Back to The Fusion: Mitofusin-2 in Alzheimer’s Disease



{kind=link}
Abstract
1. Introduction
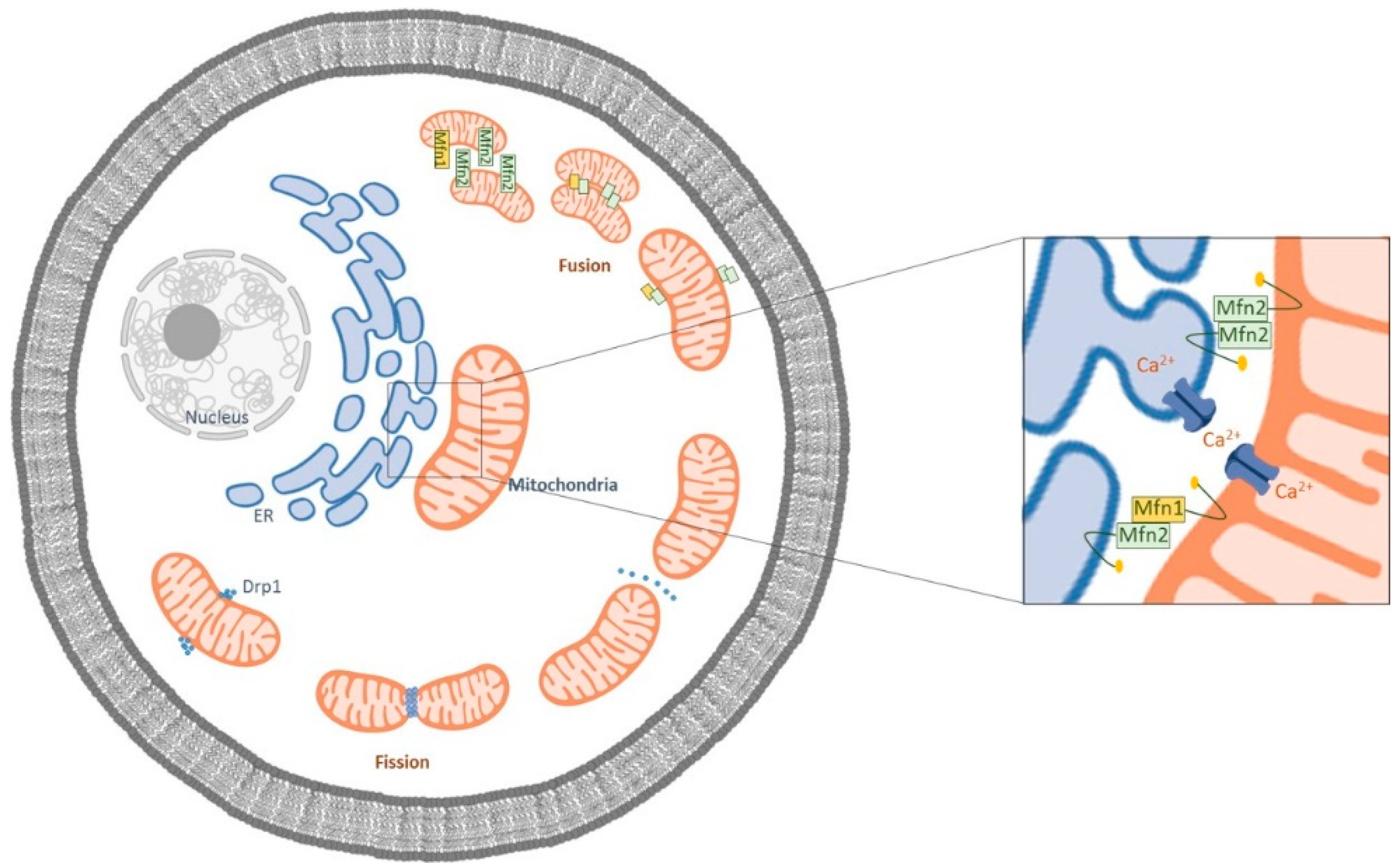
2. The Crosstalk between Mitochondria and ER in AD
3. Mitochondrial Dysfunction in Alzheimer’s Disease
4. Mfn2 as A Potential Target in Alzheimer’s Disease
5. Conclusions
Funding
Conflicts of Interest
References
- Mazat, J.-P.; Ransac, S.; Heiske, M.; Devin, A.; Rigoulet, M. Mitochondrial energetic metabolism-some general principles. IUBMB Life 2013, 65, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Langer, T. Mitofusin 2 builds a bridge between ER and mitochondria. Cell 2008, 135, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- De Brito, O.M.; Scorrano, L. An intimate liaison: Spatial organization of the endoplasmic reticulum–mitochondria relationship. EMBO J. 2010, 29, 2715–2723. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Kim, Y.J.; Park, J.K.; Kang, W.S.; Kim, S.K.; Han, C.; Na, H.R.; Park, H.J.; Kim, J.W.; Kim, Y.Y.; Park, M.H.; et al. Association between mitofusin 2 gene polymorphisms and late-onset Alzheimer’s disease in the Korean population. Psychiatry Investig. 2017, 14, 81–85. [Google Scholar] [CrossRef]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, S401–S412. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim. Biophys. Acta 2010, 1802, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Darshi, M.; Mendiola, V.L.; Mackey, M.R.; Murphy, A.N.; Koller, A.; Perkins, G.A.; Ellisman, M.H.; Taylor, S.S. ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J. Biol. Chem. 2011, 286, 2918–2932. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Perry, G.; Smith, M.A.; Wang, X. Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 33, S253–S262. [Google Scholar] [CrossRef]
- Hroudová, J.; Singh, N.; Fišar, Z. Mitochondrial dysfunctions in neurodegenerative diseases: Relevance to Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Leuner, K.; Schulz, K.; Schütt, T.; Pantel, J.; Prvulovic, D.; Rhein, V.; Savaskan, E.; Czech, C.; Eckert, A.; Müller, W.E. Peripheral mitochondrial dysfunction in Alzheimer’s disease: Focus on lymphocytes. Mol. Neurobiol. 2012, 46, 194–204. [Google Scholar] [CrossRef]
- Baloyannis, S.J. Mitochondrial alterations in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 119–126. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Fujioka, H.; Zhu, X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am. J. Pathol. 2008, 173, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.E.; Alkon, D.L. Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol. Dis. 1998, 5, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Nelson, O.; Tu, H.; Lei, T.; Bentahir, M.; De Strooper, B.; Bezprozvanny, I. Familial Alzheimer disease–linked mutations specifically disrupt Ca2+ leak function of presenilin 1. J. Clin. Investig. 2007, 117, 1230–1239. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; De Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef]
- Jiang, S.; Nandy, P.; Wang, W.; Ma, X.; Hsia, J.; Wang, C.; Wang, Z.; Niu, M.; Siedlak, S.L.; Torres, S.; et al. Mfn2 ablation causes an oxidative stress response and eventual neuronal death in the hippocampus and cortex. Mol. Neurodegener. 2018, 13, 5. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid—Overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef] [PubMed]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Ronnback, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [PubMed]
