Hitting More Birds with a Stone: Impact of TGF-β on ILC Activity in Cancer


 ,
,  and
and 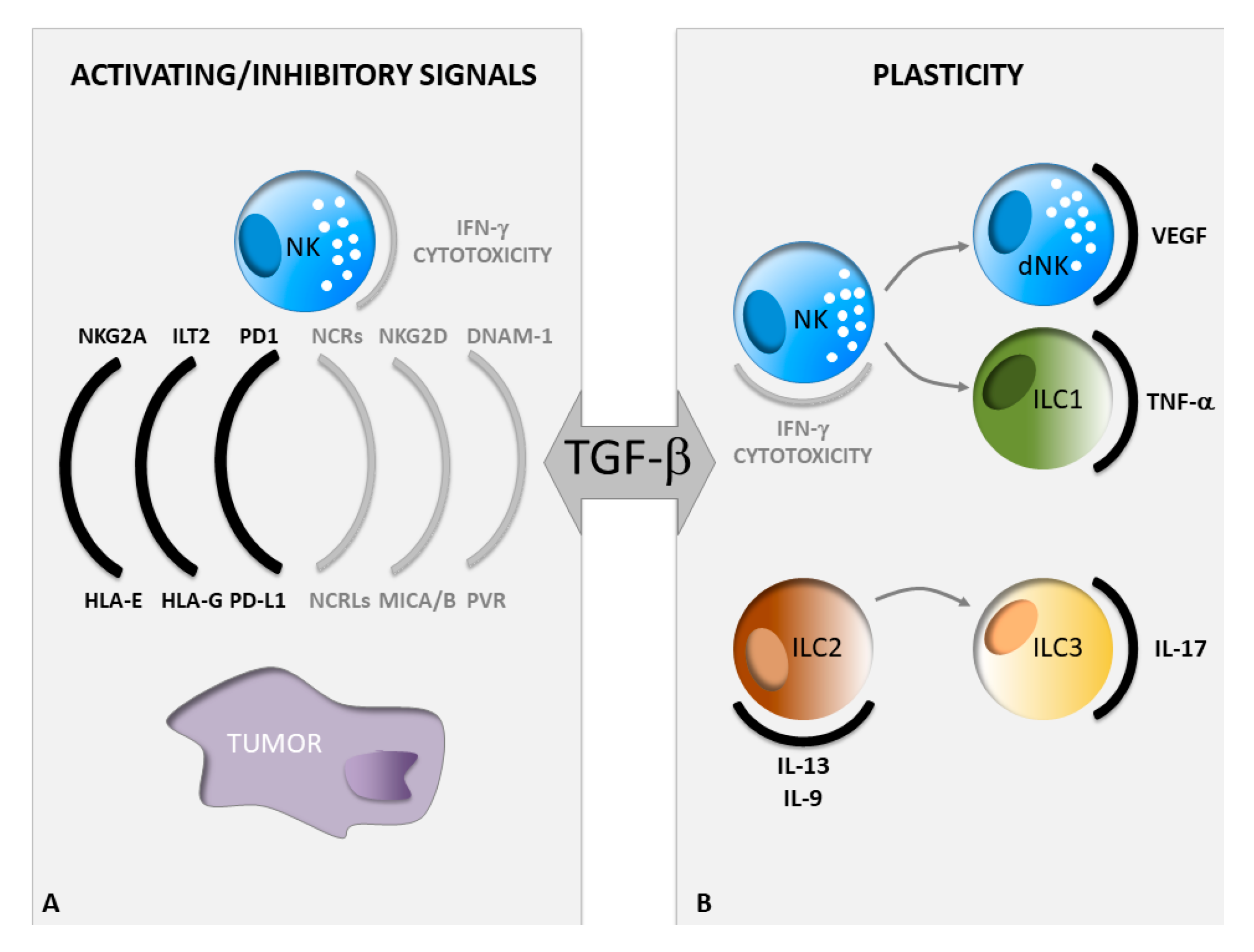{kind=link}
Abstract
1. Introduction
2. TGF-β Regulates NK Cell Activating and Inhibitory Signals in Cancer
2.1. Regulation of NK Cell Activating Signals by TGF-β
2.2. Regulation of NK Cell Inhibitory Signals by TGF-β
3. Guide of ILC Plasticity by TGF-β in Cancer
4. Targeting ILC Activity in Cancer via Inhibition of TGF-β Signaling
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFbeta in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming Growth Factor-beta Receptors and Smads: Regulatory Complexity and Functional Versatility. Trends Cell Biol. 2017, 27, 658–672. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landstrom, M. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.L.; Wang, J.; Chan, C.H.; Lee, S.W.; Campos, A.D.; Lamothe, B.; Hur, L.; Grabiner, B.C.; Lin, X.; Darnay, B.G.; et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 2009, 325, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017, 18, 1004–1015. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Pende, D.; Parolini, S.; Pessino, A.; Sivori, S.; Augugliaro, R.; Morelli, L.; Marcenaro, E.; Accame, L.; Malaspina, A.; Biassoni, R.; et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 1999, 190, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Vitale, M.; Morelli, L.; Sanseverino, L.; Augugliaro, R.; Bottino, C.; Moretta, L.; Moretta, A. P46, a novel natural killer cell-specific surface molecule that mediates cell activation. J. Exp. Med. 1997, 186, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. NKG2D Receptor and Its Ligands in Host Defense. Cancer Immunol. Res. 2015, 3, 575–582. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, N.; Lu, Y.; Davidson, D.; Colonna, M.; Veillette, A. DNAM-1 controls NK cell activation via an ITT-like motif. J. Exp. Med. 2015, 212, 2165–2182. [Google Scholar] [CrossRef]
- Trinchieri, G.; Valiante, N. Receptors for the Fc fragment of IgG on natural killer cells. Nat. Immun. 1993, 12, 218–234. [Google Scholar]
- Ndhlovu, L.C.; Lopez-Verges, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef]
- Sun, H.; Sun, C.; Xiao, W. Expression regulation of co-inhibitory molecules on human natural killer cells in response to cytokine stimulations. Cytokine 2014, 65, 33–41. [Google Scholar] [CrossRef]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346. [Google Scholar]
- Panda, S.K.; Colonna, M. Innate lymphoid cells: A potential link between microbiota and immune responses against cancer. Semin. Immunol. 2019, 41, 101271. [Google Scholar] [CrossRef]
- Tugues, S.; Ducimetiere, L.; Friebel, E.; Becher, B. Innate lymphoid cells as regulators of the tumor microenvironment. Semin. Immunol. 2019, 41, 101270. [Google Scholar] [CrossRef] [PubMed]
