ADCK2 Haploinsufficiency Reduces Mitochondrial Lipid Oxidation and Causes Myopathy Associated with CoQ Deficiency

 ,
,  , , , ,
, , , ,  , , ,
, , ,  add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Approval
2.2. Mouse Model
2.3. Cell Strains and Culture
2.4. Targeted Gene Sequencing
2.5. Measurement of Plasma Acyl-Carnitine and Urinary Organic Acids
2.6. Yeast Functional Complementation
2.7. Physical and Behavioral Tests
2.8. CoQ Determination and Biosynthesis
2.9. Mitochondrial Biochemistry
2.10. Microarray Analysis
2.11. Untargeted Metabolomics Assay
2.12. Metabolic Cages
2.13. Quantitative Real-Time PCR
2.14. ADCK2 Silencing by siRNA
2.15. Antibodies
2.16. Statistics
3. Results
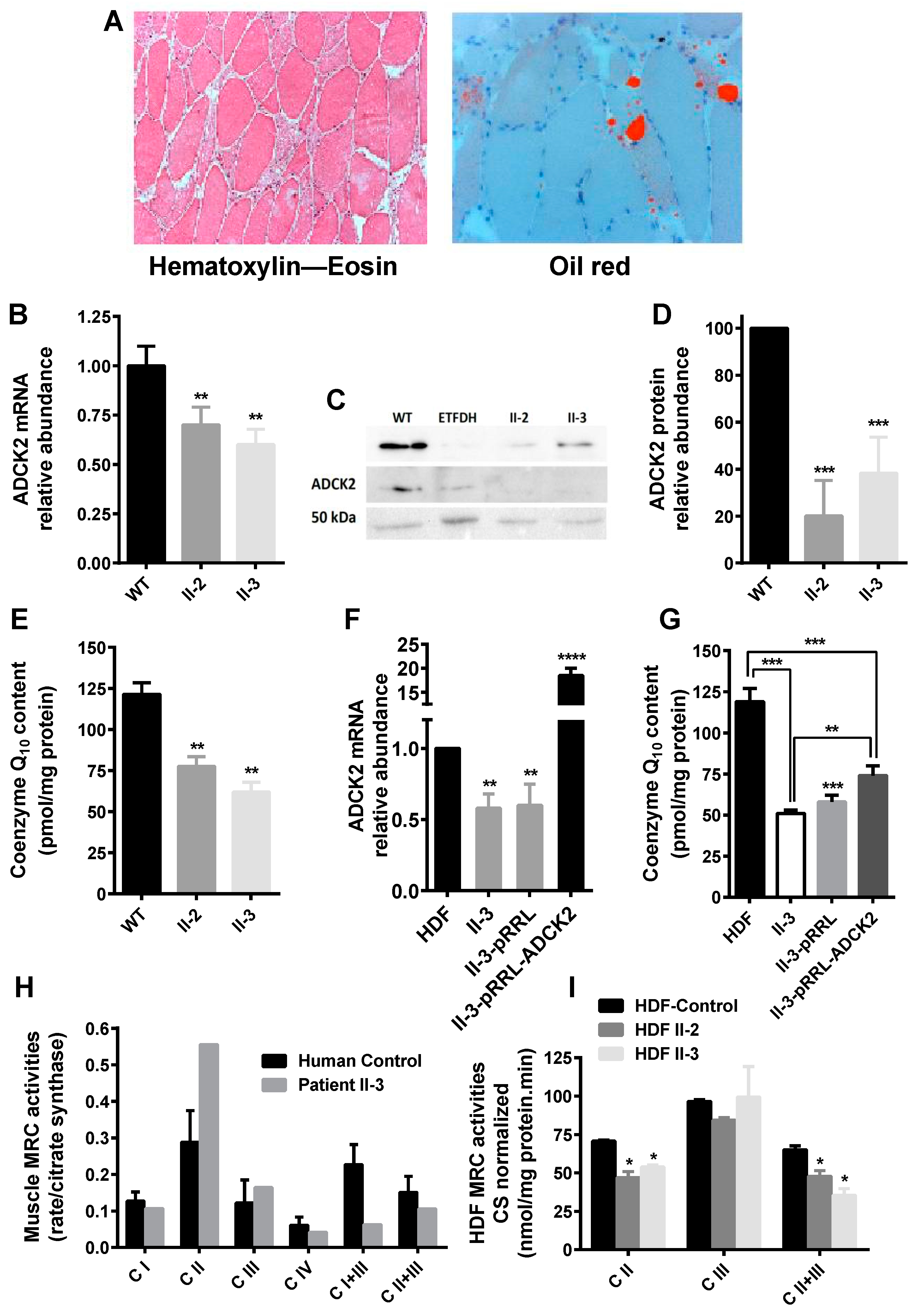
3.1. Haploinsufficiency of Human ADCK2 Caused a Defect in Lipid Catabolism and CoQ10-Deficient Myopathy
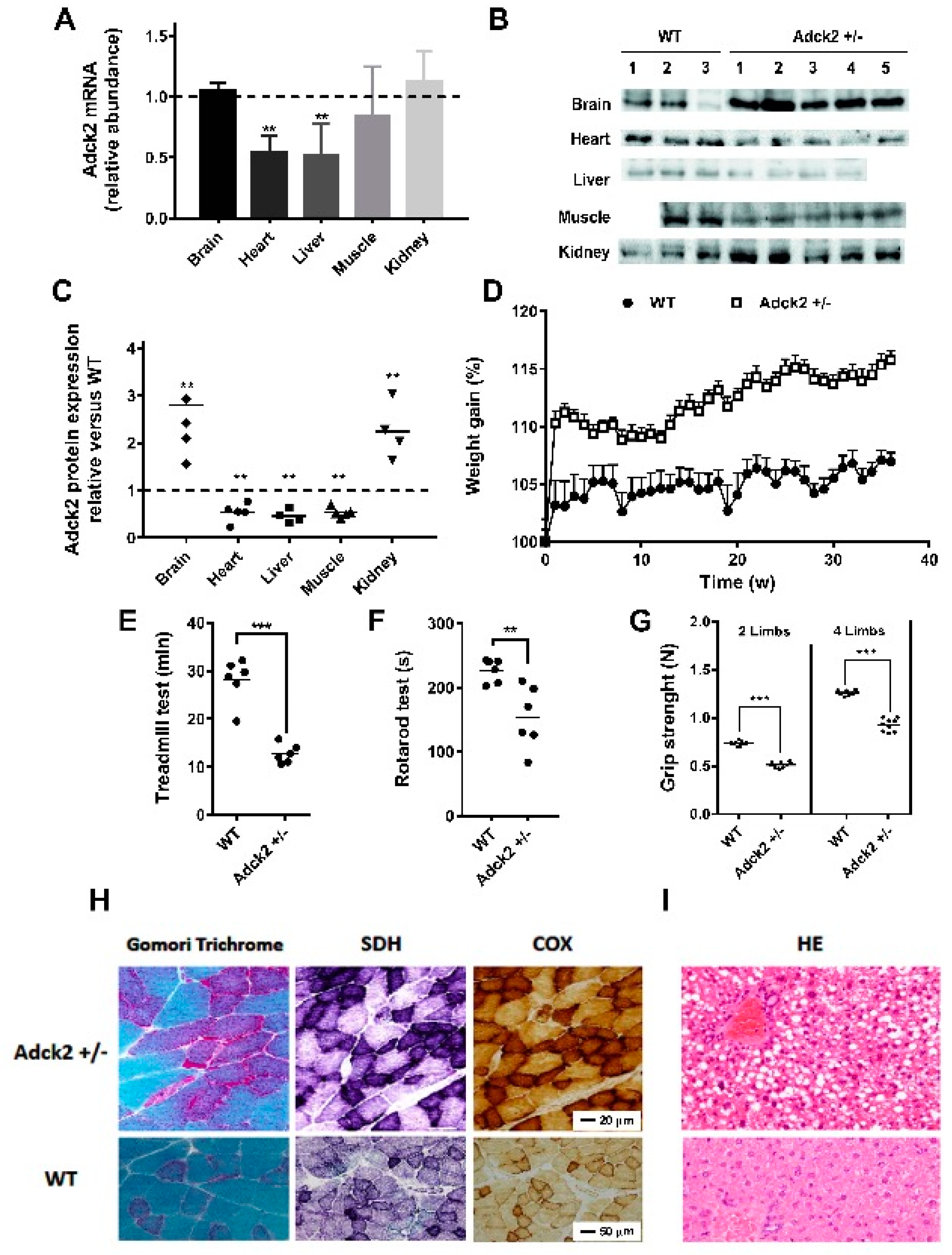
3.2. A Heterozygous Adck2 Knockout Mouse Model Recapitulated the Phenotype Observed in Patient II-3
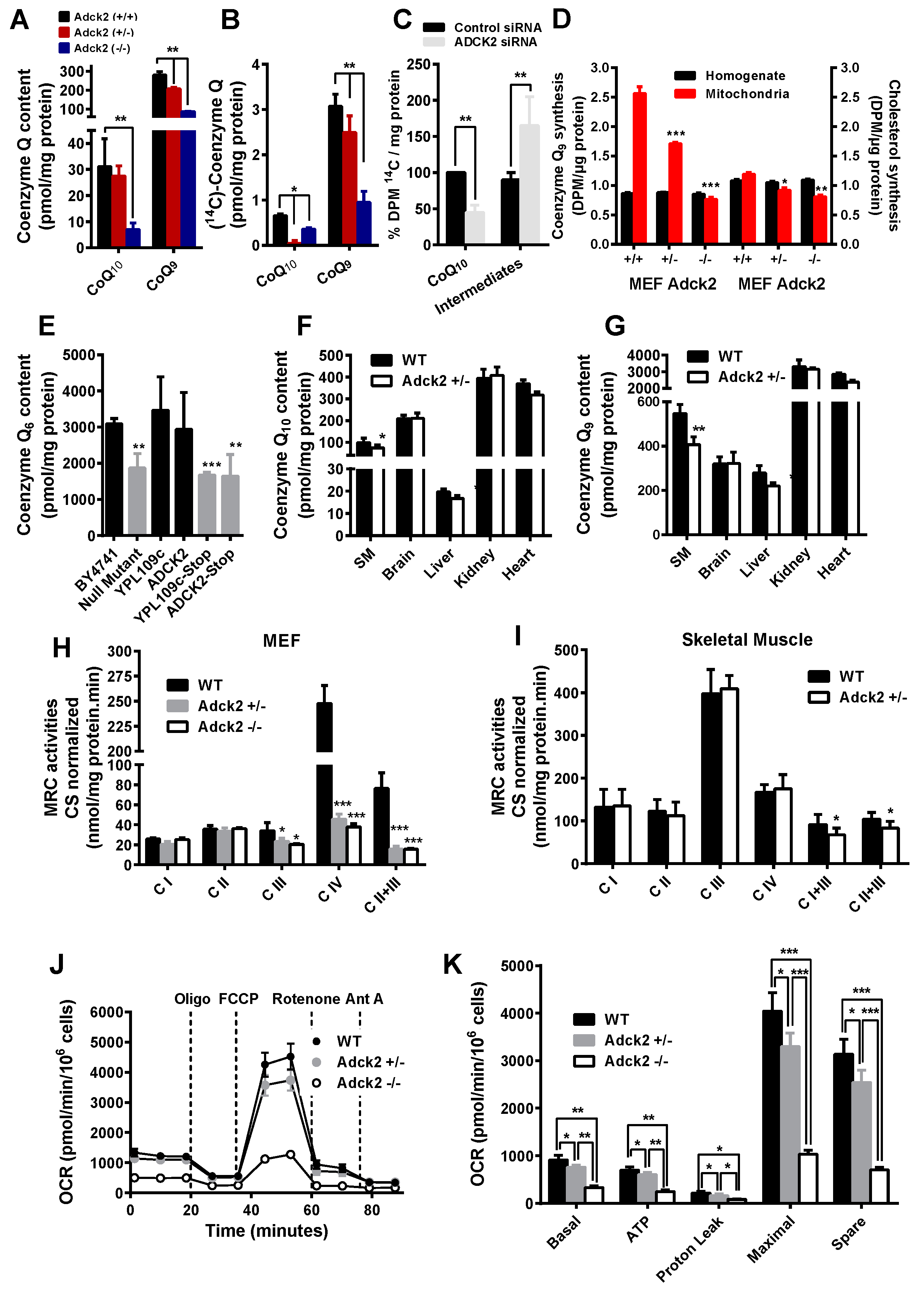
3.3. Adck2 Deficiency Caused CoQ Deficiency in MEFs and Skeletal Muscle
3.4. Adck2 Haploinsufficiency Caused Mitochondrial Dysfunction
