Ringer’s Lactate Prevents Early Organ Failure by Providing Extracellular Calcium


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. In Vitro Studies
2.2. Primary Acinar Cell Preparation
2.3. Lactate dehydrogenase (LDH) Release Assays
2.4. Fura-2/JC-1 Spectrophotometry
2.5. In Vivo Experimental Models and Parameters
2.5.1. Linoleic Acid Administration
2.5.2. Acute Pancreatitis Model
2.5.3. Monitoring
2.6. Histology, Special Stains, and Immunohistochemical Studies
2.7. Biochemical Assays
2.8. Cytokine/Chemokine Assays
2.9. Experiment Statistics
2.10. Meta-Analysis
3. Results
3.1. Calcium Binds Long-Chain Fatty Acids in an Energetically Favorable Manner
3.2. NEFA Like Linoleic Acid Cause Hypocalcemia and Organ Failure
3.3. Physiologically Relevant Extracellular Calcium (CaE), Unlike Lactate, Reduces NEFA Toxicity
3.4. Physiologically Relevant Extracellular Calcium Concentrations Do Not Affect Caerulein-Induced Injury
3.5. Antagonizing Intracellular Calcium Increase Has Little or No Effect on La-Induced Acinar Injury
3.6. Therapeutic Extracellular Supplementation Binds Fatty Acids Generated During Severe AP
3.7. Therapeutic Extracellular Calcium Supplementation Prevents Hypocalcemia, and Transiently Reduces the Increase in Serum Non-Esterified Fatty Acids and Organ Failure
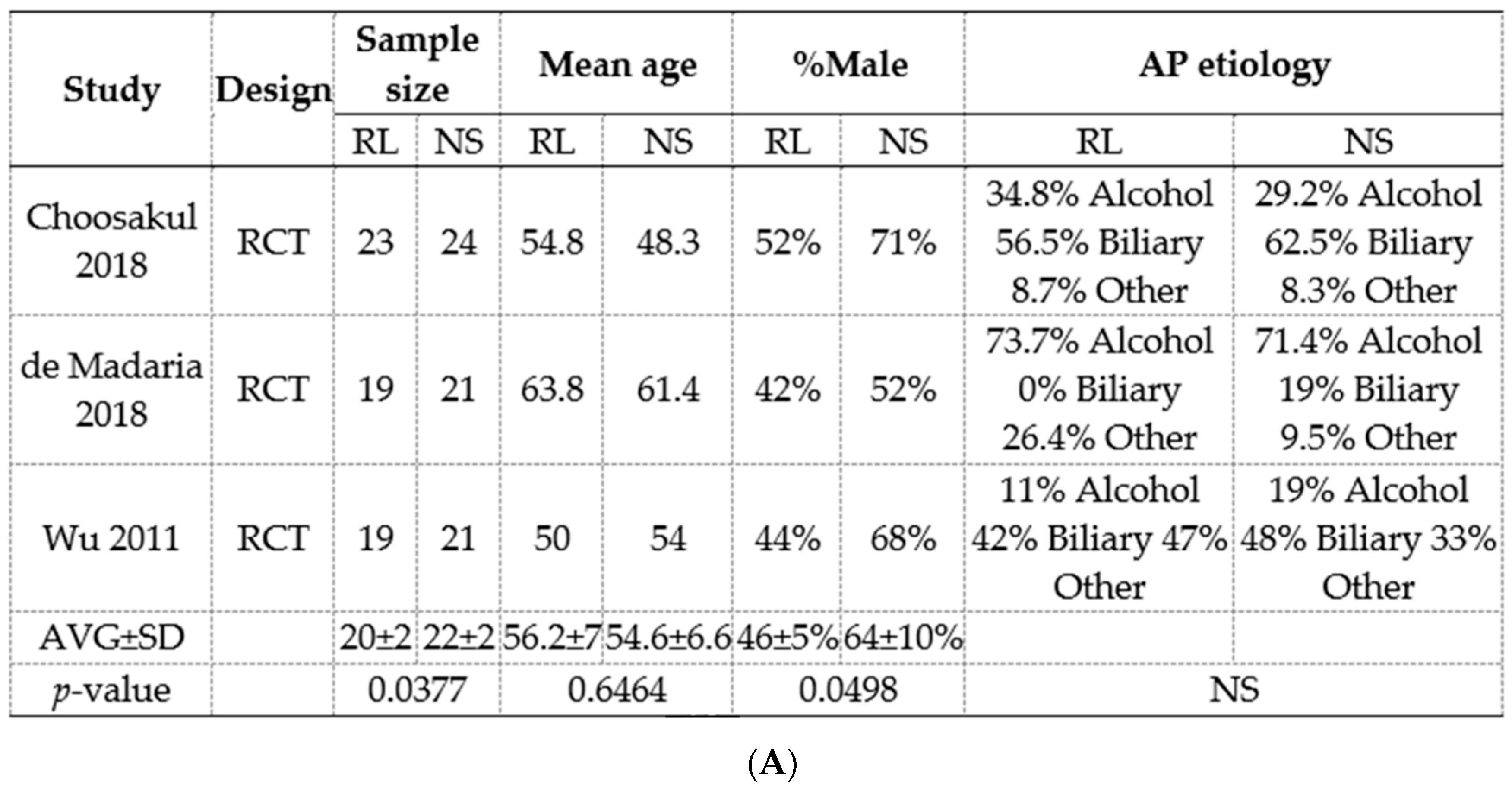
3.8. Meta-Analysis of RCTs Comparing Normal Saline (Ns) Show a Reduction In Pancreatic Necrosis, but No Improvement in Long-Term Outcomes
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S. Classification of acute pancreatitis—2012: Revision of the Atlanta classification and definitions by international consensus. Gut 2013, 62, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Mounzer, R.; Langmead, C.J.; Wu, B.U.; Evans, A.C.; Bishehsari, F.; Muddana, V.; Singh, V.K.; Slivka, A.; Whitcomb, D.C.; Yadav, D.; et al. Comparison of existing clinical scoring systems to predict persistent organ failure in patients with acute pancreatitis. Gastroenterology 2012, 142, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Gong, Y.; Ying, B.; Cheng, B. Association of Initial Serum Total Calcium Concentration with Mortality in Critical Illness. Biomed. Res. Int. 2018, 2018, 7648506. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, D.; Gong, Y.; Ying, B.; Cheng, B. Association of serum total and ionized calcium with all-cause mortality incritically ill patients with acute kidney injury. Clin. Chim. Acta 2019, 494, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Ranson, J.H.; Rifkind, K.M.; Roses, D.F.; Fink, S.D.; Eng, K.; Spencer, F.C. Prognostic signs and the role of operative management in acute pancreatitis. Surg. Gynecol. Obstet. 1974, 139, 69–81. [Google Scholar] [PubMed]