- Leal, N.S.; Schreiner, B.; Pinho, C.M.; Filadi, R.; Wiehager, B.; Karlström, H.; Pizzo, P.; Ankarcrona, M. Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid β-peptide production. J. Cell. Mol. Med. 2016, 20, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118, 5411–5419. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.-Y.; Han, S.-H.; Son, S.M.; Hong, H.-S.; Choi, Y.-J.; Byun, J.; Mook-Jung, I. Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS ONE 2012, 7, e34929. [Google Scholar] [CrossRef]
- Park, J.; Choi, H.; Min, J.-S.; Kim, B.; Lee, S.-R.; Yun, J.W.; Choi, M.-S.; Chang, K.-T.; Lee, D.-S. Loss of mitofusin 2 links beta-amyloid-mediated mitochondrial fragmentation and Cdk5-induced oxidative stress in neuron cells. J. Neurochem. 2015, 132, 687–702. [Google Scholar] [CrossRef]
- Gan, X.; Huang, S.; Wu, L.; Wang, Y.; Hu, G.; Li, G.; Zhang, H.; Yu, H.; Swerdlow, R.H.; Chen, J.X.; et al. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim. Biophys. Acta 2014, 1842, 220–231. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimer’s Dis. 2010, 20, S265–S279. [Google Scholar] [CrossRef]
- Tyumentsev, M.A.; Stefanova, N.A.; Muraleva, N.A.; Rumyantseva, Y.V.; Kiseleva, E.; Vavilin, V.A.; Kolosova, N.G. Mitochondrial dysfunction as a predictor and driver of Alzheimer’s disease-like pathology in OXYS rats. J. Alzheimer’s Dis. 2018, 63, 1075–1088. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.-G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, M.; Mannermaa, A.; Thompson, D.; Easton, D.; Pirskanen, M.; Helisalmi, S.; Koivisto, A.M.; Lehtovirta, M.; Ryynänen, M.; Soininen, H. Genome-wide linkage disequilibrium mapping of late-onset Alzheimer’s disease in Finland. Neurology 2001, 57, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Suh, B.C.; Cho, S.Y.; Choi, S.K.; Kang, S.H.; Yoo, J.H.; Hwang, J.Y.; Choi, B.O. Early-onset Charcot-Marie-Tooth patients with mitofusin 2 mutations and brain involvement. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1203–1206. [Google Scholar] [CrossRef] [PubMed]
- Bo, R.D.; Moggio, M.; Rango, M.; Bonato, S.; D’Angelo, M.G.; Ghezzi, S.; Airoldi, G.; Bassi, M.T.; Guglieri, M.; Napoli, L.; et al. Mutated mitofusin 2 presents with intrafamilial variability and brain mitochondrial dysfunction. Neurology 2008, 71, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhou, H.; Jiang, L.; Mao, Y.; Cui, X.; Xie, B.; Cui, D.; Wang, H.; Zhang, Q.; Xu, S. MiR-195 dependent roles of mitofusin2 in the mitochondrial dysfunction of hippocampal neurons in SAMP8 mice. Brain Res. 2016, 1652, 135–143. [Google Scholar] [CrossRef]
- Xu, L.-L.; Shen, Y.; Wang, X.; Wei, L.-F.; Wang, P.; Yang, H.; Wang, C.-F.; Xie, Z.-H.; Bi, J.-Z. Mitochondrial dynamics changes with age in an APPsw/PS1dE9 mouse model of Alzheimer’s disease. Neuroreport 2017, 28, 222–228. [Google Scholar] [CrossRef]
- Karakaya, T.; Fußer, F.; Schroder, J.; Pantel, J. Pharmacological treatment of mild cognitive impairment as a prodromal syndrome of Alzheimer’s disease. Curr. Neuropharmacol. 2013, 11, 102–108. [Google Scholar] [CrossRef]
- Silva, D.F.; Santana, I.; Esteves, A.R.; Baldeiras, I.; Arduino, D.M.; Oliveira, C.R.; Cardoso, S.M. Prodromal metabolic phenotype in MCI cybrids: Implications for Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 180–190. [Google Scholar] [CrossRef]