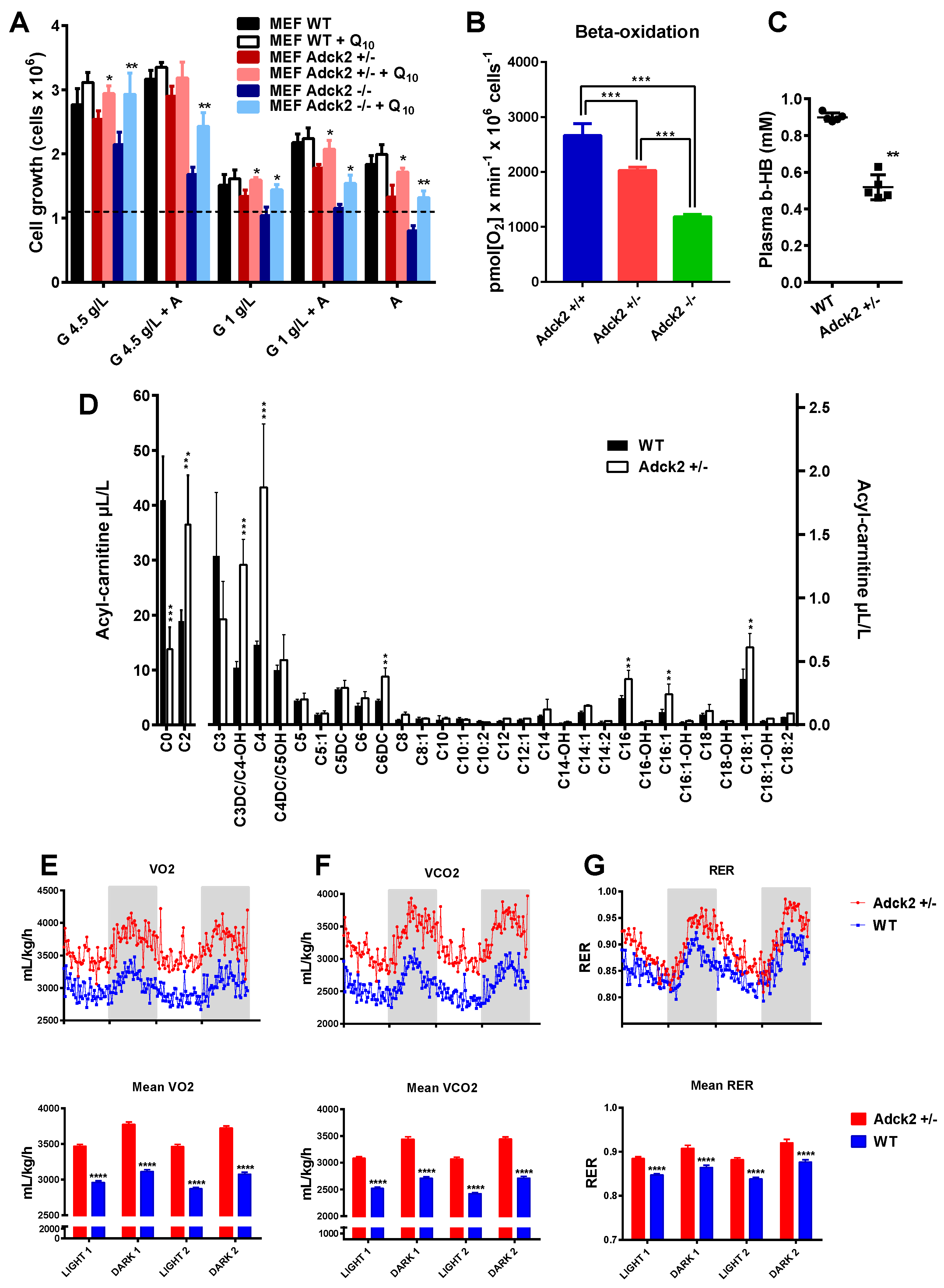
3.5. Adck2 Haploinsufficiency Decreased Mitochondrial Fatty Acid β-Oxidation Required to Promote Cellular Growth
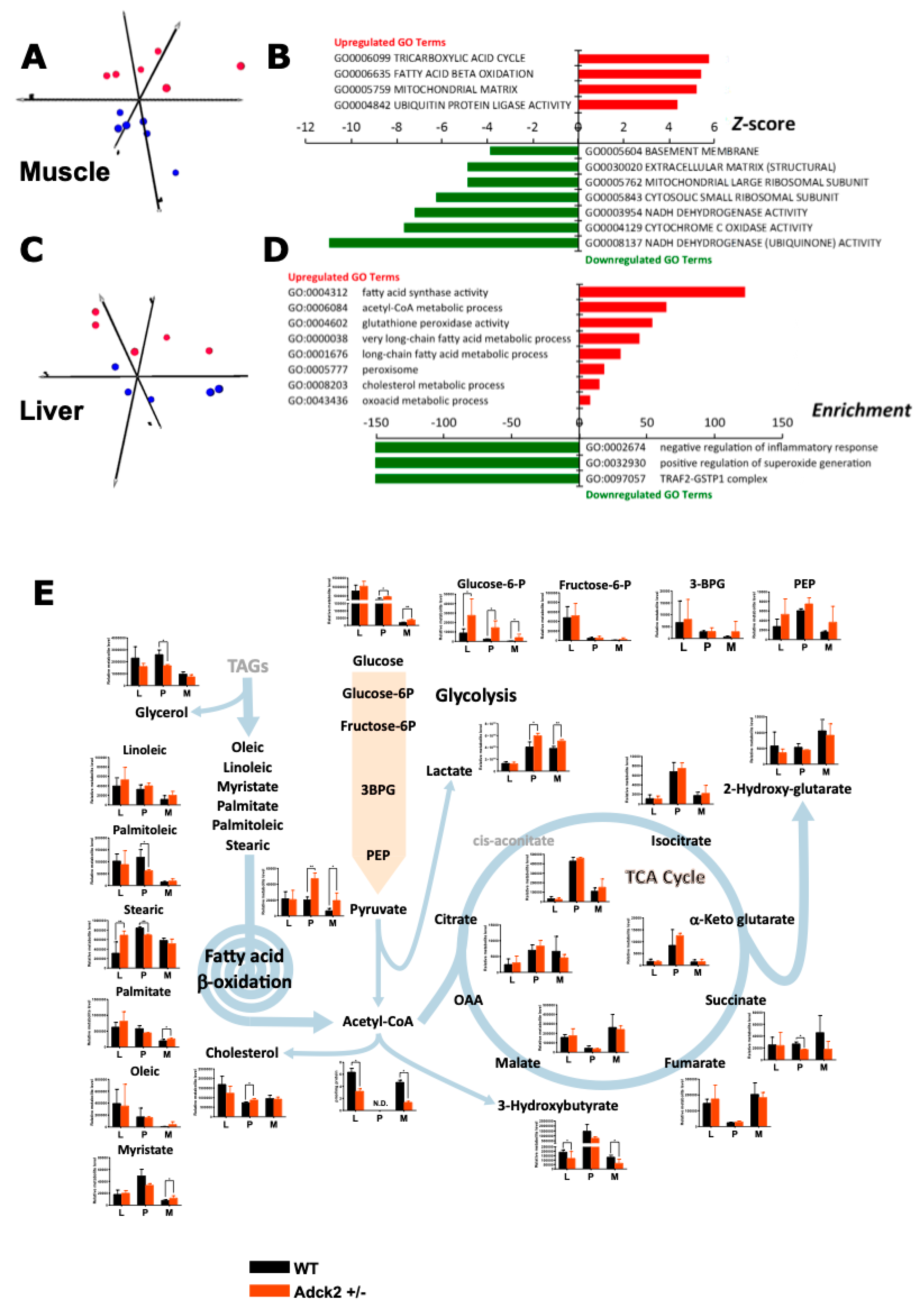
3.6. Adck2 Haploinsufficiency Affected the Gene Expression Profile in Muscle and Liver
3.7. Adck2 Haploinsufficiency Affected the Metabolomics Profile
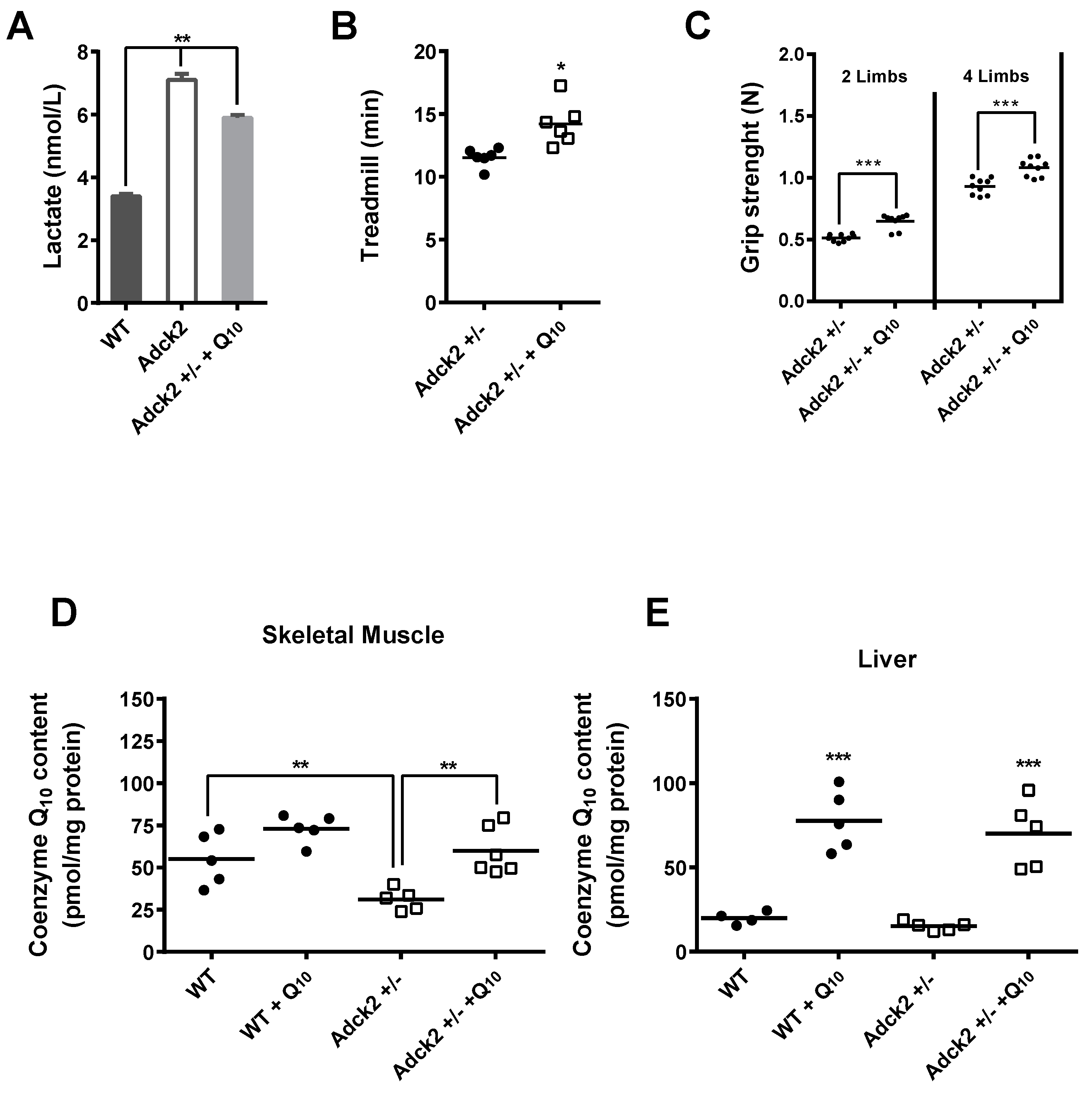
3.8. CoQ Supplementation Partially Rescued the Adck2+/− Phenotype
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, R.L.; Soeters, M.R.; Wust, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Bernier, M.; Mattison, J.A.; Aon, M.A.; Kaiser, T.A.; Anson, R.M.; Ikeno, Y.; Anderson, R.M.; Ingram, D.K.; de Cabo, R. Daily Fasting Improves Health and Survival in Male Mice Independent of Diet Composition and Calories. Cell Metab. 2019, 29, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Cortassa, S.; Aon, M.A.; O’Rourke, B.; Jacques, R.; Tseng, H.J.; Marban, E.; Winslow, R.L. A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys. J. 2006, 91, 1564–1589. [Google Scholar] [CrossRef] [PubMed]