- Stamatiades, E.G.; Li, M.O. Tissue-resident cytotoxic innate lymphoid cells in tumor immunosurveillance. Semin. Immunol. 2019, 40, 101269. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.S.; Colonna, M. Diversity and function of group 1 innate lymphoid cells. Immunol. Lett. 2016, 179, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Weizman, O.E.; Adams, N.M.; Schuster, I.S.; Krishna, C.; Pritykin, Y.; Lau, C.; Degli-Esposti, M.A.; Leslie, C.S.; Sun, J.C.; O’Sullivan, T.E. ILC1 Confer Early Host Protection at Initial Sites of Viral Infection. Cell 2017, 171, 795–808. [Google Scholar] [CrossRef]
- Kabata, H.; Moro, K.; Koyasu, S. The group 2 innate lymphoid cell (ILC2) regulatory network and its underlying mechanisms. Immunol. Rev. 2018, 286, 37–52. [Google Scholar] [CrossRef]
- Moro, K.; Ealey, K.N.; Kabata, H.; Koyasu, S. Isolation and analysis of group 2 innate lymphoid cells in mice. Nat. Protoc. 2015, 10, 792–806. [Google Scholar] [CrossRef]
- Montaldo, E.; Juelke, K.; Romagnani, C. Group 3 innate lymphoid cells (ILC3s): Origin, differentiation, and plasticity in humans and mice. Eur. J. Immunol. 2015, 45, 2171–2182. [Google Scholar] [CrossRef]
- Lim, A.I.; Verrier, T.; Vosshenrich, C.A.; Di Santo, J.P. Developmental options and functional plasticity of innate lymphoid cells. Curr. Opin. Immunol. 2017, 44, 61–68. [Google Scholar] [CrossRef]
- Bald, T.; Wagner, M.; Gao, Y.; Koyasu, S.; Smyth, M.J. Hide and seek: Plasticity of innate lymphoid cells in cancer. Semin. Immunol. 2019, 41, 101273. [Google Scholar] [CrossRef]
- Lee, J.C.; Lee, K.M.; Kim, D.W.; Heo, D.S. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef]
- Otegbeye, F.; Ojo, E.; Moreton, S.; Mackowski, N.; Lee, D.A.; de Lima, M.; Wald, D.N. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS ONE 2018, 13, e0191358. [Google Scholar]
- Wilson, E.B.; El-Jawhari, J.J.; Neilson, A.L.; Hall, G.D.; Melcher, A.A.; Meade, J.L.; Cook, G.P. Human tumour immune evasion via TGF-beta blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumour activity. PLoS ONE 2011, 6, e22842. [Google Scholar] [CrossRef]
- Castriconi, R.; Cantoni, C.; Della, C.M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef] [PubMed]
- Zingoni, A.; Fionda, C.; Borrelli, C.; Cippitelli, M.; Santoni, A.; Soriani, A. Natural Killer Cell Response to Chemotherapy-Stressed Cancer Cells: Role in Tumor Immunosurveillance. Front. Immunol. 2017, 8, 1194. [Google Scholar] [CrossRef] [PubMed]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and Its Ligands: “One for All, All for One”. Front. Immunol. 2018, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Guerra, N.; Tan, Y.X.; Joncker, N.T.; Choy, A.; Gallardo, F.; Xiong, N.; Knoblaugh, S.; Cado, D.; Greenberg, N.M.; Raulet, D.H. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008, 28, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Mao, F.Y.; Zhao, Y.L.; Lv, Y.P.; Teng, Y.S.; Duan, M.; Chen, W.; Cheng, P.; Wang, T.T.; Liang, Z.Y.; et al. Altered NKp30, NKp46, NKG2D, and DNAM-1 Expression on Circulating NK Cells Is Associated with Tumor Progression in Human Gastric Cancer. J. Immunol. Res. 2018, 2018, 6248590. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol. 2010, 12, 7–13. [Google Scholar] [CrossRef]
- Friese, M.A.; Wischhusen, J.; Wick, W.; Weiler, M.; Eisele, G.; Steinle, A.; Weller, M. RNA interference targeting transforming growth factor-beta enhances NKG2D-mediated antiglioma immune response, inhibits glioma cell migration and invasiveness, and abrogates tumorigenicity in vivo. Cancer Res. 2004, 64, 7596–7603. [Google Scholar] [CrossRef]
- Kloess, S.; Huenecke, S.; Piechulek, D.; Esser, R.; Koch, J.; Brehm, C.; Soerensen, J.; Gardlowski, T.; Brinkmann, A.; Bader, P.; et al. IL-2-activated haploidentical NK cells restore NKG2D-mediated NK-cell cytotoxicity in neuroblastoma patients by scavenging of plasma MICA. Eur. J. Immunol. 2010, 40, 3255–3267. [Google Scholar] [CrossRef]
- Kloss, S.; Chambron, N.; Gardlowski, T.; Weil, S.; Koch, J.; Esser, R.; Pogge von, S.E.; Morgan, M.A.; Arseniev, L.; Seitz, O.; et al. Cetuximab Reconstitutes Pro-Inflammatory Cytokine Secretions and Tumor-Infiltrating Capabilities of sMICA-Inhibited NK Cells in HNSCC Tumor Spheroids. Front. Immunol. 2015, 6, 543. [Google Scholar] [CrossRef] [PubMed]
- Kloss, S.; Chambron, N.; Gardlowski, T.; Arseniev, L.; Koch, J.; Esser, R.; Glienke, W.; Seitz, O.; Kohl, U. Increased sMICA and TGFbeta1 levels in HNSCC patients impair NKG2D-dependent functionality of activated NK cells. Oncoimmunology 2015, 4, e1055993. [Google Scholar] [CrossRef]
- Dasgupta, S.; Bhattacharya-Chatterjee, M.; O’Malley, B.W., Jr.; Chatterjee, S.K. Tumor metastasis in an orthotopic murine model of head and neck cancer: Possible role of TGF-beta 1 secreted by the tumor cells. J. Cell Biochem. 2006, 97, 1036–1051. [Google Scholar] [CrossRef] [PubMed]
- Vulpis, E.; Soriani, A.; Cerboni, C.; Santoni, A.; Zingoni, A. Cancer Exosomes as Conveyors of Stress-Induced Molecules: New Players in the Modulation of NK Cell Response. Int. J. Mol. Sci. 2019, 20, 611. [Google Scholar] [CrossRef] [PubMed]
- Zingoni, A.; Vulpis, E.; Cecere, F.; Amendola, M.G.; Fuerst, D.; Saribekyan, T.; Achour, A.; Sandalova, T.; Nardone, I.; Peri, A.; et al. MICA-129 Dimorphism and Soluble MICA Are Associated With the Progression of Multiple Myeloma. Front. Immunol. 2018, 9, 926. [Google Scholar] [CrossRef]
- Clayton, A.; Tabi, Z. Exosomes and the MICA-NKG2D system in cancer. Blood Cells Mol. Dis. 2005, 34, 206–213. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef]
