A Real-World Experience of Clinical, Biochemical and Genetic Assessment of Patients with Homozygous Familial Hypercholesterolemia

 ,
, 
 ,
,
Abstract
1. Introduction
2. Experimental Section
2.1. Study Population, Clinical and Biochemical Assessment
2.2. Genetic Analysis
2.3. High-Resolution Carotid Ultrasound
2.4. Statistical Analyses
3. Results
3.1. Genetic Screening
3.2. Biochemical and Clinical Features
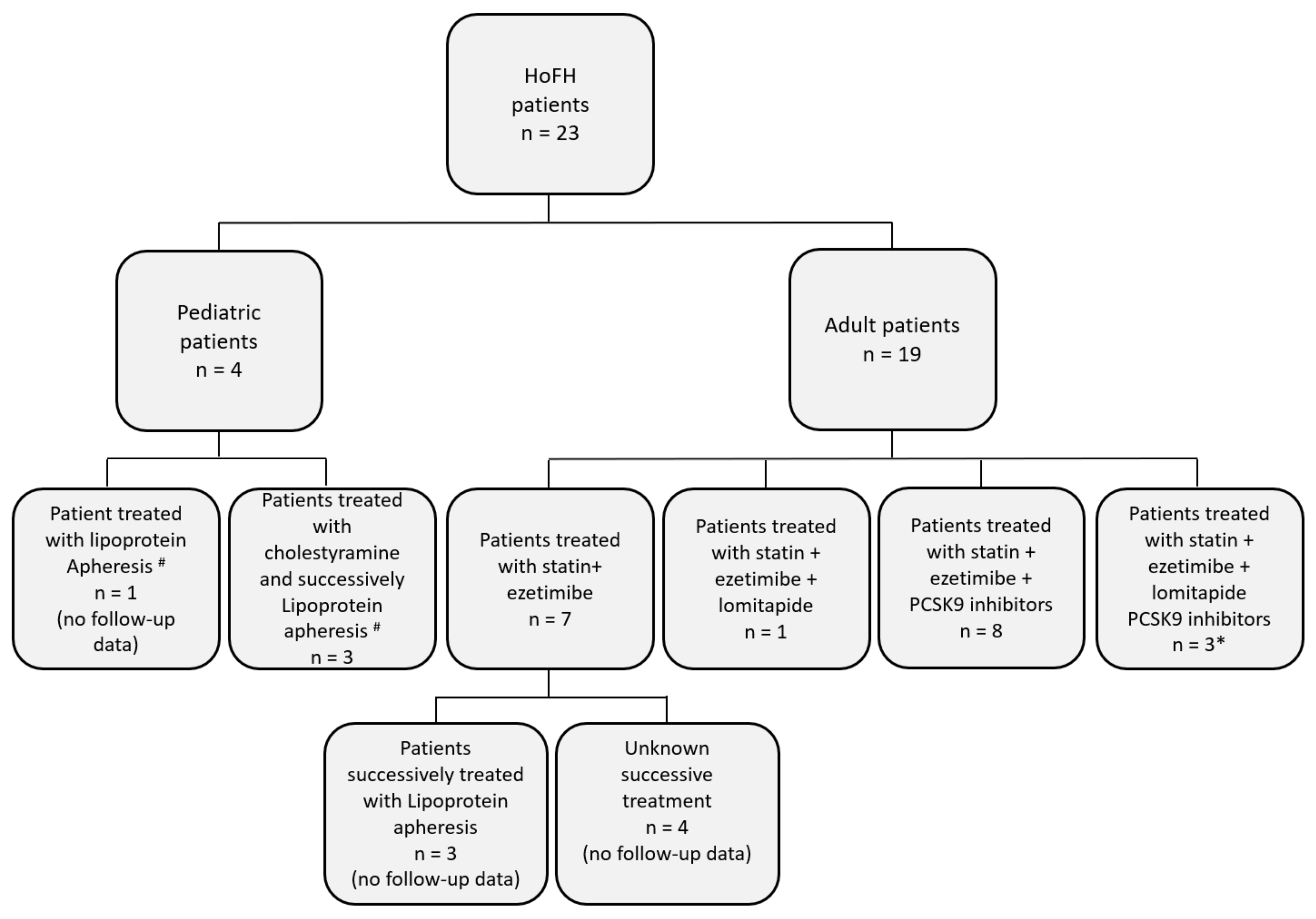
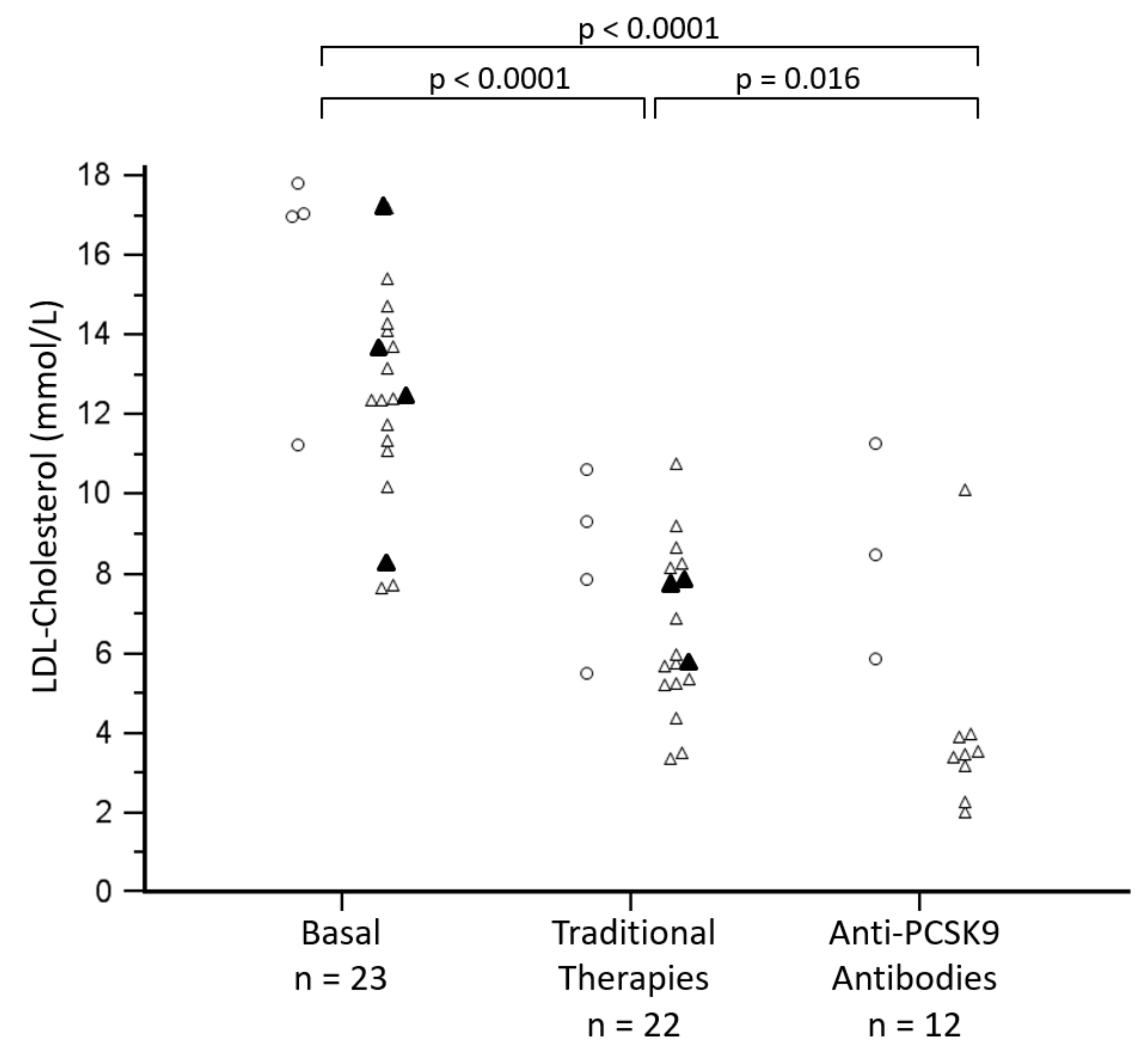
3.3. Management of Hypercholesterolemia
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Defesche, J.C.; Gidding, S.S.; Harada-Shiba, M.; Hegele, R.A.; Santos, R.D.; Wierzbicki, A.S. Familial hypercholesterolaemia. Nat. Rev. Dis. Prim. 2017, 3, 17093. [Google Scholar] [CrossRef] [PubMed]
- Sturm, A.C.; Knowles, J.W.; Gidding, S.S.; Ahmad, Z.S.; Ahmed, C.D.; Ballantyne, C.M.; Baum, S.J.; Bourbon, M.; Carrie, A.; Cuchel, M.; et al. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2018, 72, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Bruckert, E.; Ginsberg, H.N.; Raal, F.J.; Santos, R.D.; Hegele, R.A.; Kuivenhoven, J.A.; Nordestgaard, B.G.; Descamps, O.S.; Steinhagen-Thiessen, E.; et al. Homozygous familial hypercholesterolaemia: New insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 2014, 35, 2146–2157. [Google Scholar] [CrossRef] [PubMed]
- Akioyamen, L.E.; Genest, J.; Shan, S.D.; Reel, R.L.; Albaum, J.M.; Chu, A.; Tu, J.V. Estimating the prevalence of heterozygous familial hypercholesterolaemia: A systematic review and meta-analysis. BMJ Open 2017, 7, e016461. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490a. [Google Scholar] [CrossRef]
- D’Erasmo, L.; Cefalu, A.B.; Noto, D.; Giammanco, A.; Averna, M.; Pintus, P.; Medde, P.; Vigna, G.B.; Sirtori, C.; Calabresi, L.; et al. Efficacy of Lomitapide in the Treatment of Familial Homozygous Hypercholesterolemia: Results of a Real-World Clinical Experience in Italy. Adv. Ther. 2017, 34, 1200–1210. [Google Scholar] [CrossRef]
- Widhalm, K.; Benke, I.M.; Fritz, M.; Geiger, H.; Helk, O.; Fritsch, M.; Hoermann, G.; Kostner, G. Homozygous familial hypercholesterolemia: Summarized case reports. Atherosclerosis 2017, 257, 86–89. [Google Scholar] [CrossRef]
- Hartgers, M.L.; Defesche, J.C.; Langslet, G.; Hopkins, P.N.; Kastelein, J.J.P.; Baccara-Dinet, M.T.; Seiz, W.; Hamon, S.; Banerjee, P.; Stefanutti, C. Alirocumab efficacy in patients with double heterozygous, compound heterozygous, or homozygous familial hypercholesterolemia. J. Clin. Lipidol. 2018, 12, 390–396. [Google Scholar] [CrossRef]