- Blamey, S.L.; Imrie, C.W.; O’Neill, J.; Gilmour, W.H.; Carter, D.C. Prognostic factors in acute pancreatitis. Gut 1984, 25, 1340–1346. [Google Scholar] [CrossRef]
- De-Madaria, E.; Herrera-Marante, I.; Gonzalez-Camacho, V.; Bonjoch, L.; Quesada-Vazquez, N.; Almenta-Saavedra, I.; Miralles-Macia, C.; Acevedo-Piedra, N.G.; Roger-Ibanez, M.; Sanchez-Marin, C.; et al. Fluid resuscitation with lactated Ringer’s solution vs normal saline in acute pancreatitis: A triple-blind, randomized, controlled trial. United Eur. Gastroenterol. J. 2018, 6, 63–72. [Google Scholar] [CrossRef]
- Wu, B.U.; Hwang, J.Q.; Gardner, T.H.; Repas, K.; Delee, R.; Yu, S.; Smith, B.; Banks, P.A.; Conwell, D.L. Lactated Ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin. Gastroenterol. Hepatol. 2011, 9, 710–717. [Google Scholar] [CrossRef]
- Choosakul, S.; Harinwan, K.; Chirapongsathorn, S.; Opuchar, K.; Sanpajit, T.; Piyanirun, W.; Puttapitakpong, C. Comparison of normal saline versus Lactated Ringer’s solution for fluid resuscitation in patients with mild acute pancreatitis, A randomized controlled trial. Pancreatology 2018. [Google Scholar] [CrossRef]
- Iqbal, U.; Anwar, H.; Scribani, M. Ringer’s lactate versus normal saline in acute pancreatitis: A systematic review and meta-analysis. J. Dig. Dis. 2018, 19, 335–341. [Google Scholar] [CrossRef]
- Hoque, R.; Farooq, A.; Ghani, A.; Gorelick, F.; Mehal, W.Z. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 2014, 146, 1763–1774. [Google Scholar] [CrossRef] [PubMed]
- Didwania, A.; Miller, J.; Kassel, D.; Jackson, E.V., Jr.; Chernow, B. Effect of intravenous lactated Ringer’s solution infusion on the circulating lactate concentration: Part 3. Results of a prospective, randomized, double-blind, placebo-controlled trial. Crit. Care Med. 1997, 25, 1851–1854. [Google Scholar] [CrossRef] [PubMed]
- Zitek, T.; Skaggs, Z.D.; Rahbar, A.; Patel, J.; Khan, M. Does Intravenous Lactated Ringer’s Solution Raise Serum Lactate? J. Emerg. Med. 2018, 55, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Antequera Martin, A.M.; Barea Mendoza, J.A.; Muriel, A.; Saez, I.; Chico-Fernandez, M.; Estrada-Lorenzo, J.M.; Plana, M.N. Buffered solutions versus 0.9% saline for resuscitation in critically ill adults and children. Cochrane Database Syst. Rev. 2019, 7, CD012247. [Google Scholar] [CrossRef]
- Cogo, P.E.; Giordano, G.; Badon, T.; Orzali, A.; Zimmermann, I.U.; Zacchello, F.; Sauer, P.J.; Carnielli, V.P. Simultaneous measurement of the rates of appearance of palmitic and linoleic acid in critically ill infants. Pediatr. Res. 1997, 41, 178–182. [Google Scholar] [CrossRef][Green Version]