- Weil, S.; Memmer, S.; Lechner, A.; Huppert, V.; Giannattasio, A.; Becker, T.; Muller-Runte, A.; Lampe, K.; Beutner, D.; Quaas, A.; et al. Natural Killer Group 2D Ligand Depletion Reconstitutes Natural Killer Cell Immunosurveillance of Head and Neck Squamous Cell Carcinoma. Front. Immunol. 2017, 8, 387. [Google Scholar] [CrossRef]
- Baragano, R.A.; Suarez-Alvarez, B.; Lopez-Larrea, C. Secretory pathways generating immunosuppressive NKG2D ligands: New targets for therapeutic intervention. Oncoimmunology 2014, 3, e28497. [Google Scholar] [CrossRef]
- Eisele, G.; Wischhusen, J.; Mittelbronn, M.; Meyermann, R.; Waldhauer, I.; Steinle, A.; Weller, M.; Friese, M.A. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain 2006, 129, 2416–2425. [Google Scholar] [CrossRef]
- Park, Y.P.; Choi, S.C.; Kiesler, P.; Gil-Krzewska, A.; Borrego, F.; Weck, J.; Krzewski, K.; Coligan, J.E. Complex regulation of human NKG2D-DAP10 cell surface expression: Opposing roles of the gammac cytokines and TGF-beta1. Blood 2011, 118, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.L.; Takami, A.; Yoshioka, K.; Nakata, K.; Sato, T.; Kasahara, Y.; Nakao, S. Human microRNA-1245 down-regulates the NKG2D receptor in natural killer cells and impairs NKG2D-mediated functions. Haematologica 2012, 97, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Fujii, R.; Jochems, C.; Tritsch, S.R.; Wong, H.C.; Schlom, J.; Hodge, J.W. An IL-15 superagonist/IL-15Ralpha fusion complex protects and rescues NK cell-cytotoxic function from TGF-beta1-mediated immunosuppression. Cancer Immunol. Immunother. 2018, 67, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.C.; Wan, Z.; Sheard, M.A.; Sun, J.; Jackson, J.R.; Malvar, J.; Xu, Y.; Wang, L.; Sposto, R.; Kim, E.S.; et al. TGFbetaR1 Blockade with Galunisertib (LY2157299) Enhances Anti-Neuroblastoma Activity of the Anti-GD2 Antibody Dinutuximab (ch14.18) with Natural Killer Cells. Clin. Cancer Res. 2017, 23, 804–813. [Google Scholar] [CrossRef]
- Nam, J.S.; Terabe, M.; Mamura, M.; Kang, M.J.; Chae, H.; Stuelten, C.; Kohn, E.; Tang, B.; Sabzevari, H.; Anver, M.R.; et al. An anti-transforming growth factor beta antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res. 2008, 68, 3835–3843. [Google Scholar] [CrossRef]
- Rouas-Freiss, N.; Moreau, P.; LeMaoult, J.; Carosella, E.D. The dual role of HLA-G in cancer. J. Immunol. Res. 2014, 2014, 359748. [Google Scholar] [CrossRef]
- Amiot, L.; Ferrone, S.; Grosse-Wilde, H.; Seliger, B. Biology of HLA-G in cancer: A candidate molecule for therapeutic intervention? Cell Mol. Life Sci. 2011, 68, 417–431. [Google Scholar] [CrossRef]
- Wan, R.; Wang, Z.W.; Li, H.; Peng, X.D.; Liu, G.Y.; Ou, J.M.; Cheng, A.Q. Human Leukocyte Antigen-G Inhibits the Anti-Tumor Effect of Natural Killer Cells via Immunoglobulin-Like Transcript 2 in Gastric Cancer. Cell Physiol. Biochem. 2017, 44, 1828–1841. [Google Scholar] [CrossRef]
