The Vasoactive Mas Receptor in Essential Hypertension




Abstract
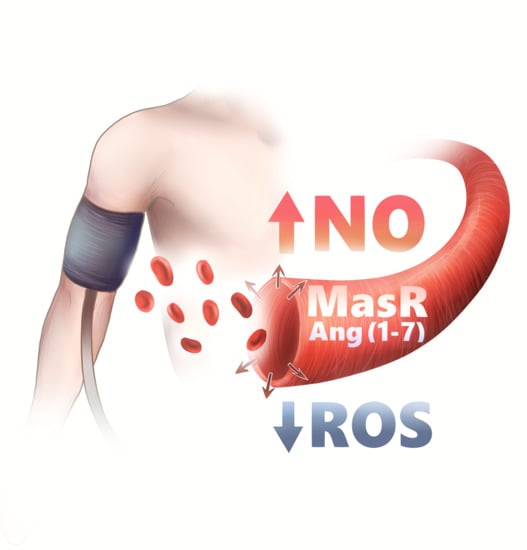
1. Introduction
2. The Renin-Angiotensin-Aldosterone System (RAAS)
2.1. The Classical RAAS
2.2. Extension of the RAAS

2.3. Blood Pressure Regulation by the RAAS
2.4. The Two Arms of the RAAS
3. Novel MasR Agonists
4. Mas Receptor in the Management of Hypertension

{kind=link}
{kind=link}
| Drug | Mechanism of Action | Effects on Blood Pressure | Status |
|---|---|---|---|
| AVE 0991 | AT2R/MasR agonist. Orally active nonpeptide drug. | AVE 0991 binding to bovine aortic endothelial cell membranes [43]. ACh-induced vasorelaxation in rats [47]. Potentiation of bradykinin through a Mas-mediated mechanism [48]. | Preclinical studies |
| CGEN-856S CGEN-857 | MasR agonist. Peptide drug. | Vasorelaxation in murine aortic rings [28]. Dose-dependent decrease in mean arterial pressure (MAP) in SHRs [28]. | Preclinical studies |
| HPβCD- Ang-(1-7) | Stable Ang-(1-7) analogue. Hydroxypropyl-β-cyclodextrin protects Ang-(1-7) from digestive tract enzymes. | Chronic oral administration lowers BP in rats following ischemia-reperfusion injury [56]. | Preclinical studies |
| Cyclic Ang-(1-7) | Peptidase resistant Ang-(1-7) analogue. | Improves endothelial function post-MI in male Sprague Dawley rats [51]. cAng-(1-7) improved peripheral endothelium-dependent vasodilation, as measured in isolated aortic rings [51]. | Preclinical studies |
| RB150/QGC001 | A central acting prodrug of the selective APA inhibitor, EC33. Orally available compound with the ability to cross the blood brain barrier. | Dose-dependent and long-lasting reduction in BP in rats, possibly following a specific blockage the of brain renin–angiotensin–aldosterone system [57]. | Preclinical and phase II studies |
| Alamandine | Vasoactive peptide derivative of AngA with selective agonism on MrgD. | Central and peripheral BP reduction [20]. Diminishes reperfusion injury after ischemia [30]. | Preclinical studies |
5. Discussion
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Bloch, M.J. Worldwide prevalence of hypertension exceeds 1.3 billion. J. Am. Soc. Hypertens. 2016, 10, 753–754. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.T.; Bundy, J.D.; Kelly, T.N.; Reed, J.E.; Kearney, P.M.; Reynolds, K.; Chen, J.; He, J. Global disparities of hypertension prevalence and control: A systematic analysis of population-based studies from 90 countries. Circulation 2016, 134, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [PubMed]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E., Jr.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2018, 138, e426–e483. [Google Scholar] [CrossRef] [PubMed]
- Rossier, B.C.; Bochud, M.; Devuyst, O. The hypertension pandemic: An evolutionary perspective. Physiology 2017, 32, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, S.; Caulfield, M.; Dominiczak, A.F. Genetic and molecular aspects of hypertension. Circ. Res. 2015, 116, 937–959. [Google Scholar] [CrossRef]
- Carretero, O.A.; Oparil, S. Essential hypertension: Part II: Treatment. Circulation 2000, 101, 446–453. [Google Scholar] [CrossRef]
- Wise, I.A.; Charchar, F.J. Epigenetic modifications in essential hypertension. Int. J. Mol. Sci. 2016, 17, 451. [Google Scholar] [CrossRef]