- Kreil, P.; Spiteller, G.; Johannes, G.; Wagner, W. Strong increase of 9-hydroxy-10,12-octadecadienoic acid in low density lipoprotein after a hemorrhagic shock. Z. Naturforsch. C 1998, 53, 876–882. [Google Scholar] [CrossRef]
- Pilitsis, J.G.; Coplin, W.M.; O’Regan, M.H.; Wellwood, J.M.; Diaz, F.G.; Fairfax, M.R.; Michael, D.B.; Phillis, J.W. Free fatty acids in cerebrospinal fluids from patients with traumatic brain injury. Neurosci. Lett. 2003, 349, 136–138. [Google Scholar] [CrossRef]
- Qi, P.; Abdullahi, A.; Stanojcic, M.; Patsouris, D.; Jeschke, M.G. Lipidomic analysis enables prediction of clinical outcomes in burn patients. Sci. Rep. 2016, 6, 38707. [Google Scholar] [CrossRef]
- Kosaka, K.; Suzuki, K.; Hayakawa, M.; Sugiyama, S.; Ozawa, T. Leukotoxin, a linoleate epoxide: Its implication in the late death of patients with extensive burns. Mol. Cell Biochem. 1994, 139, 141–148. [Google Scholar] [CrossRef]
- Sztefko, K.; Panek, J. Serum free fatty acid concentration in patients with acute pancreatitis. Pancreatology 2001, 1, 230–236. [Google Scholar] [CrossRef]
- Domschke, S.; Malfertheiner, P.; Uhl, W.; Buchler, M.; Domschke, W. Free fatty acids in serum of patients with acute necrotizing or edematous pancreatitis. Int. J. Pancreatol. 1993, 13, 105–110. [Google Scholar] [PubMed]
- Noel, P.; Patel, K.; Durgampudi, C.; Trivedi, R.N.; de Oliveira, C.; Crowell, M.D.; Pannala, R.; Lee, K.; Brand, R.; Chennat, J.; et al. Peripancreatic fat necrosis worsens acute pancreatitis independent of pancreatic necrosis via unsaturated fatty acids increased in human pancreatic necrosis collections. Gut 2016, 65, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Trivedi, R.N.; Durgampudi, C.; Noel, P.; Cline, R.A.; DeLany, J.P.; Navina, S.; Singh, V.P. Lipolysis of visceral adipocyte triglyceride by pancreatic lipases converts mild acute pancreatitis to severe pancreatitis independent of necrosis and inflammation. Am. J. Pathol. 2015, 185, 808–819. [Google Scholar] [CrossRef] [PubMed]
- Navina, S.; Acharya, C.; DeLany, J.P.; Orlichenko, L.S.; Baty, C.J.; Shiva, S.S.; Durgampudi, C.; Karlsson, J.M.; Lee, K.; Bae, K.T.; et al. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci. Transl. Med. 2011, 3, 107ra110. [Google Scholar] [CrossRef]
- De Oliveira, C.; Khatua, B.; Bag, A.; El-Kurdi, B.; Patel, K.; Mishra, V.; Navina, S.; Singh, V.P. Multimodal Transgastric Local Pancreatic Hypothermia Reduces Severity of Acute Pancreatitis in Rats and Increases Survival. Gastroenterology 2019, 156, 735–747. [Google Scholar] [CrossRef]
