Characterization and Analysis of the Skin Microbiota in Acne: Impact of Systemic Antibiotics



Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Antibiotic Treatment and Sample Collection
2.3. DNA Extraction and 16S rRNA Gene Polymerase Chain Reaction Amplification and Sequencing
2.4. Data Analysis
2.5. Statistical Analysis
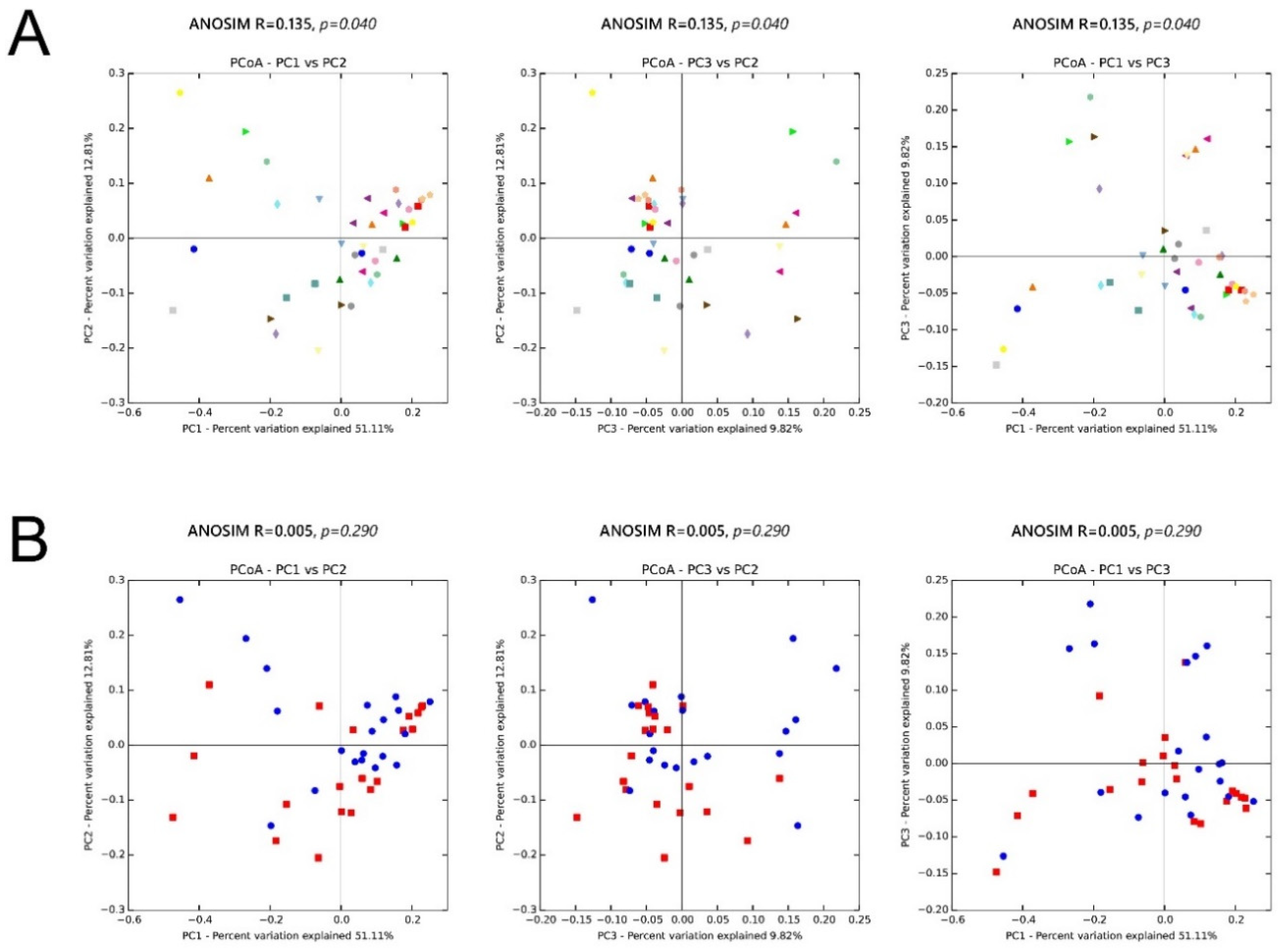
3. Results
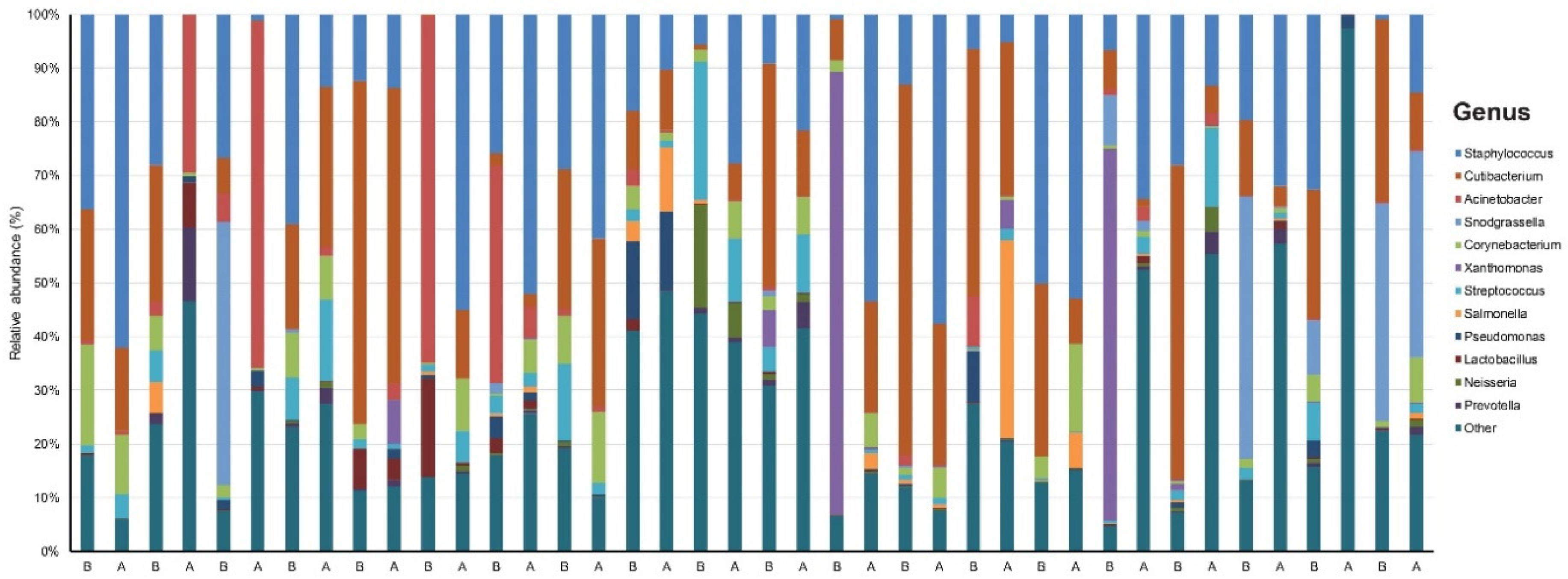
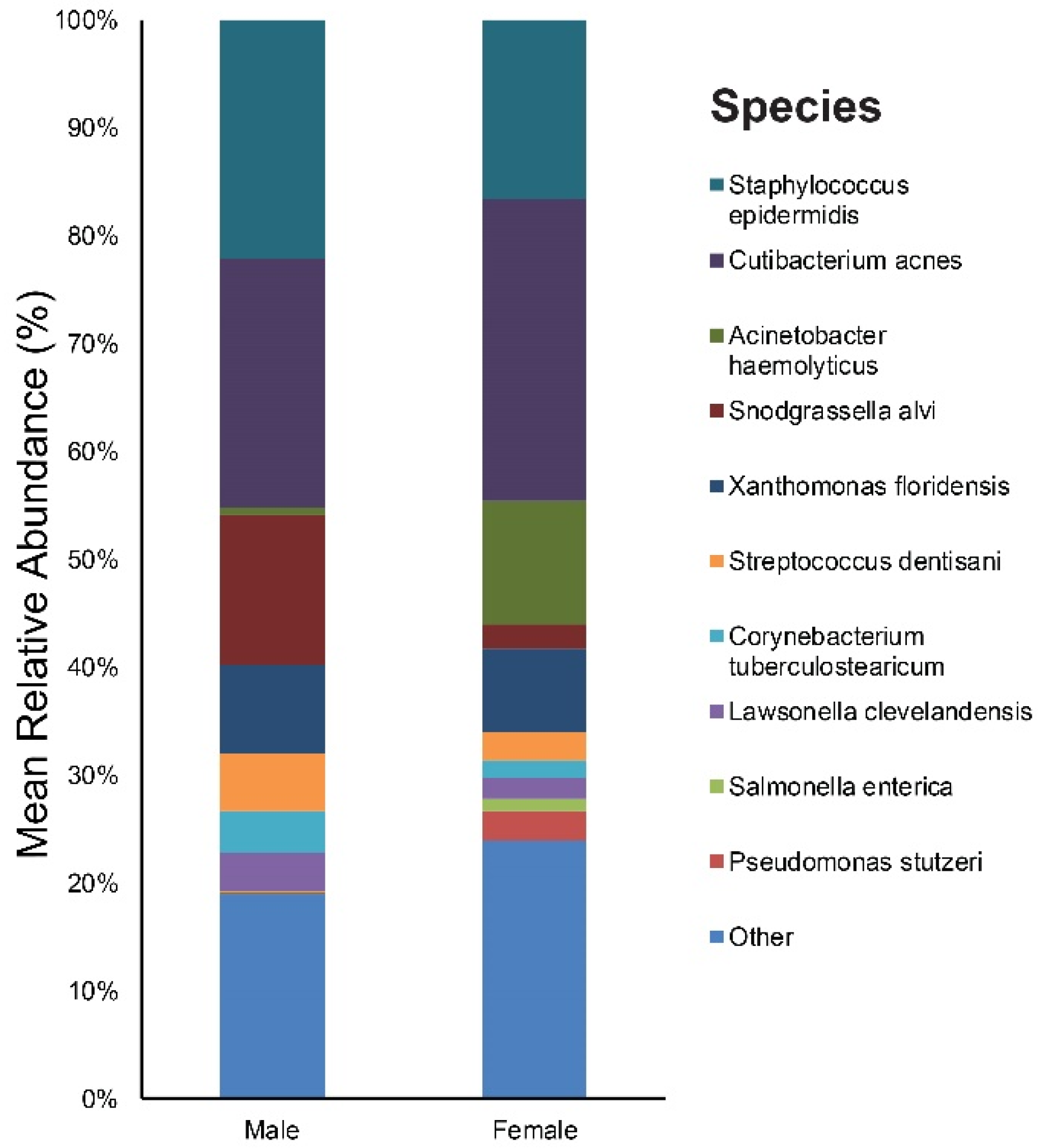
3.1. Taxonomic Assignment
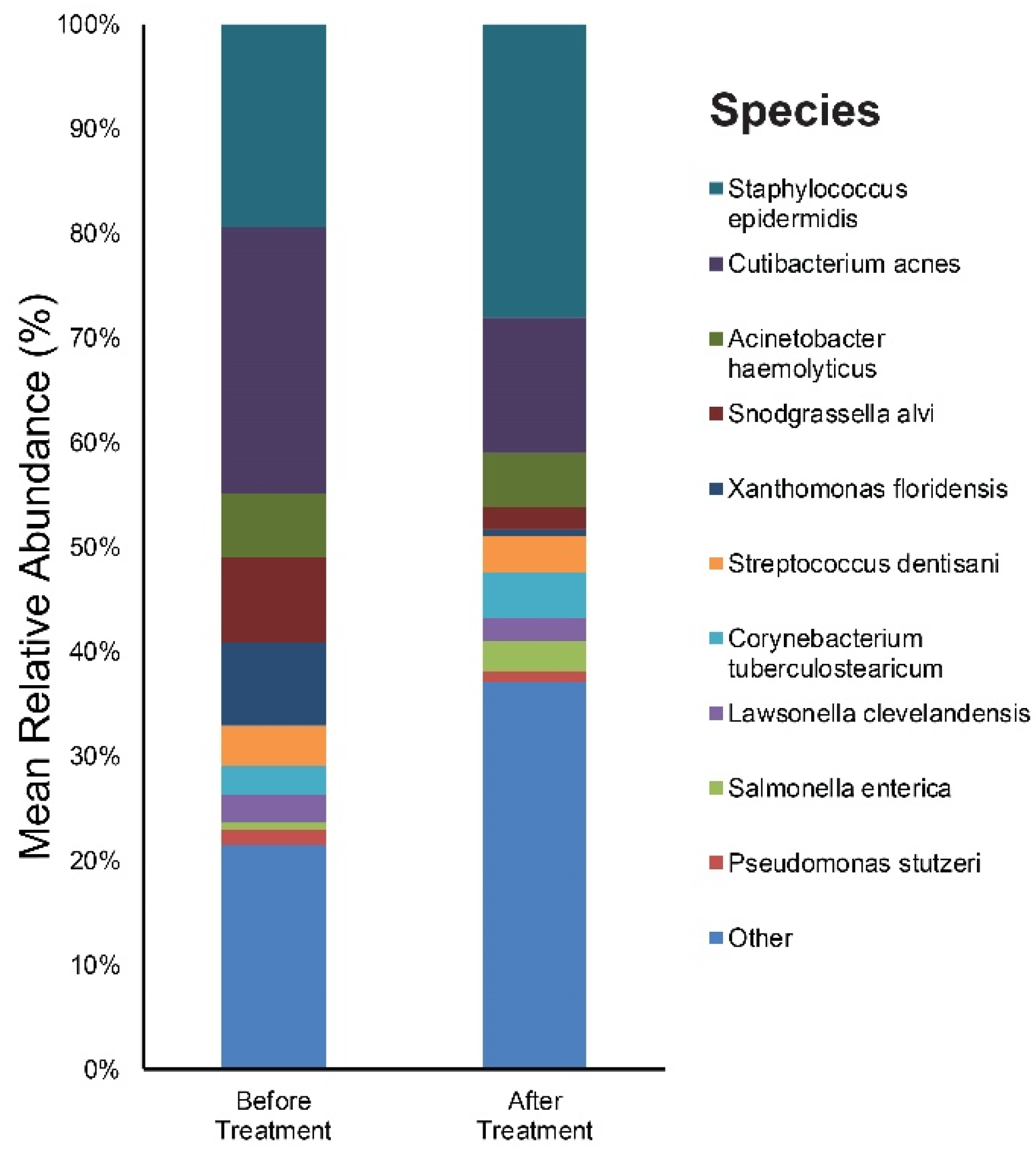
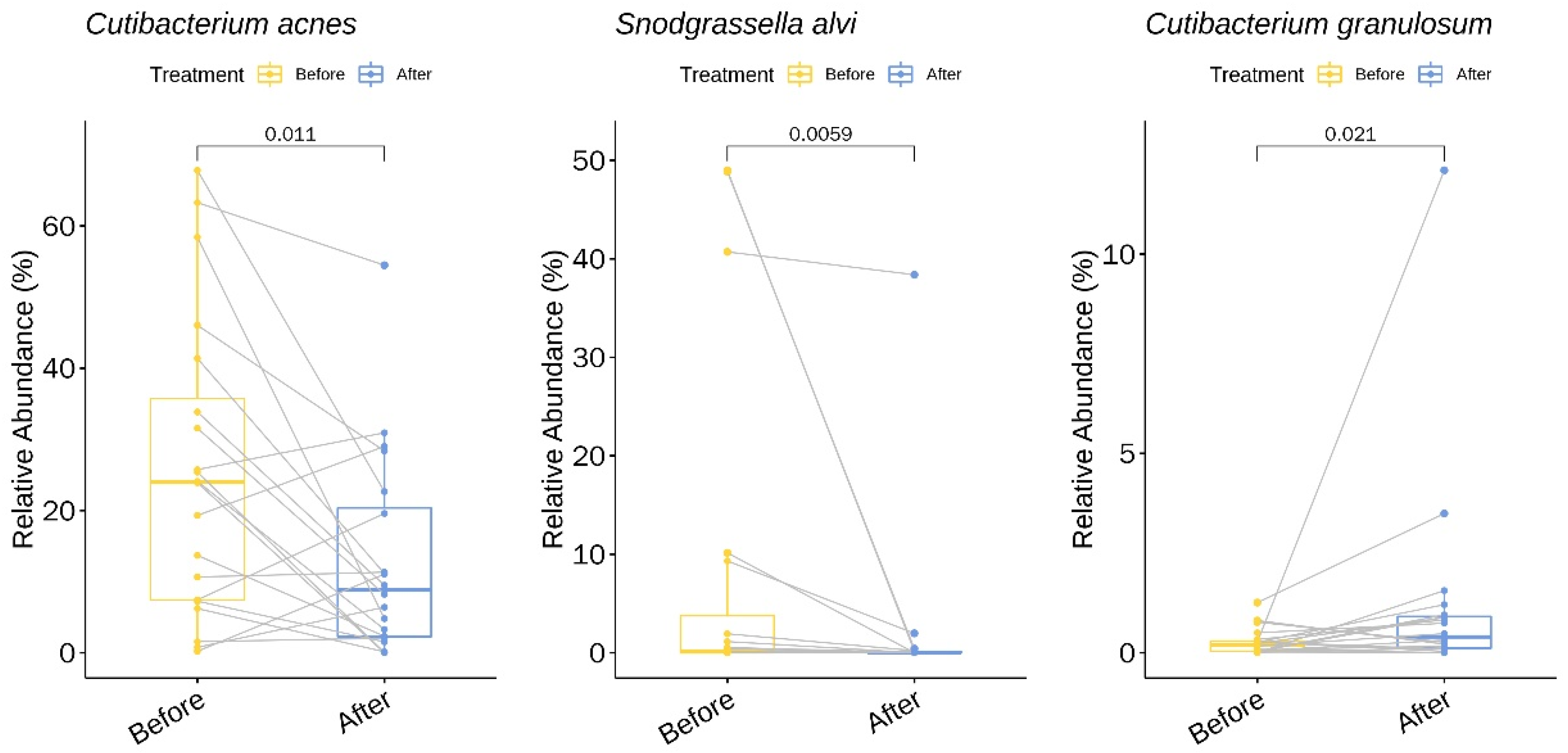
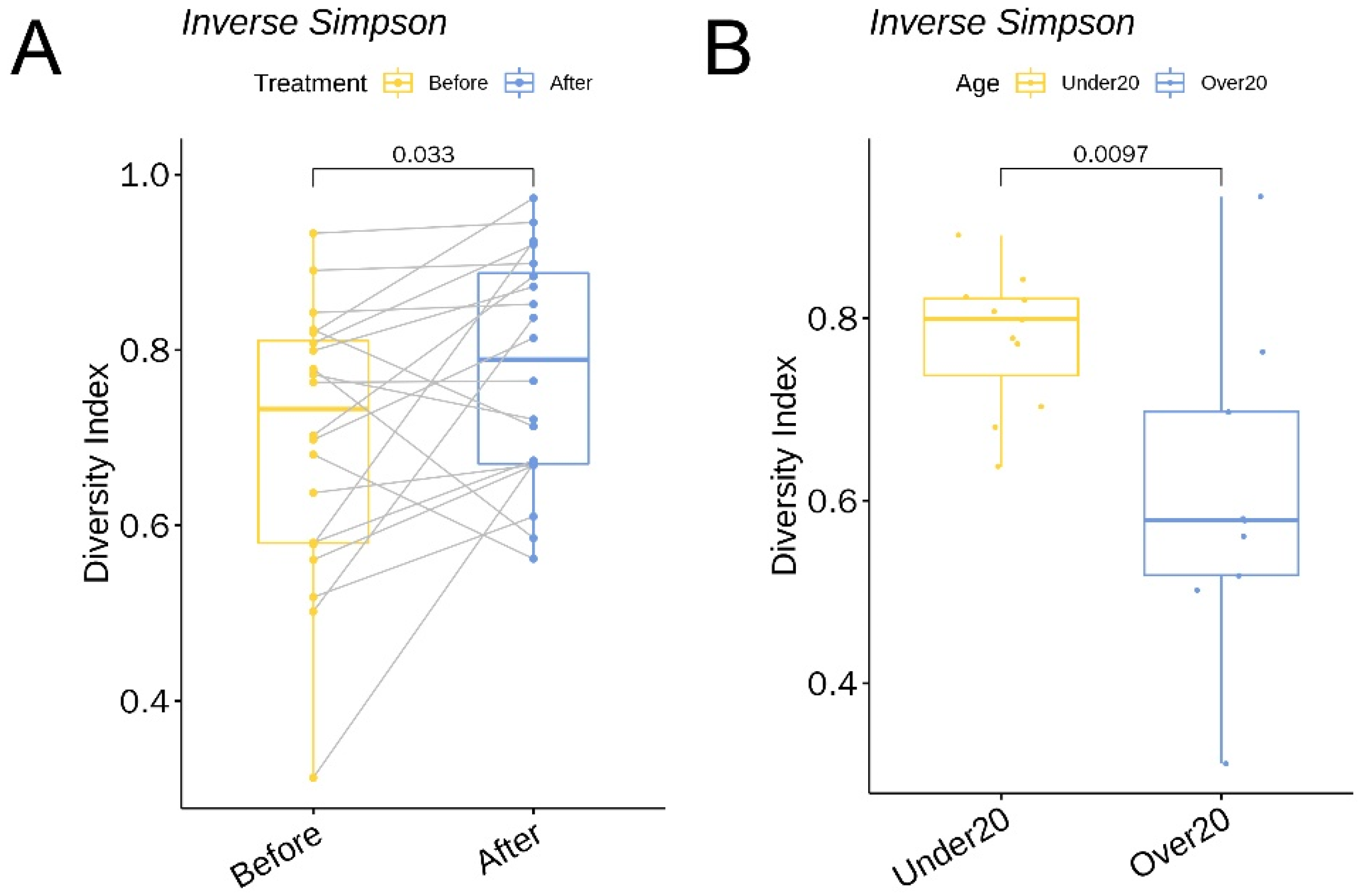
3.2. Relative Abundance of Individual Bacterial Taxa Pre- and Post-Doxycycline Treatment
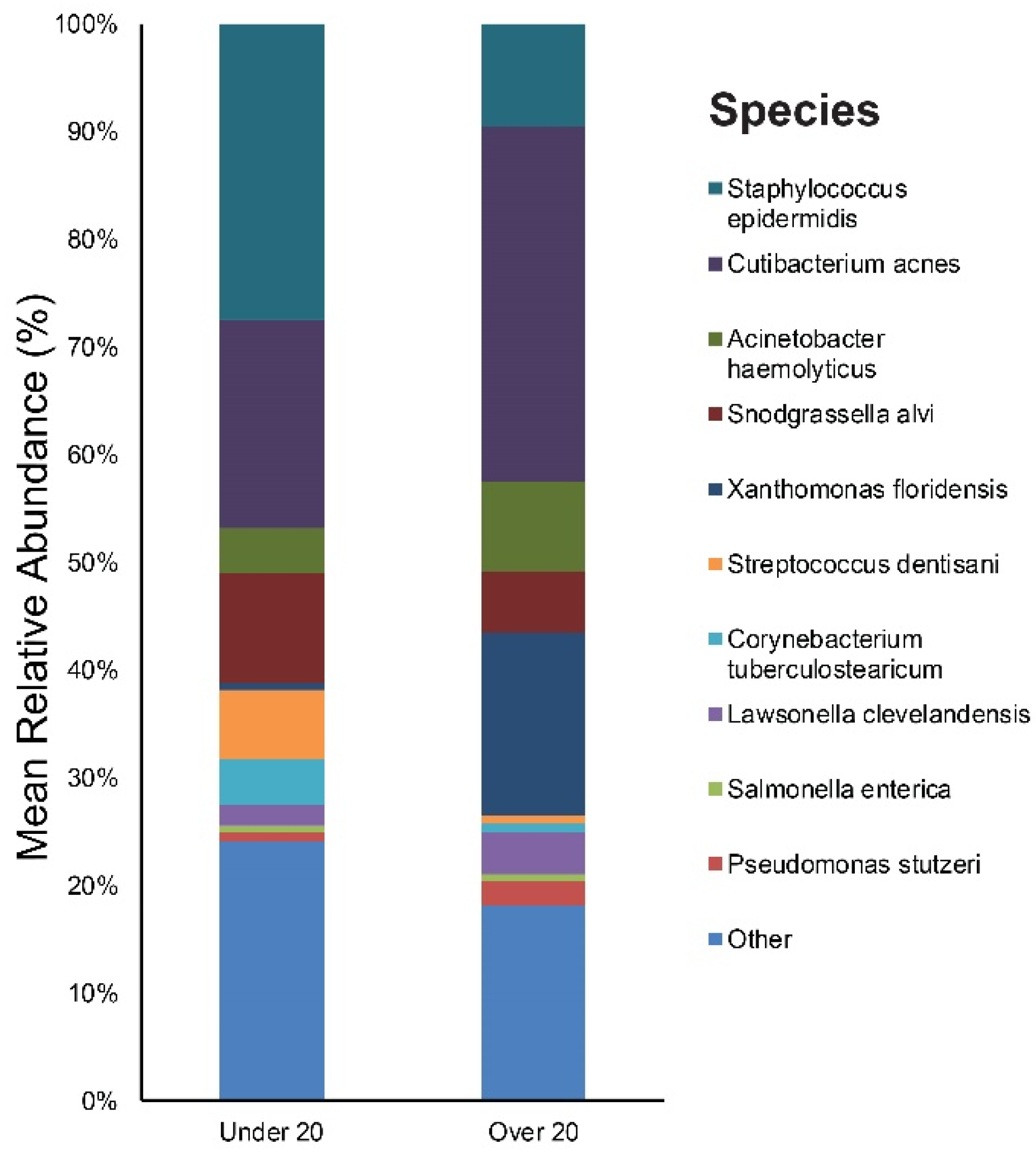
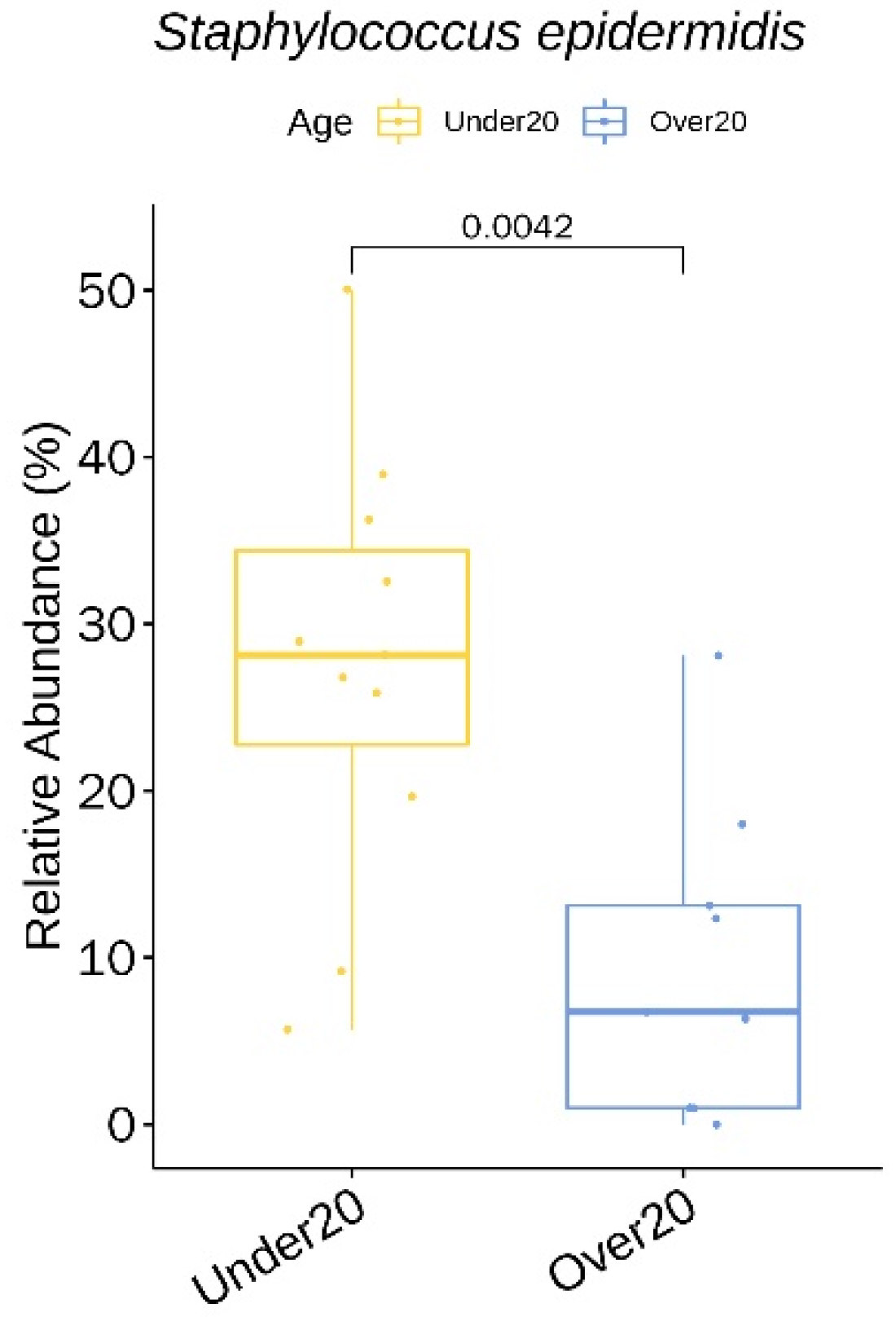
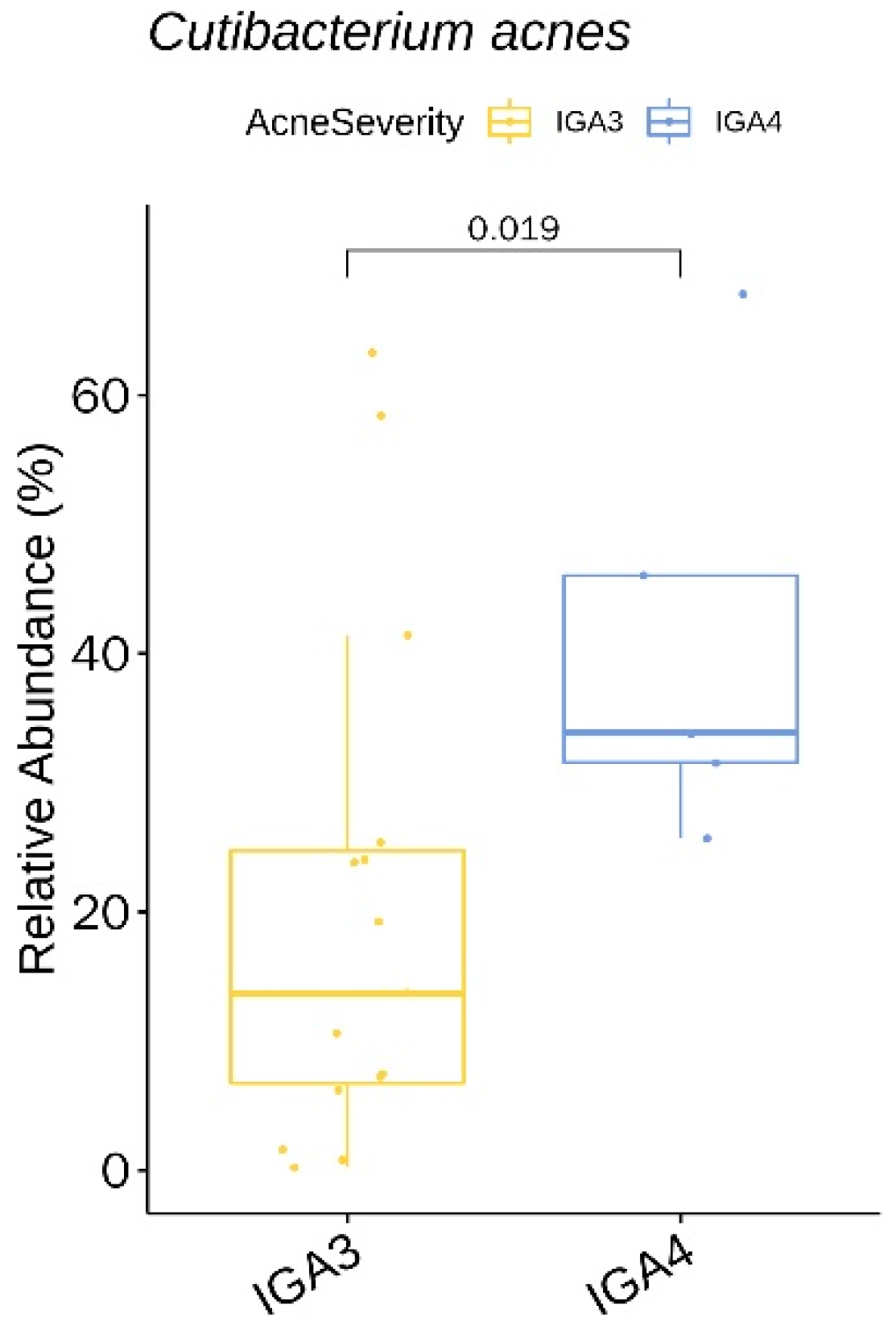
3.3. Relative Abundance of Individual Bacterial Taxa at Baseline According to Age, Sex, and Acne Severity
3.4. α Diversity
3.5. β Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Xu, H.; Li, H. Acne, the skin microbiome, and antibiotic treatment. Am. J. Clin. Derm. 2019, 20, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.B.; Byun, E.J.; Kim, H.S. Potential role of the microbiome in acne: A comprehensive review. J. Clin Med. 2019, 8, 987. [Google Scholar] [CrossRef]