- Drenjancevic-Peric, I.; Jelakovic, B.; Lombard, J.H.; Kunert, M.P.; Kibel, A.; Gros, M. High-salt diet and hypertension: Focus on the renin-angiotensin system. Kidney Blood Press. Res. 2011, 34, 1–11. [Google Scholar] [CrossRef]
- Bogdarina, I.; Welham, S.; King, P.J.; Burns, S.P.; Clark, A.J. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ. Res. 2007, 100, 520–526. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Oparil, S.; Schmieder, R.E. New approaches in the treatment of hypertension. Circ. Res. 2015, 116, 1074–1095. [Google Scholar] [CrossRef] [PubMed]
- Mirabito Colafella, K.M.; Bovee, D.M.; Danser, A.H.J. The renin-angiotensin-aldosterone system and its therapeutic targets. Exp. Eye Res. 2019, 186, 107680. [Google Scholar] [CrossRef] [PubMed]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.H. Hypertension: Renin-angiotensin-aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Seva Pessoa, B.; van der Lubbe, N.; Verdonk, K.; Roks, A.J.; Hoorn, E.J.; Danser, A.H. Key developments in renin-angiotensin-aldosterone system inhibition. Nat. Rev. Nephrol. 2013, 9, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, K.; Danser, A.H.; van Esch, J.H. Angiotensin II type 2 receptor agonists: Where should they be applied? Expert Opin. Investig. Drugs 2012, 21, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Nehme, A.; Zouein, F.A.; Zayeri, Z.D.; Zibara, K. An update on the tissue renin angiotensin system and its role in physiology and pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14. [Google Scholar] [CrossRef]
- Ahmad, S.; Varagic, J.; Groban, L.; Dell’Italia, L.J.; Nagata, S.; Kon, N.D.; Ferrario, C.M. Angiotensin-(1–12): A chymase-mediated cellular Angiotensin II substrate. Curr. Hypertens. Rep. 2014, 16, 429. [Google Scholar] [CrossRef][Green Version]
- Yugandhar, V.G.; Clark, M.A. Angiotensin III: A physiological relevant peptide of the renin angiotensin system. Peptides 2013, 46, 26–32. [Google Scholar] [CrossRef]
- Stewart, M.H.; Lavie, C.J.; Ventura, H.O. Emerging therapy in hypertension. Curr. Hypertens. Rep. 2019, 21, 23. [Google Scholar] [CrossRef]
- Smith, A.I.; Turner, A.J. What’s new in the renin-angiotensin system? Cell. Mol. Life Sci. 2004, 61, 2675–2676. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, T.A.; Vinh, A.; Jones, E.S.; Widdop, R.E. Ganging up on Angiotensin II type 1 receptors in vascular remodeling. Hypertension 2012, 60, 17–19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ocaranza, M.P.; Michea, L.; Chiong, M.; Lagos, C.F.; Lavandero, S.; Jalil, J.E. Recent insights and therapeutic perspectives of Angiotensin-(1–9) in the cardiovascular system. Clin. Sci. 2014, 127, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Ocaranza, M.P.; Moya, J.; Barrientos, V.; Alzamora, R.; Hevia, D.; Morales, C.; Pinto, M.; Escudero, N.; Garcia, L.; Novoa, U.; et al. Angiotensin-(1–9) reverses experimental hypertension and cardiovascular damage by inhibition of the angiotensin converting enzyme/ang ii axis. J. Hypertens. 2014, 32, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.J.; Santos, R.A.; Bradford, C.N.; Mecca, A.P.; Sumners, C.; Katovich, M.J.; Raizada, M.K. Therapeutic implications of the vasoprotective axis of the renin-angiotensin system in cardiovascular diseases. Hypertension 2010, 55, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Bader, M.; Alenina, N.; Young, D.; Santos, R.A.S.; Touyz, R.M. The meaning of MAS. Hypertension 2018, 72, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.; Campagnole-Santos, M.J.; Bara EHT, N.C.; Fontes, M.A.; Silva, L.C.; Neves, L.A.; Oliveira, D.R.; Caligiorne, S.M.; Rodrigues, A.R.; Gropen Junior, C.; et al. Characterization of a new angiotensin antagonist selective for Angiotensin-(1–7): Evidence that the actions of Angiotensin-(1–7) are mediated by specific angiotensin receptors. Brain Res. Bull. 1994, 35, 293–298. [Google Scholar] [CrossRef]