- Dettelbach, M.A.; Deftos, L.J.; Stewart, A.F. Intraperitoneal free fatty acids induce severe hypocalcemia in rats: A model for the hypocalcemia of pancreatitis. J. Bone Miner Res. 1990, 5, 1249–1255. [Google Scholar] [CrossRef]
- Acharya, C.; Cline, R.A.; Jaligama, D.; Noel, P.; Delany, J.P.; Bae, K.; Furlan, A.; Baty, C.J.; Karlsson, J.M.; Rosario, B.L.; et al. Fibrosis Reduces Severity of Acute-on-Chronic Pancreatitis in Humans. Gastroenterology 2013, 145, 466–475. [Google Scholar] [CrossRef]
- Herbert, F. Pancreatic Fat Necrosis: A Chemical Study. Br. J. Exp. Pathol. 1928, 9, 57–63. [Google Scholar]
- Powers, R.E.; Saluja, A.K.; Houlihan, M.J.; Steer, M.L. Diminished agonist-stimulated inositol trisphosphate generation blocks stimulus-secretion coupling in mouse pancreatic acini during diet-induced experimental pancreatitis. J. Clin. Investig. 1986, 77, 1668–1674. [Google Scholar] [CrossRef]
- Durgampudi, C.; Noel, P.; Patel, K.; Cline, R.; Trivedi, R.N.; DeLany, J.P.; Yadav, D.; Papachristou, G.I.; Lee, K.; Acharya, C.; et al. Acute Lipotoxicity Regulates Severity of Biliary Acute Pancreatitis without Affecting Its Initiation. Am. J. Pathol. 2014, 184, 1773–1784. [Google Scholar] [CrossRef]
- Patel, K.; Durgampudi, C.; Noel, P.; Trivedi, R.N.; de Oliveira, C.; Singh, V.P. Fatty Acid Ethyl Esters Are Less Toxic Than Their Parent Fatty Acids Generated during Acute Pancreatitis. Am. J. Pathol. 2016, 186, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Akella, A.; Sharma, P.; Pandey, R.; Deshpande, S.B. Characterization of oleic acid-induced acute respiratory distress syndrome model in rat. Indian J. Exp. Biol. 2014, 52, 712–719. [Google Scholar] [PubMed]
- Zhao, F.; Wang, W.; Fang, Y.; Li, X.; Shen, L.; Cao, T.; Zhu, H. Molecular mechanism of sustained inflation in acute respiratory distress syndrome. J. Trauma Acute Care Surg. 2012, 73, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Hussain, N.; Wu, F.; Zhu, L.; Thrall, R.S.; Kresch, M.J. Neutrophil apoptosis during the development and resolution of oleic acid-induced acute lung injury in the rat. Am. J. Respir. Cell Mol. Biol. 1998, 19, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Panek, J.; Sztefko, K.; Drozdz, W. Composition of free fatty acid and triglyceride fractions in human necrotic pancreatic tissue. Med. Sci. Monit. 2001, 7, 894–898. [Google Scholar] [PubMed]
- Khatua, B.; Trivedi, R.N.; Noel, P.; Patel, K.; Singh, R.; de Oliveira, C.; Trivedi, S.; Mishra, V.; Lowe, M.; Singh, V.P. Carboxyl Ester Lipase May Not Mediate Lipotoxic Injury during Severe Acute Pancreatitis. Am. J. Pathol. 2019. [Google Scholar] [CrossRef]