- Kelhala, H.L.; Aho, V.T.E.; Fyhrquist, N.; Pereira, P.A.B.; Kubin, M.E.; Paulin, L.; Palatsi, R.; Auvinen, P.; Tasanen, K.; Lauerma, A. Isotretinoin and lymecycline treatments modify the skin microbiota in acne. Exp. Derm. 2018, 27, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Dreno, B.; Martin, R.; Moyal, D.; Henley, J.B.; Khammari, A.; Seite, S. Skin microbiome and acne vulgaris: Staphylococcus, a new actor in acne. Exp. Derm. 2017, 26, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Chien, A.L.; Tsai, J.; Leung, S.; Mongodin, E.F.; Nelson, A.M.; Kang, S.; Garza, L.A. Association of systemic antibiotic treatment of acne with skin microbiota characteristics. JAMA Derm. 2019, 155, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.O.; Fricovsky, E.; Ceballos, G.; Villarreal, F. Tetracyclines: A pleitropic family of compounds with promising therapeutic properties. Review of the literature. Am. J. Physiol. Cell Physiol. 2010, 299, C539–C548. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; Green, E.D.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef]
- Panda, S.; El khader, I.; Casellas, F.; Lopez Vivancos, J.; Garcia Cors, M.; Santiago, A.; Cuenca, S.; Guarner, F.; Manichanh, C. Short-term effect of antibiotics on human gut microbiota. PLoS ONE 2014, 9, e95476. [Google Scholar] [CrossRef]
- Roghmann, M.C.; Lydecker, A.D.; Hittle, L.; DeBoy, R.T.; Nowak, R.G.; Johnson, J.K.; Mongodin, E.F. Comparison of the microbiota of older adults living in nursing homes and the community. mSphere 2017, 2. [Google Scholar] [CrossRef]
- Ishaq, H.M.; Mohammad, I.S.; Shahzad, M.; Ma, C.; Raza, M.A.; Wu, X.; Guo, H.; Shi, P.; Xu, J. Molecular alteration analysis of human gut microbial composition in graves’ disease patients. Int. J. Biol. Sci. 2018, 14, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Porcar, M.; Louie, K.B.; Kosina, S.M.; Van Goethem, M.W.; Bowen, B.P.; Tanner, K.; Northen, T.R. Microbial ecology on solar panels in Berkeley, CA, United States. Front. Microbiol. 2018, 9, 3043. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Fu, L.; Niu, B.; Wu, S.; Wooley, J. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief. Bioinform. 2012, 13, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Ramasamy, S.; Barnard, E.; Dawson, T.L., Jr.; Li, H. The role of the skin microbiota in acne pathophysiology. Br. J. Derm. 2019, 181, 691–699. [Google Scholar] [CrossRef]
- Zaenglein, A.L.; Pathy, A.L.; Schlosser, B.J.; Alikhan, A.; Baldwin, H.E.; Berson, D.S.; Bowe, W.P.; Graber, E.M.; Harper, J.C.; Kang, S.; et al. Guidelines of care for the management of acne vulgaris. J. Am. Acad. Derm. 2016, 74, 945–973.e33. [Google Scholar] [CrossRef]
- Oprica, C.; Emtestam, L.; Hagstromer, L.; Nord, C.E. Clinical and microbiological comparisons of isotretinoin vs. tetracycline in acne vulgaris. Acta Derm. Venereol. 2007, 87, 246–254. [Google Scholar] [CrossRef]
- Murillo, N.; Aubert, J.; Raoult, D. Microbiota of demodex mites from rosacea patients and controls. Microb. Pathog. 2014, 71–72, 37–40. [Google Scholar] [CrossRef]
- Akcinar, U.G.; Unal, E.; Dogruman Al, F. Demodex spp. as a possible aetiopathogenic factor of acne and relation with acne severity and type. Postepy Derm. Alergol. 2018, 35, 174–181. [Google Scholar] [CrossRef]
