Prassopoulos, P.K.; Stratakis, C.A. The complex of myxomas, spotty skin pigmentation and endocrine overactivity (Carney complex): Imaging findings with clinical and pathological correlation. Insights Imaging 2013, 4, 119–133. [Google Scholar] [CrossRef]
- Pack, S.D.; Kirschner, L.S.; Pak, E.; Zhuang, Z.; Carney, J.A.; Stratakis, C.A. Genetic and histologic studies of somatomammotropic pituitary tumors in patients with the “complex of spotty skin pigmentation, myxomas, endocrine overactivity and schwannomas” (Carney complex). J. Clin. Endocrinol. Metab. 2000, 85, 3860–3865. [Google Scholar]
- Boikos, S.A.; Stratakis, C.A. Pituitary pathology in patients with Carney Complex: Growth-hormone producing hyperplasia or tumors and their association with other abnormalities. Pituitary 2006, 9, 203–209. [Google Scholar] [CrossRef]
- Lonser, R.R.; Mehta, G.U.; Kindzelski, B.A.; Ray-Chaudhury, A.; Vortmeyer, A.O.; Dickerman, R.; Oldfield, E.H. Surgical Management of Carney Complex-Associated Pituitary Pathology. Neurosurgery 2017, 80, 780–786. [Google Scholar] [CrossRef]
- Kiefer, F.W.; Winhofer, Y.; Iacovazzo, D.; Korbonits, M.; Wolfsberger, S.; Knosp, E.; Trautinger, F.; Hoftberger, R.; Krebs, M.; Luger, A.; et al. PRKAR1A mutation causing pituitary-dependent Cushing disease in a patient with Carney complex. Eur. J. Endocrinol. 2017, 177, K7–K12. [Google Scholar] [CrossRef]
- Hernandez-Ramirez, L.C.; Tatsi, C.; Lodish, M.B.; Faucz, F.R.; Pankratz, N.; Chittiboina, P.; Lane, J.; Kay, D.M.; Valdes, N.; Dimopoulos, A.; et al. Corticotropinoma as a Component of Carney Complex. J. Endocr. Soc. 2017, 1, 918–925. [Google Scholar] [CrossRef]
- Kaltsas, G.A.; Kola, B.; Borboli, N.; Morris, D.G.; Gueorguiev, M.; Swords, F.M.; Czirjak, S.; Kirschner, L.S.; Stratakis, C.A.; Korbonits, M.; et al. Sequence analysis of the PRKAR1A gene in sporadic somatotroph and other pituitary tumours. Clin. Endocrinol. (Oxf.) 2002, 57, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, H.; Mizusawa, N.; Nagahiro, S.; Yamada, S.; Sano, T.; Itakura, M.; Yoshimoto, K. GH-secreting pituitary adenomas infrequently contain inactivating mutations of PRKAR1A and LOH of 17q23-24. Clin. Endocrinol. (Oxf.) 2003, 58, 464–470. [Google Scholar] [CrossRef]
- Dumitrescu, C.E.; Collins, M.T. McCune-Albright syndrome. Orphanet J. Rare Dis. 2008, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, L.S.; Shenker, A.; Gejman, P.V.; Merino, M.J.; Friedman, E.; Spiegel, A.M. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N. Engl. J. Med. 1991, 325, 1688–1695. [Google Scholar] [CrossRef]
- Akintoye, S.O.; Chebli, C.; Booher, S.; Feuillan, P.; Kushner, H.; Leroith, D.; Cherman, N.; Bianco, P.; Wientroub, S.; Robey, P.G.; et al. Characterization of gsp-mediated growth hormone excess in the context of McCune-Albright syndrome. J. Clin. Endocrinol. Metab. 2002, 87, 5104–5112. [Google Scholar] [CrossRef]
- Beckers, A.; Lodish, M.B.; Trivellin, G.; Rostomyan, L.; Lee, M.; Faucz, F.R.; Yuan, B.; Choong, C.S.; Caberg, J.H.; Verrua, E.; et al. X-linked acrogigantism syndrome: Clinical profile and therapeutic responses. Endocr. Relat. Cancer 2015, 22, 353–367. [Google Scholar] [CrossRef]
- Trivellin, G.; Daly, A.F.; Faucz, F.R.; Yuan, B.; Rostomyan, L.; Larco, D.O.; Schernthaner-Reiter, M.H.; Szarek, E.; Leal, L.F.; Caberg, J.H.; et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N. Engl. J. Med. 2014, 371, 2363–2374. [Google Scholar] [CrossRef]
- Daly, A.F.; Yuan, B.; Fina, F.; Caberg, J.H.; Trivellin, G.; Rostomyan, L.; de Herder, W.W.; Naves, L.A.; Metzger, D.; Cuny, T.; et al. Somatic mosaicism underlies X-linked acrogigantism syndrome in sporadic male subjects. Endocr. Relat. Cancer 2016, 23, 221–233. [Google Scholar] [CrossRef]