- Stefanutti, C.; Pang, J.; Di Giacomo, S.; Wu, X.; Wang, X.; Morozzi, C.; Watts, G.F.; Lin, J. A cross-national investigation of cardiovascular survival in homozygous familial hypercholesterolemia: The Sino-Roman Study. J. Clin. Lipidol. 2019, 13, 608–617. [Google Scholar] [CrossRef]
- Ben-Omran, T.; Masana, L.; Kolovou, G.; Ariceta, G.; Novoa, F.J.; Lund, A.M.; Bogsrud, M.P.; Araujo, M.; Hussein, O.; Ibarretxe, D.; et al. Real-World Outcomes with Lomitapide Use in Paediatric Patients with Homozygous Familial Hypercholesterolaemia. Adv. Ther. 2019, 36, 1786–1811. [Google Scholar] [CrossRef]
- Luirink, I.K.; Braamskamp, M.; Wiegman, A.; Hartgers, M.L.; Sjouke, B.; Defesche, J.C.; Hovingh, G.K. The clinical and molecular diversity of homozygous familial hypercholesterolemia in children: Results from the GeneTics of clinical homozygous hypercholesterolemia (GoTCHA) study. J. Clin. Lipidol. 2019, 13, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Staiano, A.; di Taranto, M.D.; Bloise, E.; D’Agostino, M.N.; D’Angelo, A.; Marotta, G.; Gentile, M.; Jossa, F.; Iannuzzi, A.; Rubba, P.; et al. Investigation of Single Nucleotide Polymorphisms Associated to Familial Combined Hyperlipidemia with Random Forests. In Neural Nets and Surroundings; Apolloni, B., Bassis, S., Esposito, A., Morabito, F.C., Eds.; Smart Innovation, Systems and Technologies; Springer: Berlin/Heidelberg, Germany, 2013; Volume 19, pp. 169–178. [Google Scholar]
- Di Taranto, M.D.; Staiano, A.; D’Agostino, M.N.; D’Angelo, A.; Bloise, E.; Morgante, A.; Marotta, G.; Gentile, M.; Rubba, P.; Fortunato, G. Association of USF1 and APOA5 polymorphisms with familial combined hyperlipidemia in an Italian population. Mol. Cell. Probes 2015, 29, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Averna, M.; Cefalu, A.B.; Casula, M.; Noto, D.; Arca, M.; Bertolini, S.; Calandra, S.; Catapano, A.L.; Tarugi, P. Familial hypercholesterolemia: The Italian Atherosclerosis Society Network (LIPIGEN). Atheroscler. Suppl. 2017, 29, 11–16. [Google Scholar] [CrossRef]
- Pirillo, A.; Garlaschelli, K.; Arca, M.; Averna, M.; Bertolini, S.; Calandra, S.; Tarugi, P.; Catapano, A.L. Spectrum of mutations in Italian patients with familial hypercholesterolemia: New results from the LIPIGEN study. Atheroscler. Suppl. 2017, 29, 17–24. [Google Scholar] [CrossRef]
- Casula, M.; Olmastroni, E.; Pirillo, A.; Catapano, A.L. Evaluation of the performance of Dutch Lipid Clinic Network score in an Italian FH population: The LIPIGEN study. Atherosclerosis 2018, 277, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Ruotolo, A.; Di Taranto, M.D.; D’Agostino, M.N.; Marotta, G.; Gentile, M.; Nunziata, M.; Sodano, M.; Di Noto, R.; Del Vecchio, L.; Rubba, P.; et al. The novel variant p.Ser465Leu in the PCSK9 gene does not account for the decreased LDLR activity in members of a FH family. Clin. Chem. Lab. Med. 2014, 52, e175–e178. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Di Taranto, M.D.; Benito-Vicente, A.; Giacobbe, C.; Uribe, K.B.; Rubba, P.; Etxebarria, A.; Guardamagna, O.; Gentile, M.; Martin, C.; Fortunato, G. Identification and in vitro characterization of two new PCSK9 Gain of Function variants found in patients with Familial Hypercholesterolemia. Sci. Rep. 2017, 7, 15282. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Romano, M.; Di Taranto, M.D.; Mirabelli, P.; D’Agostino, M.N.; Iannuzzi, A.; Marotta, G.; Gentile, M.; Raia, M.; Di Noto, R.; Del Vecchio, L.; et al. An improved method on stimulated T-lymphocytes to functionally characterize novel and known LDLR mutations. J. Lipid Res. 2011, 52, 2095–2100. [Google Scholar] [CrossRef]
- Romano, M.; Di Taranto, M.D.; D’Agostino, M.N.; Marotta, G.; Gentile, M.; Abate, G.; Mirabelli, P.; Di Noto, R.; Del Vecchio, L.; Rubba, P.; et al. Identification and functional characterization of LDLR mutations in familial hypercholesterolemia patients from Southern Italy. Atherosclerosis 2010, 210, 493–496. [Google Scholar] [CrossRef]
- Tang, R.; Hennig, M.; Thomasson, B.; Scherz, R.; Ravinetto, R.; Catalini, R.; Rubba, P.; Zanchetti, A.; Bond, M.G. Baseline reproducibility of B-mode ultrasonic measurement of carotid artery intima-media thickness: The European Lacidipine Study on Atherosclerosis (ELSA). J. Hypertens. 2000, 18, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Rubba, P.; Gentile, M.; Marotta, G.; Iannuzzi, A.; Sodano, M.; De Simone, B.; Jossa, F.; Iannuzzo, G.; Giacobbe, C.; Di Taranto, M.D.; et al. Causative mutations and premature cardiovascular disease in patients with heterozygous familial hypercholesterolaemia. Eur. J. Prev. Cardiol. 2017, 24, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Gentile, M.; Iannuzzo, G.; Mattiello, A.; Marotta, G.; Iannuzzi, A.; Panico, S.; Rubba, P. Association between Lp (a) and atherosclerosis in menopausal women without metabolic syndrome. Biomark. Med. 2016, 10, 397–402. [Google Scholar] [CrossRef] [PubMed]
- De Michele, M.; Panico, S.; Iannuzzi, A.; Celentano, E.; Ciardullo, A.V.; Galasso, R.; Sacchetti, L.; Zarrilli, F.; Bond, M.G.; Rubba, P. Association of obesity and central fat distribution with carotid artery wall thickening in middle-aged women. Stroke 2002, 33, 2923–2928. [Google Scholar] [CrossRef]
- Futema, M.; Whittall, R.A.; Kiley, A.; Steel, L.K.; Cooper, J.A.; Badmus, E.; Leigh, S.E.; Karpe, F.; Neil, H.A.; Humphries, S.E. Analysis of the frequency and spectrum of mutations recognised to cause familial hypercholesterolaemia in routine clinical practice in a UK specialist hospital lipid clinic. Atherosclerosis 2013, 229, 161–168. [Google Scholar] [CrossRef]