- Zhao, Y.E.; Hu, L.; Wu, L.P.; Ma, J.X. A meta-analysis of association between acne vulgaris and demodex infestation. J. Zhejiang Univ. Sci. B 2012, 13, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Bhate, K.; Williams, H.C. What’s new in acne? An analysis of systematic reviews published in 2011–2012. Clin. Exp. Derm. 2014, 39, 273–277, quiz 277–278. [Google Scholar] [CrossRef] [PubMed]
- Jahns, A.C.; Oprica, C.; Vassilaki, I.; Golovleva, I.; Palmer, R.H.; Alexeyev, O.A. Simultaneous visualization of propionibacterium acnes and propionibacterium granulosum with immunofluorescence and fluorescence in situ hybridization. Anaerobe 2013, 23, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Rainer, B.M.; Thompson, K.G.; Antonescu, C.; Florea, L.; Mongodin, E.F.; Bui, J.; Fischer, A.H.; Pasieka, H.B.; Garza, L.A.; Kang, S.; et al. Characterization and analysis of the skin microbiota in rosacea: A case-control study. Am. J. Clin. Derm. 2019. [Google Scholar] [CrossRef] [PubMed]
- Claudel, J.P.; Auffret, N.; Leccia, M.T.; Poli, F.; Corvec, S.; Dreno, B. Staphylococcus epidermidis: A potential new player in the physiopathology of acne? Dermatology 2019, 235, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kao, M.S.; Yu, J.; Huang, S.; Marito, S.; Gallo, R.L.; Huang, C.M. A precision microbiome approach using sucrose for selective augmentation of staphylococcus epidermidis fermentation against propionibacterium acnes. Int. J. Mol. Sci. 2016, 17, 1870. [Google Scholar] [CrossRef]
- Wang, Y.; Kuo, S.; Shu, M.; Yu, J.; Huang, S.; Dai, A.; Two, A.; Gallo, R.L.; Huang, C.M. Staphylococcus epidermidis in the human skin microbiome mediates fermentation to inhibit the growth of propionibacterium acnes: Implications of probiotics in acne vulgaris. Appl. Microbiol. Biotechnol. 2014, 98, 411–424. [Google Scholar] [CrossRef]
- Skabytska, Y.; Biedermann, T. Staphylococcus epidermidis sets things right again. J. Investig. Derm. 2016, 136, 559–560. [Google Scholar] [CrossRef]
- Yang, A.J.; Marito, S.; Yang, J.J.; Keshari, S.; Chew, C.H.; Chen, C.C.; Huang, C.M. A microtube array membrane (MTAM) encapsulated live fermenting staphylococcus epidermidis as a skin probiotic patch against cutibacterium acnes. Int. J. Mol. Sci. 2018, 20, 14. [Google Scholar] [CrossRef]