- Rostomyan, L.; Daly, A.F.; Petrossians, P.; Nachev, E.; Lila, A.R.; Lecoq, A.L.; Lecumberri, B.; Trivellin, G.; Salvatori, R.; Moraitis, A.G.; et al. Clinical and genetic characterization of pituitary gigantism: An international collaborative study in 208 patients. Endocr. Relat. Cancer 2015, 22, 745–757. [Google Scholar] [CrossRef]
- Naves, L.A.; Daly, A.F.; Dias, L.A.; Yuan, B.; Zakir, J.C.; Barra, G.B.; Palmeira, L.; Villa, C.; Trivellin, G.; Junior, A.J.; et al. Aggressive tumor growth and clinical evolution in a patient with X-linked acro-gigantism syndrome. Endocrine 2016, 51, 236–244. [Google Scholar] [CrossRef]
- Xekouki, P.; Szarek, E.; Bullova, P.; Giubellino, A.; Quezado, M.; Mastroyannis, S.A.; Mastorakos, P.; Wassif, C.A.; Raygada, M.; Rentia, N.; et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in humans and mice. J. Clin. Endocrinol. Metab. 2015, 100, E710–E719. [Google Scholar] [CrossRef]
- Moosavi, B.; Berry, E.A.; Zhu, X.L.; Yang, W.C.; Yang, G.F. The assembly of succinate dehydrogenase: A key enzyme in bioenergetics. Cell. Mol. Life Sci. 2019, 76, 4023–4042. [Google Scholar] [CrossRef]
- Dwight, T.; Mann, K.; Benn, D.E.; Robinson, B.G.; McKelvie, P.; Gill, A.J.; Winship, I.; Clifton-Bligh, R.J. Familial SDHA mutation associated with pituitary adenoma and pheochromocytoma/paraganglioma. J. Clin. Endocrinol. Metab. 2013, 98, E1103–E1108. [Google Scholar] [CrossRef]
- Denes, J.; Swords, F.; Rattenberry, E.; Stals, K.; Owens, M.; Cranston, T.; Xekouki, P.; Moran, L.; Kumar, A.; Wassif, C.; et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: Results from a large patient cohort. J. Clin. Endocrinol. Metab. 2015, 100, E531–E541. [Google Scholar] [CrossRef]
- Roszko, K.L.; Blouch, E.; Blake, M.; Powers, J.F.; Tischler, A.S.; Hodin, R.; Sadow, P.; Lawson, E.A. Case Report of a Prolactinoma in a Patient With a Novel MAX Mutation and Bilateral Pheochromocytomas. J. Endocr. Soc. 2017, 1, 1401–1407. [Google Scholar] [CrossRef]
- Guerrero-Perez, F.; Fajardo, C.; Torres Vela, E.; Gimenez-Palop, O.; Lisbona Gil, A.; Martin, T.; Gonzalez, N.; Diez, J.J.; Iglesias, P.; Robledo, M.; et al. 3P association (3PAs): Pituitary adenoma and pheochromocytoma/paraganglioma. A heterogeneous clinical syndrome associated with different gene mutations. Eur. J. Intern. Med. 2019, 69, 14–19. [Google Scholar] [CrossRef]
- Daly, A.F.; Castermans, E.; Oudijk, L.; Guitelman, M.A.; Beckers, P.; Potorac, I.; Neggers, S.; Sacre, N.; van der Lely, A.J.; Bours, V.; et al. Pheochromocytomas and pituitary adenomas in three patients with MAX exon deletions. Endocr. Relat. Cancer 2018, 25, L37–L42. [Google Scholar] [CrossRef]
- Solarski, M.; Rotondo, F.; Foulkes, W.D.; Priest, J.R.; Syro, L.V.; Butz, H.; Cusimano, M.D.; Kovacs, K. DICER1 gene mutations in endocrine tumors. Endocr. Relat. Cancer 2018, 25, R197–R208. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Priest, J.R.; Duchaine, T.F. DICER1: Mutations, microRNAs and mechanisms. Nat. Rev. Cancer 2014, 14, 662–672. [Google Scholar] [CrossRef]
- De Kock, L.; Sabbaghian, N.; Plourde, F.; Srivastava, A.; Weber, E.; Bouron-Dal Soglio, D.; Hamel, N.; Choi, J.H.; Park, S.H.; Deal, C.L.; et al. Pituitary blastoma: A pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol. 2014, 128, 111–122. [Google Scholar] [CrossRef]
- Tatsi, C.; Stratakis, C.A. Neonatal Cushing Syndrome: A Rare but Potentially Devastating Disease. Clin. Perinatol. 2018, 45, 103–118. [Google Scholar] [CrossRef]
- Schultz, K.A.P.; Williams, G.M.; Kamihara, J.; Stewart, D.R.; Harris, A.K.; Bauer, A.J.; Turner, J.; Shah, R.; Schneider, K.; Schneider, K.W.; et al. DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin. Cancer Res. 2018, 24, 2251–2261. [Google Scholar] [CrossRef]
- Cotton, E.; Ray, D. DICER1 mutation and pituitary prolactinoma. Endocrinol. Diabetes Metab. Case Rep. 2018, 2018. [Google Scholar] [CrossRef]