- Hobbs, H.H.; Brown, M.S.; Goldstein, J.L. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum. Mutat. 1992, 1, 445–466. [Google Scholar] [CrossRef]
- Jensen, H.K.; Jensen, L.G.; Holst, H.U.; Andreasen, P.H.; Hansen, P.S.; Larsen, M.L.; Kolvraa, S.; Bolund, L.; Gregersen, N.; Faergeman, O. Normolipidemia and hypercholesterolemia in persons heterozygous for the same 1592 + 5G --> A splice site mutation in the low-density lipoprotein receptor gene. Clin. Genet. 1999, 56, 378–388. [Google Scholar] [CrossRef]
- Holla, O.; Teie, C.; Berge, K.E.; Leren, T.P. Identification of deletions and duplications in the low density lipoprotein receptor gene by MLPA. Clin. Chim. Acta Int. J. Clin. Chem. 2005, 356, 164–171. [Google Scholar] [CrossRef]
- Bertolini, S.; Cassanelli, S.; Garuti, R.; Ghisellini, M.; Simone, M.L.; Rolleri, M.; Masturzo, P.; Calandra, S. Analysis of LDL receptor gene mutations in Italian patients with homozygous familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 408–418. [Google Scholar] [CrossRef]
- Bochmann, H.; Geisel, J.; Herrmann, W.; Purcz, T.; Reuter, W.; Julius, U.; Metzler, W.; Bergmann, S.; Jaross, W.; Gehrisch, S. Eight novel LDL receptor gene mutations among patients under LDL apheresis in Dresden and Leipzig. Hum. Mutat. 2001, 17, 76–77. [Google Scholar] [CrossRef]
- Durst, R.; Ibe, U.K.; Shpitzen, S.; Schurr, D.; Eliav, O.; Futema, M.; Whittall, R.; Szalat, A.; Meiner, V.; Knobler, H.; et al. Molecular genetics of familial hypercholesterolemia in Israel-revisited. Atherosclerosis 2017, 257, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Kawashiri, M.A.; Noguchi, T.; Mori, M.; Tsuchida, M.; Takata, M.; Nohara, A.; Inazu, A.; Kobayashi, J.; Yachie, A.; et al. A novel method for determining functional LDL receptor activity in familial hypercholesterolemia: Application of the CD3/CD28 assay in lymphocytes. Clin. Chim. Acta Int. J. Clin. Chem. 2009, 400, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Nohara, A.; Higashikata, T.; Lu, H.; Inazu, A.; Mabuchi, H. Molecular genetic analysis of familial hypercholesterolemia: Spectrum and regional difference of LDL receptor gene mutations in Japanese population. Atherosclerosis 2002, 165, 335–342. [Google Scholar] [CrossRef]
- Sjouke, B.; Kusters, D.M.; Kindt, I.; Besseling, J.; Defesche, J.C.; Sijbrands, E.J.; Roeters van Lennep, J.E.; Stalenhoef, A.F.; Wiegman, A.; de Graaf, J.; et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: Prevalence, genotype-phenotype relationship, and clinical outcome. Eur. Heart J. 2015, 36, 560–565. [Google Scholar] [CrossRef]