- Barclay, C.J. Energy demand and supply in human skeletal muscle. J. Muscle. Res. Cell Motil. 2017, 38, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Fabra, M.; Navas, P.; Brea-Calvo, G. Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim. Biophys. Acta 2016, 1857, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Kuzmiak-Glancy, S.; Willis, W.T. Skeletal muscle fuel selection occurs at the mitochondrial level. J. Exp. Biol. 2014, 217, 1993–2003. [Google Scholar] [CrossRef] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Grunert, S.C. Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency. Orph. J. Rare Dis. 2014, 9, 117. [Google Scholar] [CrossRef] [PubMed]
- Chokchaiwong, S.; Kuo, Y.T.; Hsu, S.P.; Hsu, Y.C.; Lin, S.H.; Zhong, W.B.; Lin, Y.F.; Kao, S.H. ETF-QO Mutants Uncoupled Fatty Acid beta-Oxidation and Mitochondrial Bioenergetics Leading to Lipid Pathology. Cells 2019, 8, 106. [Google Scholar] [CrossRef]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.; Hans, V.H.; Palmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef]
- Liang, W.C.; Ohkuma, A.; Hayashi, Y.K.; Lopez, L.C.; Hirano, M.; Nonaka, I.; Noguchi, S.; Chen, L.H.; Jong, Y.J.; Nishino, I. ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul. Disord. 2009, 19, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Li, D.; Shan, J.; Liu, S.; Li, W.; Zhao, Y.; Lin, P.; Zheng, J.; Li, D.; Gong, Y.; et al. Increased muscle coenzyme Q10 in riboflavin responsive MADD with ETFDH gene mutations due to secondary mitochondrial proliferation. Mol. Genet. Metab. 2013, 109, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Mourier, A.; Motori, E.; Brandt, T.; Lagouge, M.; Atanassov, I.; Galinier, A.; Rappl, G.; Brodesser, S.; Hultenby, K.; Dieterich, C.; et al. Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J. Cell Biol. 2015, 208, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nature reviews. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Salviati, L.; Trevisson, E.; Doimo, M.; Navas, P. Primary Coenzyme Q10 Deficiency. In GeneReviews (R); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Kuhl, I.; Miranda, M.; Atanassov, I.; Kuznetsova, I.; Hinze, Y.; Mourier, A.; Filipovska, A.; Larsson, N.G. Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife 2017, 6, e30952. [Google Scholar] [CrossRef]