- Rosner, M.; Hanneder, M.; Siegel, N.; Valli, A.; Fuchs, C.; Hengstschlager, M. The mTOR pathway and its role in human genetic diseases. Mutat. Res. 2008, 659, 284–292. [Google Scholar] [CrossRef]
- Tigas, S.; Carroll, P.V.; Jones, R.; Bingham, E.; Russell-Jones, D.; Powell, M.; Scobie, I.N. Simultaneous Cushing’s disease and tuberous sclerosis; a potential role for TSC in pituitary ontogeny. Clin. Endocrinol. (Oxf.) 2005, 63, 694–695. [Google Scholar] [CrossRef]
- Nandagopal, R.; Vortmeyer, A.; Oldfield, E.H.; Keil, M.F.; Stratakis, C.A. Cushing’s syndrome due to a pituitary corticotropinoma in a child with tuberous sclerosis: An association or a coincidence? Clin. Endocrinol. (Oxf.) 2007, 67, 639–641. [Google Scholar] [CrossRef]
- Dworakowska, D.; Grossman, A.B. Are neuroendocrine tumours a feature of tuberous sclerosis? A systematic review. Endocr. Relat. Cancer 2009, 16, 45–58. [Google Scholar] [CrossRef]
- Regazzo, D.; Gardiman, M.P.; Theodoropoulou, M.; Scaroni, C.; Occhi, G.; Ceccato, F. Silent gonadotroph pituitary neuroendocrine tumor in a patient with tuberous sclerosis complex: Evaluation of a possible molecular link. Endocrinol. Diabetes Metab. Case Rep. 2018, 2018. [Google Scholar] [CrossRef]
- Cambiaso, P.; Galassi, S.; Palmiero, M.; Mastronuzzi, A.; Del Bufalo, F.; Capolino, R.; Cacchione, A.; Buonuomo, P.S.; Gonfiantini, M.V.; Bartuli, A.; et al. Growth hormone excess in children with neurofibromatosis type-1 and optic glioma. Am. J. Med. Genet. A 2017, 173, 2353–2358. [Google Scholar] [CrossRef]
- Sani, I.; Albanese, A. Endocrine Long-Term Follow-Up of Children with Neurofibromatosis Type 1 and Optic Pathway Glioma. Horm. Res. Paediatr. 2017, 87, 179–188. [Google Scholar] [CrossRef]
- Hozumi, K.; Fukuoka, H.; Odake, Y.; Takeuchi, T.; Uehara, T.; Sato, T.; Inoshita, N.; Yoshida, K.; Matsumoto, R.; Bando, H.; et al. Acromegaly caused by a somatotroph adenoma in patient with neurofibromatosis type 1. Endocr. J. 2019, 66, 853–857. [Google Scholar] [CrossRef]
- Hernandez-Ramirez, L.C.; Gam, R.; Valdes, N.; Lodish, M.B.; Pankratz, N.; Balsalobre, A.; Gauthier, Y.; Faucz, F.R.; Trivellin, G.; Chittiboina, P.; et al. Loss-of-function mutations in the CABLES1 gene are a novel cause of Cushing’s disease. Endocr. Relat. Cancer 2017, 24, 379–392. [Google Scholar] [CrossRef]
- Seemann, N.; Kuhn, D.; Wrocklage, C.; Keyvani, K.; Hackl, W.; Buchfelder, M.; Fahlbusch, R.; Paulus, W. CDKN2A/p16 inactivation is related to pituitary adenoma type and size. J. Pathol. 2001, 193, 491–497. [Google Scholar] [CrossRef]
- Kirsch, M.; Morz, M.; Pinzer, T.; Schackert, H.K.; Schackert, G. Frequent loss of the CDKN2C (p18INK4c) gene product in pituitary adenomas. Genes Chromosomes Cancer 2009, 48, 143–154. [Google Scholar] [CrossRef]
- Bellodi, C.; Krasnykh, O.; Haynes, N.; Theodoropoulou, M.; Peng, G.; Montanaro, L.; Ruggero, D. Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res. 2010, 70, 6026–6035. [Google Scholar] [CrossRef]
- Brioude, F.; Nicolas, C.; Marey, I.; Gaillard, S.; Bernier, M.; Das Neves, C.; Le Bouc, Y.; Touraine, P.; Netchine, I. Hypercortisolism due to a Pituitary Adenoma Associated with Beckwith-Wiedemann Syndrome. Horm. Res. Paediatr. 2016, 86, 206–211. [Google Scholar] [CrossRef]
- Marques, P.; Spencer, R.; Morrison, P.J.; Carr, I.M.; Dang, M.N.; Bonthron, D.T.; Hunter, S.; Korbonits, M. Cantu syndrome with coexisting familial pituitary adenoma. Endocrine 2018, 59, 677–684. [Google Scholar] [CrossRef]
- Syro, L.V.; Sundsbak, J.L.; Scheithauer, B.W.; Toledo, R.A.; Camargo, M.; Heyer, C.M.; Sekiya, T.; Uribe, H.; Escobar, J.I.; Vasquez, M.; et al. Somatotroph pituitary adenoma with acromegaly and autosomal dominant polycystic kidney disease: SSTR5 polymorphism and PKD1 mutation. Pituitary 2012, 15, 342–349. [Google Scholar] [CrossRef]