- Wu, D.; Kuiaste, I.; Moreau, P.; Carosella, E.; Yotnda, P. Rescuing lymphocytes from HLA-G immunosuppressive effects mediated by the tumor microenvironment. Oncotarget 2015, 6, 37385–37397. [Google Scholar] [CrossRef]
- Ullah, M.; Azazzen, D.; Kaci, R.; Benabbou, N.; Pujade, L.E.; Pocard, M.; Mirshahi, M. High Expression of HLA-G in Ovarian Carcinomatosis: The Role of Interleukin-1beta. Neoplasia 2019, 21, 331–342. [Google Scholar] [CrossRef]
- Ostapchuk, E.O.; Kali, A.; Belyaev, N.N. Transforming Growth Factor-beta and Tumor Necrosis Factor-alpha Reduce the Sensitivity of MiaPaCa2 Pancreatic Cancer Cells to Lysis by NK Cells. Bull. Exp. Biol. Med. 2018, 165, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Roth, P.; Mittelbronn, M.; Wick, W.; Meyermann, R.; Tatagiba, M.; Weller, M. Malignant glioma cells counteract antitumor immune responses through expression of lectin-like transcript-1. Cancer Res. 2007, 67, 3540–3544. [Google Scholar] [CrossRef] [PubMed]
- Wilky, B.A. Immune checkpoint inhibitors: The linchpins of modern immunotherapy. Immunol. Rev. 2019, 290, 6–23. [Google Scholar] [CrossRef]
- Escors, D.; Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Garcia-Granda, M.J.; Vera, R.; Kochan, G. The intracellular signalosome of PD-L1 in cancer cells. Signal. Transduct. Target. Ther. 2018, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Tsai, Y.; Zhu, R.; Keng, P.C.; Chen, Y.; Chen, Y.; Lee, S.O. FASN-TGF-beta1-PD-L1 axis contributes to the development of resistance to NK cell cytotoxicity of cisplatin-resistant lung cancer cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 313–322. [Google Scholar] [CrossRef]
- AlHossiny, M.; Luo, L.; Frazier, W.R.; Steiner, N.; Gusev, Y.; Kallakury, B.; Glasgow, E.; Creswell, K.; Madhavan, S.; Kumar, R.; et al. Ly6E/K Signaling to TGFbeta Promotes Breast Cancer Progression, Immune Escape, and Drug Resistance. Cancer Res. 2016, 76, 3376–3386. [Google Scholar] [CrossRef]
- David, J.M.; Dominguez, C.; McCampbell, K.K.; Gulley, J.L.; Schlom, J.; Palena, C. A novel bifunctional anti-PD-L1/TGF-beta Trap fusion protein (M7824) efficiently reverts mesenchymalization of human lung cancer cells. Oncoimmunology 2017, 6, e1349589. [Google Scholar] [CrossRef]
- Evanno, E.; Godet, J.; Piccirilli, N.; Guilhot, J.; Milin, S.; Gombert, J.M.; Fouchaq, B.; Roche, J. Tri-methylation of H3K79 is decreased in TGF-beta1-induced epithelial-to-mesenchymal transition in lung cancer. Clin. Epigenetics 2017, 9, 80. [Google Scholar] [CrossRef]
- Hsu, J.M.; Xia, W.; Hsu, Y.H.; Chan, L.C.; Yu, W.H.; Cha, J.H.; Chen, C.T.; Liao, H.W.; Kuo, C.W.; Khoo, K.H.; et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef]