- Bertolini, S.; Pisciotta, L.; Rabacchi, C.; Cefalu, A.B.; Noto, D.; Fasano, T.; Signori, A.; Fresa, R.; Averna, M.; Calandra, S. Spectrum of mutations and phenotypic expression in patients with autosomal dominant hypercholesterolemia identified in Italy. Atherosclerosis 2013, 227, 342–348. [Google Scholar] [CrossRef]
- Sanchez-Hernandez, R.M.; Civeira, F.; Stef, M.; Perez-Calahorra, S.; Almagro, F.; Plana, N.; Novoa, F.J.; Saenz-Aranzubia, P.; Mosquera, D.; Soler, C.; et al. Homozygous Familial Hypercholesterolemia in Spain: Prevalence and Phenotype-Genotype Relationship. Circ. Cardiovasc. Genet. 2016, 9, 504–510. [Google Scholar] [CrossRef]
- Di Taranto, M.D.; de Falco, R.; Guardamagna, O.; Massini, G.; Giacobbe, C.; Auricchio, R.; Malamisura, B.; Proto, M.; Palma, D.; Greco, L.; et al. Lipid profile and genetic status in a familial hypercholesterolemia pediatric population: Exploring the LDL/HDL ratio. Clin. Chem. Lab. Med. 2019, 57, 1102–1110. [Google Scholar] [CrossRef]
- Moriguchi, E.H.; Tamachi, H.; Goto, Y. Hepatic lipase activity and high density lipoproteins in familial hypercholesterolemia: Adaptational mechanisms for LDL-receptor deficient state. Tokai J. Exp. Clin. Med. 1990, 15, 401–406. [Google Scholar]
- Ganjali, S.; Momtazi, A.A.; Banach, M.; Kovanen, P.T.; Stein, E.A.; Sahebkar, A. HDL abnormalities in familial hypercholesterolemia: Focus on biological functions. Prog. Lipid Res. 2017, 67, 16–26. [Google Scholar] [CrossRef]
- Chora, J.R.; Medeiros, A.M.; Alves, A.C.; Bourbon, M. Analysis of publicly available LDLR, APOB, and PCSK9 variants associated with familial hypercholesterolemia: Application of ACMG guidelines and implications for familial hypercholesterolemia diagnosis. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 591–598. [Google Scholar] [CrossRef]
- Di Taranto, M.D.; D’Agostino, M.N.; Fortunato, G. Functional characterization of mutant genes associated with autosomal dominant familial hypercholesterolemia: Integration and evolution of genetic diagnosis. Nutr. Metab. Cardiovasc. Dis. NMCD 2015, 25, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Chan, K.C.; Tarabocchia, M.; Benito-Vicente, A.; Alves, A.C.; Uribe, K.B.; Bourbon, M.; Skiba, P.J.; Pordy, R.; Gipe, D.A.; et al. Functional Analysis of LDLR (Low-Density Lipoprotein Receptor) Variants in Patient Lymphocytes to Assess the Effect of Evinacumab in Homozygous Familial Hypercholesterolemia Patients With a Spectrum of LDLR Activity. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2248–2260. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Patient | Age (Years) | Gender | Age of the First CHD Event (Years) | Genetic Status | LDLR Variant (Nucleotide) | LDLR Variant (Protein) | Variant Classification | Variant References |