- Yubero, D.; Montero, R.; Martin, M.A.; Montoya, J.; Ribes, A.; Grazina, M.; Trevisson, E.; Rodriguez-Aguilera, J.C.; Hargreaves, I.P.; Salviati, L.; et al. Secondary coenzyme Q10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion 2016, 30, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Vazquez Fonseca, L.; Doimo, M.; Calderan, C.; Desbats, M.A.; Acosta, M.J.; Cerqua, C.; Cassina, M.; Ashraf, S.; Hildebrandt, F.; Sartori, G.; et al. Mutations in COQ8B (ADCK4) found in patients with steroid-resistant nephrotic syndrome alter COQ8B function. Hum. Mut. 2018, 39, 406–414. [Google Scholar] [CrossRef]
- Xie, L.X.; Hsieh, E.J.; Watanabe, S.; Allan, C.M.; Chen, J.Y.; Tran, U.C.; Clarke, C.F. Expression of the human atypical kinase ADCK3 rescues coenzyme Q biosynthesis and phosphorylation of Coq polypeptides in yeast coq8 mutants. Biochim. Biophys. Acta 2011, 1811, 348–360. [Google Scholar] [CrossRef]
- Stefely, J.A.; Licitra, F.; Laredj, L.; Reidenbach, A.G.; Kemmerer, Z.A.; Grangeray, A.; Jaeg-Ehret, T.; Minogue, C.E.; Ulbrich, A.; Hutchins, P.D.; et al. Cerebellar Ataxia and Coenzyme Q Deficiency through Loss of Unorthodox Kinase Activity. Mol. Cell 2016, 63, 608–620. [Google Scholar] [CrossRef]