- Orabi, A.I.; Shah, A.U.; Ahmad, M.U.; Choo-Wing, R.; Parness, J.; Jain, D.; Bhandari, V.; Husain, S.Z. Dantrolene mitigates caerulein-induced pancreatitis in vivo in mice. Am. J. Physiol. Gastrointest Liver Physiol. 2010, 299, G196–G204. [Google Scholar] [CrossRef]
- Tenner, S.; Baillie, J.; Dewitt, J.; Vege, S.S. American College of Gastroenterology Guidelines: Management of Acute Pancreatitis. Am. J. Gastroenterol. 2013. [Google Scholar] [CrossRef]
- Besselink, M.; van Santvoort, H.; Freeman, M.; Gardner, T.; Mayerle, J.; Vege, S.S.; Werner, J.; Banks, P.; McKay, C.; Fernandez-del Castillo, C.; et al. IAP/APA evidence-based guidelines for the management of acute pancreatitis. Pancreatology 2013, 13, e1–e15. [Google Scholar] [CrossRef]
- Wells, H.G. Experimental Fat Necrosis. J. Med. Res. 1903, 9, 70–116. [Google Scholar] [CrossRef]
- Handojo, L.; Indarto, A.; Shofinita, D.; Meitha, A.; Nabila, R.; Triharyogi, H. Calcium soap from palm fatty acid distillate (PFAD) for ruminant feed: Quality of calcium source. MATEC Web Conf. 2018, 156, 02007. [Google Scholar] [CrossRef]
- Puchtler, H.; Meloan, S.N. Demonstration of phosphates in calcium deposits: A modification of von Kossa’s reaction. Histochemistry 1978, 56, 177–185. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, C.; Khatua, B.; Noel, P.; Kostenko, S.; Bag, A.; Balakrishnan, B.; Patel, K.S.; Guerra, A.A.; Martinez, M.N.; Trivedi, S.; et al. Pancreatic triglyceride lipase mediates lipotoxic systemic inflammation. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Pezzilli, R.; Billi, P.; Morselli-Labate, A.M. Severity of acute pancreatitis: Relationship with etiology, sex and age. Hepatogastroenterology 1998, 45, 1859–1864. [Google Scholar] [PubMed]
- Malik, A.M. Acute pancreatitis. A more common and severe complication of gallstones in males. Int. J. Health Sci. 2015, 9, 141–145. [Google Scholar] [CrossRef]
- Shen, H.N.; Wang, W.C.; Lu, C.L.; Li, C.Y. Effects of gender on severity, management and outcome in acute biliary pancreatitis. PLoS ONE 2013, 8, e57504. [Google Scholar] [CrossRef]
- Bota, S.; Sporea, I.; Sirli, R.; Popescu, A.; Strain, M.; Focsa, M.; Danila, M.; Chisevescu, D. Predictive factors for severe evolution in acute pancreatitis and a new score for predicting a severe outcome. Ann. Gastroenterol. 2013, 26, 156–162. [Google Scholar]
- Shyu, J.Y.; Sainani, N.I.; Sahni, V.A.; Chick, J.F.; Chauhan, N.R.; Conwell, D.L.; Clancy, T.E.; Banks, P.A.; Silverman, S.G. Necrotizing pancreatitis: Diagnosis, imaging, and intervention. Radiographics 2014, 34, 1218–1239. [Google Scholar] [CrossRef]
- Nordback, I.; Lauslahti, K. Clinical pathology of acute necrotising pancreatitis. J. Clin. Pathol. 1986, 39, 68–74. [Google Scholar] [CrossRef]