|---|---|---|---|---|---|---|---|---|
| HoFH-1 | 29 | Female | - | Comp. Heter. | c.1135T > C + c.1567G > A | p.(Cys379Arg) + p.(Val523Met) | Defective/Defective | Both variants [20] |
| HoFH-2 a | 53 | Male | - | Comp. Heter. | c.1130G > T + c.2476C > A | p.(Cys377Phe) + p.(Pro826Thr) | Defective/Defective | [20]/[26] |
| HoFH-3 | 33 | Female | 30 | Comp. Heter. | c.1646G > A + c.1739C > T | p.(Gly549Asp) + p.(Ser580Phe) | Defective/Defective | Both variants [20] |
| HoFH-4 | 29 | Male | 32 | Comp. Heter. | c.367T > C + c.1478_1479delCT | p.(Ser123Pro) + p.(Ser493Cysfs*42) | Defective/Null | [20] (functional activity was assayed in cells from this patient) |
| HoFH-5 | 46 | Male | - | Comp. Heter. | c.304C > T and c.718G > A | p.(Gln102*) and p.(Glu240Lys) | Null/Defective | [27]/[15] |
| HoFH-6 | 35 | Male | 24 | Homoz. | c.1135T > C | p.(Cys379Arg) | Defective | [20] |
| HoFH-7 b | 20 | Female | - | Homoz. | c.1775G > A | p.(Gly592Glu) | Defective | [20] |
| HoFH-8 b | 22 | Male | - | Homoz. | c.1775G > A | p.(Gly592Glu) | Defective | [20] |
| HoFH-9 | 48 | Female | - | Comp. Heter. | c.727T > C + c.1775G > A | p.(Cys243Arg) + p.(Gly592Glu) | Defective/Defective | [28]/[20] |
| HoFH-10 | 55 | Female | 55 | Comp. Heter. | c.974G > A + c.(940 + 1_941-1)_(2311 + 1_2312-1)dup | p.(Cys325Tyr) + p.(Gly314_Gln770dup) | Defective/Null | [20]/[29] |
| HoFH-11 | 36 | Male | - | Comp. Heter. | c.352G > T + c.1646G > A | p.(Asp118Tyr) + p.(Gly549Asp) | Defective/Defective | [30]/[20] |
| HoFH-12 | 49 | Female | - | Comp. Heter. | c.323C > T + c.1586 + 1G > A | p.(Thr108Met) + p.(Thr454_Gly529de) and p.(Gly529_Phe530ins22) | Defective/Null | [31]/[30] |
| HoFH-13 | 62 | Female | 61 | Comp. Heter. | c.1135T > C + c.1586 + 5G > A | p.(Cys379Arg) + p.(Thr454_Gly529de) | Defective/Null | [20]/[28] |
| HoFH-14 | 46 | Male | 34 | Homoz. | c.-156C > T | p.(?) | Defective | [32] |
| HoFH-15 a | 44 | Female | - | Comp. Heter. | c.1130G > T + c.2476C > A | p.(Cys377Phe) + p.(Pro826Thr) | Defective/Defective | [20]/[26] |
| HoFH-16 | 64 | Female | 49 | Comp. Heter. | c.1775G > A + c.2054C > T | p.(Gly592Glu) + p.(Pro685Leu) | Defective/Defective | [20]/[33] |
| HoFH-17 | 28 | Female | - | Comp. Heter. | c.1775G > A + c.2054C > T | p.(Gly592Glu) + p.(Pro685Leu) | Defective/Defective | [20]/[33] |
| HoFH-18 | 63 | Male | 32 | Comp. Heter. | c.463T > C + c.1567G > A | p.(Cys155Arg) + p.(Val523Met) | Defective/Defective | [34]/[20] |