- De Menis, E.; Roncaroli, F.; Calvari, V.; Chiarini, V.; Pauletto, P.; Camerino, G.; Cremonini, N. Corticotroph adenoma of the pituitary in a patient with X-linked adrenal hypoplasia congenita due to a novel mutation of the DAX-1 gene. Eur. J. Endocrinol. 2005, 153, 211–215. [Google Scholar] [CrossRef]
- Reincke, M.; Sbiera, S.; Hayakawa, A.; Theodoropoulou, M.; Osswald, A.; Beuschlein, F.; Meitinger, T.; Mizuno-Yamasaki, E.; Kawaguchi, K.; Saeki, Y.; et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet. 2015, 47, 31–38. [Google Scholar] [CrossRef]
- Ma, Z.Y.; Song, Z.J.; Chen, J.H.; Wang, Y.F.; Li, S.Q.; Zhou, L.F.; Mao, Y.; Li, Y.M.; Hu, R.G.; Zhang, Z.Y.; et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015, 25, 306–317. [Google Scholar] [CrossRef]
- Perez-Rivas, L.G.; Theodoropoulou, M.; Ferrau, F.; Nusser, C.; Kawaguchi, K.; Stratakis, C.A.; Faucz, F.R.; Wildemberg, L.E.; Assie, G.; Beschorner, R.; et al. The Gene of the Ubiquitin-Specific Protease 8 Is Frequently Mutated in Adenomas Causing Cushing’s Disease. J. Clin. Endocrinol. Metab. 2015, 100, E997–E1004. [Google Scholar] [CrossRef]
- Faucz, F.R.; Tirosh, A.; Tatsi, C.; Berthon, A.; Hernandez-Ramirez, L.C.; Settas, N.; Angelousi, A.; Correa, R.; Papadakis, G.Z.; Chittiboina, P.; et al. Somatic USP8 Gene Mutations Are a Common Cause of Pediatric Cushing Disease. J. Clin. Endocrinol. Metab. 2017, 102, 2836–2843. [Google Scholar] [CrossRef]
- Ballmann, C.; Thiel, A.; Korah, H.E.; Reis, A.C.; Saeger, W.; Stepanow, S.; Kohrer, K.; Reifenberger, G.; Knobbe-Thomsen, C.B.; Knappe, U.J.; et al. USP8 Mutations in Pituitary Cushing Adenomas-Targeted Analysis by Next-Generation Sequencing. J. Endocr. Soc. 2018, 2, 266–278. [Google Scholar] [CrossRef]
- Losa, M.; Mortini, P.; Pagnano, A.; Detomas, M.; Cassarino, M.F.; Pecori Giraldi, F. Clinical characteristics and surgical outcome in USP8-mutated human adrenocorticotropic hormone-secreting pituitary adenomas. Endocrine 2019, 63, 240–246. [Google Scholar] [CrossRef]
- Albani, A.; Perez-Rivas, L.G.; Dimopoulou, C.; Zopp, S.; Colon-Bolea, P.; Roeber, S.; Honegger, J.; Flitsch, J.; Rachinger, W.; Buchfelder, M.; et al. The USP8 mutational status may predict long-term remission in patients with Cushing’s disease. Clin. Endocrinol. (Oxf.) 2018, 89, 454–458. [Google Scholar] [CrossRef]
- Cohen, M.; Persky, R.; Stegemann, R.; Hernandez-Ramirez, L.C.; Zeltser, D.; Lodish, M.B.; Chen, A.; Keil, M.F.; Tatsi, C.; Faucz, F.; et al. Germline USP8 mutation associated with pediatric Cushing disease and other clinical features: A new syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 4676–4682. [Google Scholar] [CrossRef]
- Chen, J.; Jian, X.; Deng, S.; Ma, Z.; Shou, X.; Shen, Y.; Zhang, Q.; Song, Z.; Li, Z.; Peng, H.; et al. Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat. Commun. 2018, 9, 3171. [Google Scholar] [CrossRef]
- Ronchi, C.L.; Peverelli, E.; Herterich, S.; Weigand, I.; Mantovani, G.; Schwarzmayr, T.; Sbiera, S.; Allolio, B.; Honegger, J.; Appenzeller, S.; et al. Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur. J. Endocrinol. 2016, 174, 363–372. [Google Scholar] [CrossRef]
- Freda, P.U.; Chung, W.K.; Matsuoka, N.; Walsh, J.E.; Kanibir, M.N.; Kleinman, G.; Wang, Y.; Bruce, J.N.; Post, K.D. Analysis of GNAS mutations in 60 growth hormone secreting pituitary tumors: Correlation with clinical and pathological characteristics and surgical outcome based on highly sensitive GH and IGF-I criteria for remission. Pituitary 2007, 10, 275–282. [Google Scholar] [CrossRef]
- Hayward, B.E.; Barlier, A.; Korbonits, M.; Grossman, A.B.; Jacquet, P.; Enjalbert, A.; Bonthron, D.T. Imprinting of the Gsα gene GNAS1 in the pathogenesis of acromegaly. J. Clin. Investig. 2001, 107, R31–R36. [Google Scholar] [CrossRef]
- Riminucci, M.; Collins, M.T.; Lala, R.; Corsi, A.; Matarazzo, P.; Gehron Robey, P.; Bianco, P. An R201H activating mutation of the GNAS1 (Gsalpha) gene in a corticotroph pituitary adenoma. Mol. Pathol. 2002, 55, 58–60. [Google Scholar] [CrossRef]