- Savergnini, S.Q.; Beiman, M.; Lautner, R.Q.; de Paula-Carvalho, V.; Allahdadi, K.; Pessoa, D.C.; Costa-Fraga, F.P.; Fraga-Silva, R.A.; Cojocaru, G.; Cohen, Y.; et al. Vascular relaxation, antihypertensive effect, and cardioprotection of a novel peptide agonist of the MAS receptor. Hypertension 2010, 56, 112–120. [Google Scholar] [CrossRef]
- Galandrin, S.; Denis, C.; Boularan, C.; Marie, J.; M’Kadmi, C.; Pilette, C.; Dubroca, C.; Nicaise, Y.; Seguelas, M.H.; N’Guyen, D.; et al. Cardioprotective Angiotensin-(1–7) peptide acts as a natural-biased ligand at the angiotensin II type 1 receptor. Hypertension 2016, 68, 1365–1374. [Google Scholar] [CrossRef]
- Hrenak, J.; Paulis, L.; Simko, F. Angiotensin A/Alamandine/MrgD axis: Another clue to understanding cardiovascular pathophysiology. Int. J. Mol. Sci. 2016, 17, 1098. [Google Scholar] [CrossRef]
- Qaradakhi, T.; Apostolopoulos, V.; Zulli, A. Angiotensin (1–7) and alamandine: Similarities and differences. Pharmacol. Res. 2016, 111, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.A.; Schiffrin, E.L.; White, W.B.; Mann, S.; Lindholm, L.H.; Kenerson, J.G.; Flack, J.M.; Carter, B.L.; Materson, B.J.; Ram, C.V.; et al. Clinical practice guidelines for the management of hypertension in the community: A statement by the american society of hypertension and the international society of hypertension. J. Clin. Hypertens. 2014, 16, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, M.T.; Santos, R.A.; Brosnihan, K.B.; Khosla, M.C.; Ferrario, C.M. Release of vasopressin from the rat hypothalamo-neurohypophysial system by Angiotensin-(1–7) heptapeptide. Proc. Natl. Acad. Sci. USA 1988, 85, 4095–4098. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.W.; Mizutani, S.; Harding, J.W. Pathways involved in the transition from hypertension to hypertrophy to heart failure. Treatment strategies. Heart Fail. Rev. 2008, 13, 367–375. [Google Scholar] [CrossRef]
- Sampaio, W.O.; Henrique de Castro, C.; Santos, R.A.; Schiffrin, E.L.; Touyz, R.M. Angiotensin-(1–7) counterregulates Angiotensin II signaling in human endothelial cells. Hypertension 2007, 50, 1093–1098. [Google Scholar] [CrossRef]
- Ferrario, C.M. ACE2: More of Ang-(1–7) or less Ang II? Curr. Opin. Nephrol. Hypertens. 2011, 20, 1–6. [Google Scholar] [CrossRef]
- Villela, D.; Leonhardt, J.; Patel, N.; Joseph, J.; Kirsch, S.; Hallberg, A.; Unger, T.; Bader, M.; Santos, R.A.; Sumners, C.; et al. Angiotensin type 2 receptor (AT2R) and receptor mas: A complex liaison. Clin. Sci. 2015, 128, 227–234. [Google Scholar] [CrossRef]
- Kostenis, E.; Milligan, G.; Christopoulos, A.; Sanchez-Ferrer, C.F.; Heringer-Walther, S.; Sexton, P.M.; Gembardt, F.; Kellett, E.; Martini, L.; Vanderheyden, P.; et al. G-protein-coupled receptor MAS is a physiological antagonist of the Angiotensin II type 1 receptor. Circulation 2005, 111, 1806–1813. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1–7)/MAS axis of the renin-angiotensin system: Focus on angiotensin-(1–7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef]