- Sun, H.; Huang, Q.; Huang, M.; Wen, H.; Lin, R.; Zheng, M.; Qu, K.; Li, K.; Wei, H.; Xiao, W.; et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology 2019, 70, 168–183. [Google Scholar] [CrossRef]
- Mamessier, E.; Sylvain, A.; Thibult, M.L.; Houvenaeghel, G.; Jacquemier, J.; Castellano, R.; Goncalves, A.; Andre, P.; Romagne, F.; Thibault, G.; et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Investig. 2011, 121, 3609–3622. [Google Scholar] [CrossRef] [PubMed]
- Salimi, M.; Wang, R.; Yao, X.; Li, X.; Wang, X.; Hu, Y.; Chang, X.; Fan, P.; Dong, T.; Ogg, G. Activated innate lymphoid cell populations accumulate in human tumour tissues. BMC Cancer 2018, 18, 341. [Google Scholar] [CrossRef] [PubMed]
- Bruchard, M.; Ghiringhelli, F. Deciphering the Roles of Innate Lymphoid Cells in Cancer. Front. Immunol. 2019, 10, 656. [Google Scholar] [CrossRef] [PubMed]
- Stabile, H.; Fionda, C.; Santoni, A.; Gismondi, A. Impact of bone marrow-derived signals on NK cell development and functional maturation. Cytokine Growth Factor Rev. 2018, 42, 13–19. [Google Scholar] [CrossRef]
- Sciume, G.; Fionda, C.; Stabile, H.; Gismondi, A.; Santoni, A. Negative regulation of innate lymphoid cell responses in inflammation and cancer. Immunol. Lett. 2019, 215, 28–34. [Google Scholar] [CrossRef]
- Stabile, H.; Fionda, C.; Gismondi, A.; Santoni, A. Role of Distinct Natural Killer Cell Subsets in Anticancer Response. Front. Immunol. 2017, 8, 293. [Google Scholar] [CrossRef]
- Rocca, Y.S.; Roberti, M.P.; Arriaga, J.M.; Amat, M.; Bruno, L.; Pampena, M.B.; Huertas, E.; Loria, F.S.; Pairola, A.; Bianchini, M.; et al. Altered phenotype in peripheral blood and tumor-associated NK cells from colorectal cancer patients. Innate Immun. 2013, 19, 76–85. [Google Scholar] [CrossRef]
- Bosi, A.; Zanellato, S.; Bassani, B.; Albini, A.; Musco, A.; Cattoni, M.; Desio, M.; Nardecchia, E.; D’Urso, D.G.; Imperatori, A.; et al. Natural Killer Cells from Malignant Pleural Effusion Are Endowed with a Decidual-Like Proangiogenic Polarization. J. Immunol. Res. 2018, 2018, 2438598. [Google Scholar] [CrossRef]
- Bruno, A.; Focaccetti, C.; Pagani, A.; Imperatori, A.S.; Spagnoletti, M.; Rotolo, N.; Cantelmo, A.R.; Franzi, F.; Capella, C.; Ferlazzo, G.; et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia 2013, 15, 133–142. [Google Scholar] [CrossRef]
- Carrega, P.; Bonaccorsi, I.; Di, C.E.; Morandi, B.; Paul, P.; Rizzello, V.; Cipollone, G.; Navarra, G.; Mingari, M.C.; Moretta, L.; et al. CD56(bright)perforin(low) noncytotoxic human NK cells are abundant in both healthy and neoplastic solid tissues and recirculate to secondary lymphoid organs via afferent lymph. J. Immunol. 2014, 192, 3805–3815. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Takeda, K.; Yagita, H.; Smyth, M.J.; Van, K.L.; Okumura, K.; Saiki, I. IFN-gamma-mediated inhibition of tumor angiogenesis by natural killer T-cell ligand, alpha-galactosylceramide. Blood 2002, 100, 1728–1733. [Google Scholar] [PubMed]