- Williamson, E.A.; Ince, P.G.; Harrison, D.; Kendall-Taylor, P.; Harris, P.E. G-protein mutations in human pituitary adrenocorticotrophic hormone-secreting adenomas. Eur. J. Clin. Investig. 1995, 25, 128–131. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, X.; Shen, Y.; Li, M.; Ma, H.; Xing, M.; Lu, Y. Frequent mutations and amplifications of the PIK3CA gene in pituitary tumors. Endocr. Relat. Cancer 2009, 16, 301–310. [Google Scholar] [CrossRef]
- Murat, C.B.; Braga, P.B.; Fortes, M.A.; Bronstein, M.D.; Correa-Giannella, M.L.; Giorgi, R.R. Mutation and genomic amplification of the PIK3CA proto-oncogene in pituitary adenomas. Braz. J. Med. Biol. Res. 2012, 45, 851–855. [Google Scholar] [CrossRef]
- Raverot, G.; Burman, P.; McCormack, A.; Heaney, A.; Petersenn, S.; Popovic, V.; Trouillas, J.; Dekkers, O.M.; European Society of Endocrinology. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. Eur. J. Endocrinol. 2018, 178, G1–G24. [Google Scholar] [CrossRef]
- Levy, A.; Hall, L.; Yeudall, W.A.; Lightman, S.L. P53 gene mutations in pituitary adenomas: Rare events. Clin. Endocrinol. (Oxf.) 1994, 41, 809–814. [Google Scholar] [CrossRef]
- Herman, V.; Drazin, N.Z.; Gonsky, R.; Melmed, S. Molecular screening of pituitary adenomas for gene mutations and rearrangements. J. Clin. Endocrinol. Metab. 1993, 77, 50–55. [Google Scholar]
- Kawashima, S.T.; Usui, T.; Sano, T.; Iogawa, H.; Hagiwara, H.; Tamanaha, T.; Tagami, T.; Naruse, M.; Hojo, M.; Takahashi, J.A.; et al. P53 gene mutation in an atypical corticotroph adenoma with Cushing’s disease. Clin. Endocrinol. (Oxf.) 2009, 70, 656–657. [Google Scholar] [CrossRef]
- Guo, F.; Wang, G.; Wang, F.; Xu, D.; Liu, X. Identification of Novel Genes Involved in the Pathogenesis of an ACTH-Secreting Pituitary Carcinoma: A Case Report and Literature Review. Front. Oncol. 2018, 8, 510. [Google Scholar] [CrossRef]
- Tanizaki, Y.; Jin, L.; Scheithauer, B.W.; Kovacs, K.; Roncaroli, F.; Lloyd, R.V. P53 gene mutations in pituitary carcinomas. Endocr. Pathol. 2007, 18, 217–222. [Google Scholar] [CrossRef]
- Szymas, J.; Schluens, K.; Liebert, W.; Petersen, I. Genomic instability in pituitary adenomas. Pituitary 2002, 5, 211–219. [Google Scholar] [CrossRef]
- Pack, S.D.; Qin, L.X.; Pak, E.; Wang, Y.; Ault, D.O.; Mannan, P.; Jaikumar, S.; Stratakis, C.A.; Oldfield, E.H.; Zhuang, Z.; et al. Common genetic changes in hereditary and sporadic pituitary adenomas detected by comparative genomic hybridization. Genes Chromosomes Cancer 2005, 43, 72–82. [Google Scholar] [CrossRef]
- Bi, W.L.; Greenwald, N.F.; Ramkissoon, S.H.; Abedalthagafi, M.; Coy, S.M.; Ligon, K.L.; Mei, Y.; MacConaill, L.; Ducar, M.; Min, L.; et al. Clinical Identification of Oncogenic Drivers and Copy-Number Alterations in Pituitary Tumors. Endocrinology 2017, 158, 2284–2291. [Google Scholar] [CrossRef]
- Bi, W.L.; Horowitz, P.; Greenwald, N.F.; Abedalthagafi, M.; Agarwalla, P.K.; Gibson, W.J.; Mei, Y.; Schumacher, S.E.; Ben-David, U.; Chevalier, A.; et al. Landscape of Genomic Alterations in Pituitary Adenomas. Clin. Cancer Res. 2017, 23, 1841–1851. [Google Scholar] [CrossRef]
- Song, Z.J.; Reitman, Z.J.; Ma, Z.Y.; Chen, J.H.; Zhang, Q.L.; Shou, X.F.; Huang, C.X.; Wang, Y.F.; Li, S.Q.; Mao, Y.; et al. The genome-wide mutational landscape of pituitary adenomas. Cell Res. 2016, 26, 1255–1259. [Google Scholar] [CrossRef]
- Hage, M.; Viengchareun, S.; Brunet, E.; Villa, C.; Pineau, D.; Bouligand, J.; Teglas, J.P.; Adam, C.; Parker, F.; Lombes, M.; et al. Genomic Alterations and Complex Subclonal Architecture in Sporadic GH-Secreting Pituitary Adenomas. J. Clin. Endocrinol. Metab. 2018, 103, 1929–1939. [Google Scholar] [CrossRef]