- Stefely, J.A.; Reidenbach, A.G.; Ulbrich, A.; Oruganty, K.; Floyd, B.J.; Jochem, A.; Saunders, J.M.; Johnson, I.E.; Minogue, C.E.; Wrobel, R.L.; et al. Mitochondrial ADCK3 Employs an Atypical Protein Kinase-like Fold to Enable Coenzyme Q Biosynthesis. Mol. Cell 2015, 57, 83–94. [Google Scholar] [CrossRef]
- Blumkin, L.; Leshinsky-Silver, E.; Zerem, A.; Yosovich, K.; Lerman-Sagie, T.; Lev, D. Heterozygous Mutations in the ADCK3 Gene in Siblings with Cerebellar Atrophy and Extreme Phenotypic Variability. JIMD Rep. 2014, 12, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Gerards, M.; van den Bosch, B.; Calis, C.; Schoonderwoerd, K.; van Engelen, K.; Tijssen, M.; de Coo, R.; van der Kooi, A.; Smeets, H. Nonsense mutations in CABC1/ADCK3 cause progressive cerebellar ataxia and atrophy. Mitochondrion 2010, 10, 510–515. [Google Scholar] [CrossRef]
- Horvath, R.; Czermin, B.; Gulati, S.; Demuth, S.; Houge, G.; Pyle, A.; Dineiger, C.; Blakely, E.L.; Hassani, A.; Foley, C.; et al. Adult-onset cerebellar ataxia due to mutations in CABC1/ADCK3. J. Neurol. Neurosurg. Psychiatry 2012, 83, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Tazir, M.; Lopez, L.C.; Quinzii, C.M.; Assoum, M.; Drouot, N.; Busso, C.; Makri, S.; Ali-Pacha, L.; Benhassine, T.; et al. ADCK3, an ancestral kinase, is mutated in a form of recessive ataxia associated with coenzyme Q10 deficiency. Am. J. Hum. Genet. 2008, 82, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.T.; Hersheson, J.; Plagnol, V.; Fawcett, K.; Duberley, K.E.; Preza, E.; Hargreaves, I.P.; Chalasani, A.; Laura, M.; Wood, N.W.; et al. Autosomal-recessive cerebellar ataxia caused by a novel ADCK3 mutation that elongates the protein: Clinical, genetic and biochemical characterisation. J. Neurol. Neurosurg. Psychiatry 2014, 85, 493–498. [Google Scholar] [CrossRef]
- Mignot, C.; Apartis, E.; Durr, A.; Marques Lourenco, C.; Charles, P.; Devos, D.; Moreau, C.; de Lonlay, P.; Drouot, N.; Burglen, L.; et al. Phenotypic variability in ARCA2 and identification of a core ataxic phenotype with slow progression. Orphanet J. Rare Dis. 2013, 8, 173. [Google Scholar] [CrossRef]
- Mollet, J.; Delahodde, A.; Serre, V.; Chretien, D.; Schlemmer, D.; Lombes, A.; Boddaert, N.; Desguerre, I.; de Lonlay, P.; de Baulny, H.O.; et al. CABC1 gene mutations cause ubiquinone deficiency with cerebellar ataxia and seizures. Am. J. Hum. Genet. 2008, 82, 623–630. [Google Scholar] [CrossRef]