- Ikeda, H.; Old, L.J.; Schreiber, R.D. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002, 13, 95–109. [Google Scholar] [CrossRef]
- Waters, J.P.; Pober, J.S.; Bradley, J.R. Tumour necrosis factor and cancer. J. Pathol. 2013, 230, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.S.; Ulland, T.K.; Cervantes-Barragan, L.; Bando, J.K.; Robinette, M.L.; Wang, Q.; White, A.J.; Gilfillan, S.; Cella, M.; Colonna, M. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-beta signaling. Nat. Immunol. 2017, 18, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chu, J.; Yi, P.; Dong, W.; Saultz, J.; Wang, Y.; Wang, H.; Scoville, S.; Zhang, J.; Wu, C.L.; et al. SMAD4 promotes TGF-beta-independent NK cell homeostasis and maturation and antitumor immunity. J. Clin. Investig. 2018, 128, 5123–5136. [Google Scholar] [CrossRef] [PubMed]
- Bie, Q.; Zhang, P.; Su, Z.; Zheng, D.; Ying, X.; Wu, Y.; Yang, H.; Chen, D.; Wang, S.; Xu, H. Polarization of ILC2s in peripheral blood might contribute to immunosuppressive microenvironment in patients with gastric cancer. J. Immunol. Res. 2014, 2014, 923135. [Google Scholar] [CrossRef]
- Wynn, T.A. Type 2 cytokines: Mechanisms and therapeutic strategies. Nat. Rev. Immunol. 2015, 15, 271–282. [Google Scholar] [CrossRef]
- Jovanovic, I.P.; Pejnovic, N.N.; Radosavljevic, G.D.; Arsenijevic, N.N.; Lukic, M.L. IL-33/ST2 axis in innate and acquired immunity to tumors. Oncoimmunology 2012, 1, 229–231. [Google Scholar] [CrossRef]
- Purwar, R.; Schlapbach, C.; Xiao, S.; Kang, H.S.; Elyaman, W.; Jiang, X.; Jetten, A.M.; Khoury, S.J.; Fuhlbrigge, R.C.; Kuchroo, V.K.; et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat. Med. 2012, 18, 1248–1253. [Google Scholar] [CrossRef]
- Schmitt, E.; Klein, M.; Bopp, T. Th9 cells, new players in adaptive immunity. Trends Immunol. 2014, 35, 61–68. [Google Scholar] [CrossRef]
- Wilhelm, C.; Hirota, K.; Stieglitz, B.; Van, S.J.; Tolaini, M.; Lahl, K.; Sparwasser, T.; Helmby, H.; Stockinger, B. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat. Immunol. 2011, 12, 1071–1077. [Google Scholar] [CrossRef]
- Bernink, J.H.; Ohne, Y.; Teunissen, M.B.M.; Wang, J.; Wu, J.; Krabbendam, L.; Guntermann, C.; Volckmann, R.; Koster, J.; van Tol, S.; et al. c-Kit-positive ILC2s exhibit an ILC3-like signature that may contribute to IL-17-mediated pathologies. Nat. Immunol. 2019, 20, 992–1003. [Google Scholar] [CrossRef]
- Ngiow, S.F.; Teng, M.W.; Smyth, M.J. A balance of interleukin-12 and -23 in cancer. Trends Immunol. 2013, 34, 548–555. [Google Scholar] [CrossRef]
- Kirchberger, S.; Royston, D.J.; Boulard, O.; Thornton, E.; Franchini, F.; Szabady, R.L.; Harrison, O.; Powrie, F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 2013, 210, 917–931. [Google Scholar] [CrossRef]