- Tatsi, C.; Pankratz, N.; Lane, J.; Faucz, F.R.; Hernandez-Ramirez, L.C.; Keil, M.; Trivellin, G.; Chittiboina, P.; Mills, J.L.; Stratakis, C.A.; et al. Large Genomic Aberrations in Corticotropinomas Are Associated With Greater Aggressiveness. J. Clin. Endocrinol. Metab. 2019, 104, 1792–1801. [Google Scholar] [CrossRef]
- Duong, C.V.; Emes, R.D.; Wessely, F.; Yacqub-Usman, K.; Clayton, R.N.; Farrell, W.E. Quantitative, genome-wide analysis of the DNA methylome in sporadic pituitary adenomas. Endocr. Relat. Cancer 2012, 19, 805–816. [Google Scholar] [CrossRef]
- Kober, P.; Boresowicz, J.; Rusetska, N.; Maksymowicz, M.; Goryca, K.; Kunicki, J.; Bonicki, W.; Siedlecki, J.A.; Bujko, M. DNA methylation profiling in nonfunctioning pituitary adenomas. Mol. Cell Endocrinol. 2018, 473, 194–204. [Google Scholar] [CrossRef]
- Garcia-Martinez, A.; Sottile, J.; Sanchez-Tejada, L.; Fajardo, C.; Camara, R.; Lamas, C.; Barbera, V.M.; Pico, A. DNA Methylation of Tumor Suppressor Genes in Pituitary Neuroendocrine Tumors. J. Clin. Endocrinol. Metab. 2019, 104, 1272–1282. [Google Scholar] [CrossRef]
- Gu, Y.; Zhou, X.; Hu, F.; Yu, Y.; Xie, T.; Huang, Y.; Zhao, X.; Zhang, X. Differential DNA methylome profiling of nonfunctioning pituitary adenomas suggesting tumour invasion is correlated with cell adhesion. J. Neurooncol. 2016, 129, 23–31. [Google Scholar] [CrossRef]
- Yoshino, A.; Katayama, Y.; Ogino, A.; Watanabe, T.; Yachi, K.; Ohta, T.; Komine, C.; Yokoyama, T.; Fukushima, T. Promoter hypermethylation profile of cell cycle regulator genes in pituitary adenomas. J. Neurooncol. 2007, 83, 153–162. [Google Scholar] [CrossRef]
- Ling, C.; Pease, M.; Shi, L.; Punj, V.; Shiroishi, M.S.; Commins, D.; Weisenberger, D.J.; Wang, K.; Zada, G. A pilot genome-scale profiling of DNA methylation in sporadic pituitary macroadenomas: Association with tumor invasion and histopathological subtype. PLoS ONE 2014, 9, e96178. [Google Scholar] [CrossRef]
- Pease, M.; Ling, C.; Mack, W.J.; Wang, K.; Zada, G. The role of epigenetic modification in tumorigenesis and progression of pituitary adenomas: A systematic review of the literature. PLoS ONE 2013, 8, e82619. [Google Scholar] [CrossRef]
- Buslei, R.; Kreutzer, J.; Hofmann, B.; Schmidt, V.; Siebzehnrubl, F.; Hahnen, E.; Eyupoglu, I.Y.; Fahlbusch, R.; Blumcke, I. Abundant hypermethylation of SOCS-1 in clinically silent pituitary adenomas. Acta Neuropathol. 2006, 111, 264–271. [Google Scholar] [CrossRef]
- Vandeva, S.; Jaffrain-Rea, M.L.; Daly, A.F.; Tichomirowa, M.; Zacharieva, S.; Beckers, A. The genetics of pituitary adenomas. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 461–476. [Google Scholar] [CrossRef]
- Zhu, X.; Lee, K.; Asa, S.L.; Ezzat, S. Epigenetic silencing through DNA and histone methylation of fibroblast growth factor receptor 2 in neoplastic pituitary cells. Am. J. Pathol. 2007, 170, 1618–1628. [Google Scholar] [CrossRef]
- Zhu, X.; Mao, X.; Hurren, R.; Schimmer, A.D.; Ezzat, S.; Asa, S.L. Deoxyribonucleic acid methyltransferase 3B promotes epigenetic silencing through histone 3 chromatin modifications in pituitary cells. J. Clin. Endocrinol. Metab. 2008, 93, 3610–3617. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Abbass, S.A.; Asa, S.L.; Ezzat, S. Altered expression of fibroblast growth factor receptors in human pituitary adenomas. J. Clin. Endocrinol. Metab. 1997, 82, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, S.; Zhu, X.; Loeper, S.; Fischer, S.; Asa, S.L. Tumor-derived Ikaros 6 acetylates the Bcl-XL promoter to up-regulate a survival signal in pituitary cells. Mol. Endocrinol. 2006, 20, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, S. Epigenetic control in pituitary tumors. Endocr. J. 2008, 55, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Karp, X.; Ambros, V. Developmental biology. Encountering microRNAs in cell fate signaling. Science 2005, 310, 1288–1289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yang, Z.; Gao, H. Advancements in the study of miRNA regulation during the cell cycle in human pituitary adenomas. J. Neurooncol. 2017, 134, 253–258. [Google Scholar] [CrossRef]