- Young, D.; Waitches, G.; Birchmeier, C.; Fasano, O.; Wigler, M. Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell 1986, 45, 711–719. [Google Scholar] [CrossRef]
- Santos, R.A.; Brosnihan, K.B.; Jacobsen, D.W.; DiCorleto, P.E.; Ferrario, C.M. Production of Angiotensin-(1–7) by human vascular endothelium. Hypertension 1992, 19, II56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, Z.; Dang, H.; Chen, R.; Liaw, C.; Tran, T.A.; Boatman, P.D.; Connolly, D.T.; Adams, J.W. Inhibition of mas g-protein signaling improves coronary blood flow, reduces myocardial infarct size, and provides long-term cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H299–H311. [Google Scholar] [CrossRef] [PubMed]
- Wiemer, G.; Dobrucki, L.W.; Louka, F.R.; Malinski, T.; Heitsch, H. AVE 0991, a nonpeptide mimic of the effects of Angiotensin-(1–7) on the endothelium. Hypertension 2002, 40, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Cassis, P.; Locatelli, M.; Corna, D.; Villa, S.; Rottoli, D.; Cerullo, D.; Abbate, M.; Remuzzi, G.; Benigni, A.; Zoja, C. Addition of cyclic Angiotensin-(1–7) to angiotensin-converting enzyme inhibitor therapy has a positive add-on effect in experimental diabetic nephropathy. Kidney Int. 2019, 96, 906–917. [Google Scholar] [CrossRef]
- Hernandez Prada, J.A.; Ferreira, A.J.; Katovich, M.J.; Shenoy, V.; Qi, Y.; Santos, R.A.; Castellano, R.K.; Lampkins, A.J.; Gubala, V.; Ostrov, D.A.; et al. Structure-based identification of small-molecule angiotensin-converting enzyme 2 activators as novel antihypertensive agents. Hypertension 2008, 51, 1312–1317. [Google Scholar] [CrossRef]
- Lemos, V.S.; Silva, D.M.; Walther, T.; Alenina, N.; Bader, M.; Santos, R.A. The endothelium-dependent vasodilator effect of the nonpeptide Ang (1–7) mimic AVE 0991 is abolished in the aorta of Mas-knockout mice. J. Cardiovasc. Pharmacol. 2005, 46, 274–279. [Google Scholar] [CrossRef]
- Faria-Silva, R.; Duarte, F.V.; Santos, R.A. Short-term Angiotensin (1–7) receptor mas stimulation improves endothelial function in normotensive rats. Hypertension 2005, 46, 948–952. [Google Scholar] [CrossRef]
- Carvalho, M.B.; Duarte, F.V.; Faria-Silva, R.; Fauler, B.; da Mata Machado, L.T.; de Paula, R.D.; Campagnole-Santos, M.J.; Santos, R.A. Evidence for Mas-mediated bradykinin potentiation by the Angiotensin-(1–7) nonpeptide mimic AVE 0991 in normotensive rats. Hypertension 2007, 50, 762–767. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, J.; Li, S.F.; Song, J.X.; Ren, J.Y.; Chen, H. Angiotensin-(1–7): New perspectives in atherosclerosis treatment. J. Geriatr. Cardiol. 2015, 12, 676–682. [Google Scholar] [CrossRef]
- Marques, F.D.; Ferreira, A.J.; Sinisterra, R.D.; Jacoby, B.A.; Sousa, F.B.; Caliari, M.V.; Silva, G.A.; Melo, M.B.; Nadu, A.P.; Souza, L.E.; et al. An oral formulation of Angiotensin-(1–7) produces cardioprotective effects in infarcted and isoproterenol-treated rats. Hypertension 2011, 57, 477–483. [Google Scholar] [CrossRef]