- Sproston, N.R.; Ashworth, J.J. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front. Immunol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Lau, D.C.; Dhillon, B.; Yan, H.; Szmitko, P.E.; Verma, S. Adipokines: Molecular links between obesity and atheroslcerosis. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2031–H2041. [Google Scholar] [CrossRef] [PubMed]
- Ku, N.O.; Mortensen, R.F. The mouse C-reactive protein (CRP) gene is expressed in response to IL-1 but not IL-6. Cytokine 1993, 5, 319–326. [Google Scholar] [CrossRef]
- Husain, S.Z.; Prasad, P.; Grant, W.M.; Kolodecik, T.R.; Nathanson, M.H.; Gorelick, F.S. The ryanodine receptor mediates early zymogen activation in pancreatitis. Proc. Natl. Acad. Sci. USA 2005, 102, 14386–14391. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Moormann, P. Comparative radiological and morphological study of the human pancreas. IV. Acute necrotizing pancreatitis in man. Pathol. Res. Pract. 1981, 171, 325–335. [Google Scholar] [CrossRef]
- Hotchkiss, L.W. VIII. Acute Pancreatitis with Very Extensive Fat Necrosis. Ann. Surg. 1912, 56, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Aho, H.J.; Sternby, B.; Nevalainen, T.J. Fat necrosis in human acute pancreatitis. An immunohistological study. Acta Pathol. Microbiol. Immunol. Scand. A 1986, 94, 101–105. [Google Scholar]
- Wieloch, T.; Borgstrom, B.; Pieroni, G.; Pattus, F.; Verger, R. Product activation of pancreatic lipase. Lipolytic enzymes as probes for lipid/water interfaces. J. Biol. Chem. 1982, 257, 11523–11528. [Google Scholar]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khatua, B.; Yaron, J.R.; El-Kurdi, B.; Kostenko, S.; Papachristou, G.I.; Singh, V.P. Ringer’s Lactate Prevents Early Organ Failure by Providing Extracellular Calcium. J. Clin. Med. 2020, 9, 263. https://doi.org/10.3390/jcm9010263
Khatua B, Yaron JR, El-Kurdi B, Kostenko S, Papachristou GI, Singh VP. Ringer’s Lactate Prevents Early Organ Failure by Providing Extracellular Calcium. Journal of Clinical Medicine. 2020; 9(1):263. https://doi.org/10.3390/jcm9010263
Chicago/Turabian StyleKhatua, Biswajit, Jordan R. Yaron, Bara El-Kurdi, Sergiy Kostenko, Georgios I. Papachristou, and Vijay P. Singh. 2020. "Ringer’s Lactate Prevents Early Organ Failure by Providing Extracellular Calcium" Journal of Clinical Medicine 9, no. 1: 263. https://doi.org/10.3390/jcm9010263
APA StyleKhatua, B., Yaron, J. R., El-Kurdi, B., Kostenko, S., Papachristou, G. I., & Singh, V. P. (2020). Ringer’s Lactate Prevents Early Organ Failure by Providing Extracellular Calcium. Journal of Clinical Medicine, 9(1), 263. https://doi.org/10.3390/jcm9010263