- D’Angelo, D.; Palmieri, D.; Mussnich, P.; Roche, M.; Wierinckx, A.; Raverot, G.; Fedele, M.; Croce, C.M.; Trouillas, J.; Fusco, A. Altered microRNA expression profile in human pituitary GH adenomas: Down-regulation of miRNA targeting HMGA1, HMGA2, and E2F1. J. Clin. Endocrinol. Metab. 2012, 97, E1128–E1138. [Google Scholar] [CrossRef]
- Nemeth, K.; Darvasi, O.; Liko, I.; Szucs, N.; Czirjak, S.; Reiniger, L.; Szabo, B.; Krokker, L.; Pallinger, E.; Igaz, P.; et al. Comprehensive analysis of circulating microRNAs in plasma of patients with pituitary adenomas. J. Clin. Endocrinol. Metab. 2019. [Google Scholar] [CrossRef]
- Cui, M.; Zhang, M.; Liu, H.F.; Wang, J.P. Effects of microRNA-21 targeting PITX2 on proliferation and apoptosis of pituitary tumor cells. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2995–3004. [Google Scholar]
- Roche, M.; Wierinckx, A.; Croze, S.; Rey, C.; Legras-Lachuer, C.; Morel, A.P.; Fusco, A.; Raverot, G.; Trouillas, J.; Lachuer, J. Deregulation of miR-183 and KIAA0101 in Aggressive and Malignant Pituitary Tumors. Front. Med. (Lausanne) 2015, 2, 54. [Google Scholar] [CrossRef]


{kind=link}
| Familial Syndrome | Gene | Chromosomal Locus | Suggested Mechanism of Pituitary Tumorigenesis | Most Common Functional Status | Frequency of PAs |
|---|---|---|---|---|---|
| Familial Isolated Pituitary Adenomas | AIP (15–30% of cases) | 11q13.2 | Interaction in cAMP synthesis | GH | 100% |
| Multiple Endocrine Neoplasia type 1 | MEN1 | 11q13.1 | Tumor suppressor; Involved in cell proliferation, genome stability and gene transcription | PRL or non-functioning | 40% |
| Multiple Endocrine Neoplasia type 2A/2B | RET | 10q11.21 | Proto-oncogene; Transmembrane receptor with tyrosine kinase activity | Rare | Rare |
| Multiple Endocrine Neoplasia type 4 | CDKN1B | 12p13.1 | Tumor suppressor; Cell cycle regulation | Rare | Rare |
| McCune-Albright | GNAS | 20q13.32 | cAMP-regulating protein Gsa; activation leads to increased cAMP levels and activation of protein kinase A (PKA) | GH excess | Up to 20% |
| Carney complex | PRKAR1A | 17q24.2 | Alpha regulatory subunit of PKA; inactivation of PRKAR1A leads to dissociation of the regulatory from the catalytic subunit and aberrant PKA activity | GH | 15% |
| DICER1 | DICER1 | 14q32.13 | RNA processing endoribonuclease that cleaves double stranded RNA into small interfering RNAs and mature miRNAs | ACTH | Rare |
| 3 P association | SDHx, VHL, MEN1, RET and MAX | 5p15.33 (SDHA) 1p36.13 (SDHB) 11q23.1 (SDHD) 11q12.2 (SDHAF2) 3p25.3 (VHL) 11q13.1 (MEN1) 11q13.1 (RET) 14q23.3 (MAX) | Several functions depending on identified gene. SDHx: participate in the mitochondrial complex II, energy production through the Krebs cycle and respiratory chain through electron transfer. VHL: tumor suppressor; oxygen sensing and interaction with hypoxia-inducible factors. MAX: Myc associated factor X; involved in cell proliferation, differentiation and apoptosis | PRL or GH | 100% |
| Tuberous sclerosis | TSC1, TSC2 | 9q34.13 (TSC1) 16p13.3 (TSC2) | Mediate PI3K/Akt activation and lead to inhibition of mTOR pathway | ACTH | Rare |
| X-Linked Acrogigantism | GPR101 | Xq26.3 | G-protein-coupled receptor; defects lead to constitutive activation of the cAMP-PKA pathway | GH | 85% |
| Neurofibromatosis type 1 | NF1 | 17q11.2 | Ras-GTPase-activating protein involved in Ras-dependent pathways (Ras/Raf/MEK and Ras/PI3K/TSC/mTOR) | GH | Rare |
| Familial Syndrome | Gene | Presentation |
|---|---|---|
| Familial Isolated Pituitary Adenomas | AIP (15–30% of cases) | Presence of at least two family members with pituitary adenomas, either of the same functional status (homologous) or of different (heterologous), without extra-pituitary findings. |