- Gan, X.; Wu, L.; Huang, S.; Zhong, C.; Shi, H.; Li, G.; Yu, H.; Swerdlow, R.H.; Chen, J.X.; Yan, S.S. Oxidative stress-mediated activation of extracellular signal-regulated kinase contributes to mild cognitive impairment-related mitochondrial dysfunction. Free Radic. Biol. Med. 2014, 75, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Sugioka, R.; Shimizu, S.; Tsujimoto, Y. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J. Biol. Chem. 2004, 279, 52726–52734. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Neutzner, A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol. 2012, 123, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X.L.; Reddy, A.P. Synergistic protective effects of mitochondrial division inhibitor 1 and mitochondria-targeted small peptide SS31 in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 1549–1565. [Google Scholar] [CrossRef]
- Alexiou, A.; Soursou, G.; Chatzichronis, S.; Gasparatos, E.; Kamal, M.A.; Yarla, N.S.; Perveen, A.; Barreto, G.E.; Ashraf, G.M. Role of GTPases in the regulation of mitochondrial dynamics in Alzheimer’s disease and CNS-related disorders. Mol. Neurobiol. 2019, 56, 4530–4538. [Google Scholar] [CrossRef]
- Chen, Y.; Han, S.; Huang, X.; Ni, J.; He, X. The protective effect of icariin on mitochondrial transport and distribution in primary hippocampal neurons from 3× Tg-AD mice. Int. J. Mol. Sci. 2016, 17, 163. [Google Scholar] [CrossRef]
- Ma, H.; He, X.; Yang, Y.; Li, M.; Hao, D.; Jia, Z. The genus Epimedium: An ethnopharmacological and phytochemical review. J. Ethnopharmacol. 2011, 134, 519–541. [Google Scholar] [CrossRef]
- Chimeh, U.; Zimmerman, M.A.; Gilyazova, N.; Li, P.A. B355252, a novel small molecule, confers neuroprotection against cobalt chloride toxicity in mouse hippocampal cells through altering mitochondrial dynamics and limiting autophagy induction. Int. J. Med. Sci. 2018, 15, 1384–1396. [Google Scholar] [CrossRef]
- Zafeer, M.F.; Firdaus, F.; Anis, E.; Mobarak Hossain, M. Prolong treatment with Trans-ferulic acid mitigates bioenergetics loss and restores mitochondrial dynamics in streptozotocin-induced sporadic dementia of Alzheimer’s type. Neurotoxicology 2019, 73, 246–257. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Yin, X. Mitochondria-division inhibitor 1 protects against amyloid-β induced mitochondrial fragmentation and synaptic damage in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 58, 147–162. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sita, G.; Hrelia, P.; Graziosi, A.; Morroni, F. Back to The Fusion: Mitofusin-2 in Alzheimer’s Disease. J. Clin. Med. 2020, 9, 126. https://doi.org/10.3390/jcm9010126
Sita G, Hrelia P, Graziosi A, Morroni F. Back to The Fusion: Mitofusin-2 in Alzheimer’s Disease. Journal of Clinical Medicine. 2020; 9(1):126. https://doi.org/10.3390/jcm9010126
Chicago/Turabian StyleSita, Giulia, Patrizia Hrelia, Agnese Graziosi, and Fabiana Morroni. 2020. "Back to The Fusion: Mitofusin-2 in Alzheimer’s Disease" Journal of Clinical Medicine 9, no. 1: 126. https://doi.org/10.3390/jcm9010126
APA StyleSita, G., Hrelia, P., Graziosi, A., & Morroni, F. (2020). Back to The Fusion: Mitofusin-2 in Alzheimer’s Disease. Journal of Clinical Medicine, 9(1), 126. https://doi.org/10.3390/jcm9010126