| HoFH-19 | 40 | Female | 40 | Comp. Heter. | c.1567G > A + c.2054C > T | p.(Val523Met) + p.(Pro685Leu) | Defective/Defective | [20]/[33] |
| HoFH-20 c | 7 | Female | - | Comp. Heter. | c.407A > T + c.1775G > A | p.(Asp136Val) + p.(Gly592Glu) | Defective/Defective | [20] (functional activity was assayed in cells from this patient) |
| HoFH-21 | 10 | Male | - | Comp. Heter. | c.1135T > C + c.1775G > A | p.(Cys379Arg) + p.(Gly592Glu) | Defective/Defective | Both variants [20] |
| HoFH-22 c | 4 | Male | - | Comp. Heter. | c.1135T > C + c.1775G > A | p.(Cys379Arg) + p.(Gly592Glu) | Defective/Defective | Both variants [20] |
| HoFH-23 | 9 | Male | - | Comp. Heter. | c.1739C > T + c.1775G > A | p.(Ser580Phe) + p.(Gly592Glu) | Defective/Defective | [20] (functional activity was assayed in cells from this patient) |
| Parameter | Total n = 23 | Homozygotes n = 4 | Compound Heterozygotes n = 19 | Statistical Significance |
|---|---|---|---|---|
| Age at genetic diagnosis, years | 36.2 ± 18.2 | 30.7 ± 12.1 | 37.3 ± 19.3 | ns |
| Pre-therapy Total cholesterol, mmol/L | 15.2 ± 2.8 | 18.9 ± 0.4 | 14.4 ± 2.5 | p = 0.009 |
| Pre-therapy LDL-cholesterol, mmol/L | 12.9 ± 2.9 | 15.8 ± 3.0 | 12.3 ± 2.5 | p = 0.026 |
| Pre-therapy HDL-cholesterol, mmol/L | 1.1 ± 0.2 | 1.1 ± 0.1 | 1.1 ± 0.2 | ns |
| Pre-therapy Non-HDL cholesterol, mmol/L | 14.1 ± 2.9 | 17.7 ± 0.4 | 13.4 ± 2.5 | p = 0.011 |
| Pre-therapy LDL/HDL cholesterol ratio | 13.9 ± 5.7 | 15.4 ± 0.6 | 13.6 ± 6.3 | ns |
| Pre-therapy Triglycerides, mmol/L | 1.2 ± 0.6 | 1.0 ± 0.1 | 1.3 ± 0.6 | ns |
| Post-therapy 1 Total cholesterol, mmol/L | 8.8 ± 2.0 | 9.8 ± 2.4 | 8.2 ± 2.2 | ns |
| Post-therapy 1 LDL-cholesterol, mmol/L | 7.2 ± 1.8 | 8.3 ± 2.2 | 6.6 ± 2.0 | ns |
| Post-therapy 1 HDL-cholesterol, mmol/L | 1.1 ± 0.2 | 1.1 ± 0.1 | 1.1 ± 0.2 | ns |
| Post-therapy 1 Non-HDL cholesterol, mmol/L | 7.7 ± 2.0 | 8.7 ± 2.3 | 7.1 ± 2.2 | ns |
| Post-therapy 1 LDL/HDL cholesterol ratio | 6.8 ± 2.2 | 7.7 ± 1.4 | 6.2 ± 2.0 | ns |
| Post-therapy 1 Triglycerides, mmol/L | 1.1 ± 0.7 | 0.9 ± 0.4 | 1.1 ± 0.7 | ns |
| Post-therapy 2 Total cholesterol, mmol/L | 6.8 ± 3.1 | 10.1 ± 2.9 | 5.7 ± 2.4 | p = 0.025 |
| Post-therapy 2 LDL-cholesterol, mmol/L | 5.1 ± 3.1 | 8.6 ± 2.7 | 4.0 ± 2.4 | p = 0.020 |
| Post-therapy 2 HDL-cholesterol, mmol/L | 1.2 ± 0.2 | 1.1 ± 0.2 | 1.2 ± 0.2 | ns |
| Post-therapy 2 Non-HDL cholesterol, mmol/L | 5.6 ± 3.1 | 8.9 ± 2.8 | 4.5 ± 2.4 | p = 0.023 |
| Post-therapy 2 LDL/HDL cholesterol ratio | 4.2 ± 2.6 | 7.4 ± 1.7 | 3.3 ± 2.1 | p = 0.012 |
| Post-therapy 2 Triglycerides, mmol/L | 1.0 ± 0.5 | 0.8 ± 0.2 | 1.1 ± 0.6 | ns |
| Tendon xanthomas, n (%) | 19/23 (82.6%) | 4/4 (100%) | 15/19 (78.9%) | ns |