- Durik, M.; van Veghel, R.; Kuipers, A.; Rink, R.; Haas Jimoh Akanbi, M.; Moll, G.; Danser, A.H.; Roks, A.J. The effect of the thioether-bridged, stabilized Angiotensin-(1–7) analogue cyclic Ang-(1–7) on cardiac remodeling and endothelial function in rats with myocardial infarction. Int. J. Hypertens. 2012, 2012, 536426. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A. Angiotensin-(1–7). Hypertension 2014, 63, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Yang, J.; Zhang, Y.; Dong, M.; Wang, S.; Zhang, Q.; Liu, F.F.; Zhang, K.; Zhang, C. Angiotensin-converting enzyme 2 and Angiotensin 1–7: Novel therapeutic targets. Nat. Rev. Cardiol. 2014, 11, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, J.; Villela, D.C.; Teichmann, A.; Munter, L.M.; Mayer, M.C.; Mardahl, M.; Kirsch, S.; Namsolleck, P.; Lucht, K.; Benz, V.; et al. Evidence for heterodimerization and functional interaction of the Angiotensin type 2 receptor and the receptor mas. Hypertension 2017, 69, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, B.D.; Carretero, O.A.; Janic, B.; Grecco, H.E.; Gironacci, M.M. Heteromerization between the bradykinin b2 receptor and the Angiotensin-(1–7) mas receptor: Functional consequences. Hypertension 2016, 68, 1039–1048. [Google Scholar] [CrossRef]
- Marques, F.D.; Melo, M.B.; Souza, L.E.; Irigoyen, M.C.; Sinisterra, R.D.; de Sousa, F.B.; Savergnini, S.Q.; Braga, V.B.; Ferreira, A.J.; Santos, R.A. Beneficial effects of long-term administration of an oral formulation of Angiotensin-(1–7) in infarcted rats. Int. J. Hypertens. 2012, 2012, 795452. [Google Scholar] [CrossRef] [PubMed]
- Fournie-Zaluski, M.C.; Fassot, C.; Valentin, B.; Djordjijevic, D.; Reaux-Le Goazigo, A.; Corvol, P.; Roques, B.P.; Llorens-Cortes, C. Brain renin-angiotensin system blockade by systemically active aminopeptidase a inhibitors: A potential treatment of salt-dependent hypertension. Proc. Natl. Acad. Sci. USA 2004, 101, 7775–7780. [Google Scholar] [CrossRef]
- Llorens-Cortes, C.; Touyz, R.M. Evolution of a new class of antihypertensive drugs: Targeting the brain renin-angiotensin system. Hypertension 2020, 75, 6–15. [Google Scholar] [CrossRef]
- Jesus, I.C.G.; Scalzo, S.; Alves, F.; Marques, K.; Rocha-Resende, C.; Bader, M.; Santos, R.A.S.; Guatimosim, S. Alamandine acts via MrgD to induce AMPK/NO activation against Ang II hypertrophy in cardiomyocytes. Am. J. Physiol. Cell Physiol. 2018, 314, C702–C711. [Google Scholar] [CrossRef]
- Park, B.M.; Phuong, H.T.A.; Yu, L.; Kim, S.H. Alamandine protects the heart against reperfusion injury via the MrgD receptor. Circ. J. 2018, 82, 2584–2593. [Google Scholar] [CrossRef]
- Liu, C.; Yang, C.X.; Chen, X.R.; Liu, B.X.; Li, Y.; Wang, X.Z.; Sun, W.; Li, P.; Kong, X.Q. Alamandine attenuates hypertension and cardiac hypertrophy in hypertensive rats. Amino Acids 2018, 50, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Povlsen, A.L.; Grimm, D.; Wehland, M.; Infanger, M.; Krüger, M. The Vasoactive Mas Receptor in Essential Hypertension. J. Clin. Med. 2020, 9, 267. https://doi.org/10.3390/jcm9010267
Povlsen AL, Grimm D, Wehland M, Infanger M, Krüger M. The Vasoactive Mas Receptor in Essential Hypertension. Journal of Clinical Medicine. 2020; 9(1):267. https://doi.org/10.3390/jcm9010267
Chicago/Turabian StylePovlsen, Amalie L., Daniela Grimm, Markus Wehland, Manfred Infanger, and Marcus Krüger. 2020. "The Vasoactive Mas Receptor in Essential Hypertension" Journal of Clinical Medicine 9, no. 1: 267. https://doi.org/10.3390/jcm9010267
APA StylePovlsen, A. L., Grimm, D., Wehland, M., Infanger, M., & Krüger, M. (2020). The Vasoactive Mas Receptor in Essential Hypertension. Journal of Clinical Medicine, 9(1), 267. https://doi.org/10.3390/jcm9010267