- Zhai, W.; Huang, Y.; Zhang, X.; Fei, W.; Chang, Y.; Cheng, S.; Zhou, Y.; Gao, J.; Tang, X.; Zhang, X.; et al. Profile of the skin microbiota in a healthy Chinese population. J. Derm. 2018, 45, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.B.; Cong, Z.; Imamura-Kawasawa, Y.; Kidd, B.A.; Dudley, J.T.; Thiboutot, D.M.; Nelson, A.M. Isolation and identification of the follicular microbiome: Implications for acne research. J. Investig. Derm. 2018, 138, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Fitz-Gibbon, S.; Tomida, S.; Chiu, B.H.; Nguyen, L.; Du, C.; Liu, M.; Elashoff, D.; Erfe, M.C.; Loncaric, A.; Kim, J.; et al. Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J. Investig. Derm. 2013, 133, 2152–2160. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| General Characteristics (n = 20) | |
|---|---|
| Sex (M/F), n (%) | 10/10 (50%) |
| Age (years), median (range) | 18 (11–44) |
| Fitzpatrick Skin Type, median (range) | 4 (3–5) |
| Duration of acne (years), median (range) | 2 (less than a year—10) |
| Baseline Acne severity (IGA), median (range) | 3 (3–4) |
| Acne severity (IGA) after 6 weeks of oral doxycycline, median (range) | 2 (1–3) |
| Baseline Inflamed Lesion Count, median (range) | 15 (6–30) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.-Y.; Kim, H.S.; Lee, S.H.; Kim, S. Characterization and Analysis of the Skin Microbiota in Acne: Impact of Systemic Antibiotics. J. Clin. Med. 2020, 9, 168. https://doi.org/10.3390/jcm9010168
Park S-Y, Kim HS, Lee SH, Kim S. Characterization and Analysis of the Skin Microbiota in Acne: Impact of Systemic Antibiotics. Journal of Clinical Medicine. 2020; 9(1):168. https://doi.org/10.3390/jcm9010168
Chicago/Turabian StylePark, Seo-Yeon, Hei Sung Kim, Se Hoon Lee, and Sungjoo Kim. 2020. "Characterization and Analysis of the Skin Microbiota in Acne: Impact of Systemic Antibiotics" Journal of Clinical Medicine 9, no. 1: 168. https://doi.org/10.3390/jcm9010168
APA StylePark, S.-Y., Kim, H. S., Lee, S. H., & Kim, S. (2020). Characterization and Analysis of the Skin Microbiota in Acne: Impact of Systemic Antibiotics. Journal of Clinical Medicine, 9(1), 168. https://doi.org/10.3390/jcm9010168