| Multiple Endocrine Neoplasia type 1 | MEN1 | Autosomal dominant syndrome presenting with multiple endocrine neoplasias, including anterior pituitary adenomas, hyperparathyroidism, enteropancreatic tumors, neuroendocrine tumors and others. |
| Multiple Endocrine Neoplasia type 2A/2B | RET | Autosomal dominant syndromes presenting with medullary thyroid carcinoma (MEN2A/B), pheochromocytoma (MEN2A/B), hyperparathyroidism (MEN2A), mucosal ganglioneuromas (MEN2B), and rare occurrence of anterior pituitary adenomas. |
| Multiple Endocrine Neoplasia type 4 | CDKN1B | Autosomal dominant MEN1-like syndrome, without genetic confirmation of MEN1 gene defect. |
| McCune-Albright | GNAS | Classic triad of polyostotic fibrous dysplasia, café-au-lait macules, and precocious puberty. Additional features include GH excess, hyperthyroidism, neonatal Cushing syndrome, and hypophosphatemia. |
| Carney complex | PRKAR1A | Autosomal dominant syndrome presenting with the constellation of cardiac and skin myxomas, spotty skin pigmentation, endocrine overactivity, including anterior pituitary adenomas and ACTH-independent Cushing syndrome, breast and testicular tumors, thyroid nodules, psammomatous melanotic schwannomas, and osteochondromyxomas. |
| DICER1 | DICER1 | Autosomal dominant syndrome presenting with pleuropulmonary blastomas, cystic nephromas, Sertoli-Leydig cell tumors, multinodular goiter, pituitary blastomas and other tumors. |
| 3 P association | SDHx, VHL, MEN1, RET and MAX | Combination of pituitary adenomas, pheochromocytomas and/or paragangliomas. |
| Tuberous sclerosis | TSC1, TSC2 | Autosomal dominant syndrome characterized by hamartomas, epilepsy, mental retardation, and rare occurrence of pituitary neuroendocrine tumors |
| X-Linked Acrogigantism | GPR101 | GH excess with onset of symptoms in most cases younger than 2 years of age. |
| Neurofibromatosis type 1 | NF1 | Autosomal dominant syndrome presenting with neurofibromas, skin findings (café-au-lait macules and freckling), Lisch (iris) nodules, optic pathway gliomas with consequent precocious puberty and GH excess, and other rare tumors. |
| Gene | Chromosomal Locus | Suggested Mechanism of Pituitary Tumorigenesis | Most Common Functional Status |
|---|---|---|---|
| GNAS | 20q13.32 | cAMP-regulating protein Gsa; activation leads to increased cAMP levels and activation of protein kinase A (PKA) | GH |
| USP8 | 15q21.2 | Involved in deubiquitination of EGFR; gain of functions mutations cause increase EGFR, and POMC expression | ACTH |
| AIP | 11q13.2 | Interaction in cAMP synthesis | GH |
| USP48 | 1p36.12 | Deubiquitination; activation of MAPK and increased POMC expression | ACTH |
| BRAF | 7q34 | Proto-oncogene with tyrosine kinase activity; activation of MAPK and increased POMC expression | ACTH |
| PIK3CA | 3q26.32 | Involved in PI3K/AKT pathway which regulates several cellular functions, including cell survival, growth, proliferation and metabolism | Non-functioning |
| TP53 | 17p13.1 | Tumor suppressor; involved in cell cycle, apoptosis and genomic stability | ACTH(also associated with atypical adenomas and pituitary carcinomas) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tatsi, C.; Stratakis, C.A. The Genetics of Pituitary Adenomas. J. Clin. Med. 2020, 9, 30. https://doi.org/10.3390/jcm9010030
Tatsi C, Stratakis CA. The Genetics of Pituitary Adenomas. Journal of Clinical Medicine. 2020; 9(1):30. https://doi.org/10.3390/jcm9010030
Chicago/Turabian StyleTatsi, Christina, and Constantine A. Stratakis. 2020. "The Genetics of Pituitary Adenomas" Journal of Clinical Medicine 9, no. 1: 30. https://doi.org/10.3390/jcm9010030
APA StyleTatsi, C., & Stratakis, C. A. (2020). The Genetics of Pituitary Adenomas. Journal of Clinical Medicine, 9(1), 30. https://doi.org/10.3390/jcm9010030