- Nussbaum, K.; Burkhard, S.H.; Ohs, I.; Mair, F.; Klose, C.S.N.; Arnold, S.J.; Diefenbach, A.; Tugues, S.; Becher, B. Tissue microenvironment dictates the fate and tumor-suppressive function of type 3 ILCs. J. Exp. Med. 2017, 214, 2331–2347. [Google Scholar] [CrossRef]
- Anderton, M.J.; Mellor, H.R.; Bell, A.; Sadler, C.; Pass, M.; Powell, S.; Steele, S.J.; Roberts, R.R.; Heier, A. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol. Pathol. 2011, 39, 916–924. [Google Scholar] [CrossRef]
- Biswas, S.; Guix, M.; Rinehart, C.; Dugger, T.C.; Chytil, A.; Moses, H.L.; Freeman, M.L.; Arteaga, C.L. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J. Clin. Investig. 2007, 117, 1305–1313. [Google Scholar] [CrossRef]
- Drabsch, Y.; Ten, D.P. TGF-beta signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 2012, 31, 553–568. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFbeta pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef]
- Ng, S.; Deng, J.; Chinnadurai, R.; Yuan, S.; Pennati, A.; Galipeau, J. Stimulation of Natural Killer Cell-Mediated Tumor Immunity by an IL15/TGFbeta-Neutralizing Fusion Protein. Cancer Res. 2016, 76, 5683–5695. [Google Scholar] [CrossRef]
- Knudson, K.M.; Hicks, K.C.; Luo, X.; Chen, J.Q.; Schlom, J.; Gameiro, S.R. M7824, a novel bifunctional anti-PD-L1/TGFbeta Trap fusion protein, promotes anti-tumor efficacy as monotherapy and in combination with vaccine. Oncoimmunology 2018, 7, e1426519. [Google Scholar] [CrossRef]
- Wang, Q.M.; Tang, P.M.; Lian, G.Y.; Li, C.; Li, J.; Huang, X.R.; To, K.F.; Lan, H.Y. Enhanced Cancer Immunotherapy with Smad3-Silenced NK-92 Cells. Cancer Immunol. Res. 2018, 6, 965–977. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fionda, C.; Stabile, H.; Cerboni, C.; Soriani, A.; Gismondi, A.; Cippitelli, M.; Santoni, A. Hitting More Birds with a Stone: Impact of TGF-β on ILC Activity in Cancer. J. Clin. Med. 2020, 9, 143. https://doi.org/10.3390/jcm9010143
Fionda C, Stabile H, Cerboni C, Soriani A, Gismondi A, Cippitelli M, Santoni A. Hitting More Birds with a Stone: Impact of TGF-β on ILC Activity in Cancer. Journal of Clinical Medicine. 2020; 9(1):143. https://doi.org/10.3390/jcm9010143
Chicago/Turabian StyleFionda, Cinzia, Helena Stabile, Cristina Cerboni, Alessandra Soriani, Angela Gismondi, Marco Cippitelli, and Angela Santoni. 2020. "Hitting More Birds with a Stone: Impact of TGF-β on ILC Activity in Cancer" Journal of Clinical Medicine 9, no. 1: 143. https://doi.org/10.3390/jcm9010143
APA StyleFionda, C., Stabile, H., Cerboni, C., Soriani, A., Gismondi, A., Cippitelli, M., & Santoni, A. (2020). Hitting More Birds with a Stone: Impact of TGF-β on ILC Activity in Cancer. Journal of Clinical Medicine, 9(1), 143. https://doi.org/10.3390/jcm9010143