| Corneal arcus, n (%) | 11/23 (47.8%) | 3/4 (75.0%) | 8/19 (42.1%) | ns |
| Premature coronary heart disease *, n (%) | 9/19 (47.4%) | 2/4 (50%) | 7/15 (46.7%) | ns |
| Premature cerebral or peripheral vascular disease *, n (%) | 3/19 (15.8%) | 1/4 (25.0%) | 2/15 (13.3%) | ns |
| Presence of carotid plaque *, n (%) | 13/19 (68.4%) | 2/4 (50.0%) | 11/15 (73.3%) | ns |
| First degree with Tendon xanthoma and/or Corneal arcus, n (%) | 7/23 (30.4%) | 4/4 (100%) | 3/19 (15.8%) | p = 0.001 |
| First degree relative with premature coronary heart disease, n (%) | 13/23 (56.5%) | 2/4 (50.0%) | 11/19 (57.9%) | ns |
| First degree with LDL-cholesterol higher than 4.9 mmol/L, n (%) | 18/23 (78.3%) | 4/4 (100%) | 14/19 (73.7%) | ns |
| Minor relatives with LDL-cholesterol higher than 4.1 mmol/L, n (%) | 9/23 (39.1%) | 4/4 (100%) | 5/19 (26.3%) | p = 0.006 |
| BMI, kg/m2 | 25.4 ± 5.2 | 22.9 ± 2.1 | 26.1 ± 5.7 | ns |
| DLCN score (without genetics) * | 15.6 ± 2.6 range 8–18 | 17.0 ± 1.2 range 16–18 | 15.2 ± 2.8 range 8–18 | ns |
| DLCN score (with genetics) * | 23.6 ± 2.6 range 16–26 | 25.0 ± 1.2 range 24–26 | 23.2 ± 2.8 range 16–26 | ns |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Taranto, M.D.; Giacobbe, C.; Buonaiuto, A.; Calcaterra, I.; Palma, D.; Maione, G.; Iannuzzo, G.; Di Minno, M.N.D.; Rubba, P.; Fortunato, G. A Real-World Experience of Clinical, Biochemical and Genetic Assessment of Patients with Homozygous Familial Hypercholesterolemia. J. Clin. Med. 2020, 9, 219. https://doi.org/10.3390/jcm9010219
Di Taranto MD, Giacobbe C, Buonaiuto A, Calcaterra I, Palma D, Maione G, Iannuzzo G, Di Minno MND, Rubba P, Fortunato G. A Real-World Experience of Clinical, Biochemical and Genetic Assessment of Patients with Homozygous Familial Hypercholesterolemia. Journal of Clinical Medicine. 2020; 9(1):219. https://doi.org/10.3390/jcm9010219
Chicago/Turabian StyleDi Taranto, Maria Donata, Carola Giacobbe, Alessio Buonaiuto, Ilenia Calcaterra, Daniela Palma, Giovanna Maione, Gabriella Iannuzzo, Matteo Nicola Dario Di Minno, Paolo Rubba, and Giuliana Fortunato. 2020. "A Real-World Experience of Clinical, Biochemical and Genetic Assessment of Patients with Homozygous Familial Hypercholesterolemia" Journal of Clinical Medicine 9, no. 1: 219. https://doi.org/10.3390/jcm9010219
APA StyleDi Taranto, M. D., Giacobbe, C., Buonaiuto, A., Calcaterra, I., Palma, D., Maione, G., Iannuzzo, G., Di Minno, M. N. D., Rubba, P., & Fortunato, G. (2020). A Real-World Experience of Clinical, Biochemical and Genetic Assessment of Patients with Homozygous Familial Hypercholesterolemia. Journal of Clinical Medicine, 9(1), 219. https://doi.org/10.3390/jcm9010219