- Ashraf, S.; Gee, H.Y.; Woerner, S.; Xie, L.X.; Vega-Warner, V.; Lovric, S.; Fang, H.; Song, X.; Cattran, D.C.; Avila-Casado, C.; et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Investig. 2013, 123, 5179–5189. [Google Scholar] [CrossRef]
- Simpson, K.J.; Selfors, L.M.; Bui, J.; Reynolds, A.; Leake, D.; Khvorova, A.; Brugge, J.S. Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat. Cell biol. 2008, 10, 1027–1038. [Google Scholar] [CrossRef]
- Wiedemeyer, W.R.; Dunn, I.F.; Quayle, S.N.; Zhang, J.; Chheda, M.G.; Dunn, G.P.; Zhuang, L.; Rosenbluh, J.; Chen, S.; Xiao, Y.; et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc. Natl. Acad. Sci. USA 2010, 107, 11501–11506. [Google Scholar] [CrossRef]
- Brough, R.; Frankum, J.R.; Sims, D.; Mackay, A.; Mendes-Pereira, A.M.; Bajrami, I.; Costa-Cabral, S.; Rafiq, R.; Ahmad, A.S.; Cerone, M.A.; et al. Functional viability profiles of breast cancer. Cancer Discov. 2011, 1, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Schoolmeesters, A.; Brown, D.D.; Fedorov, Y. Kinome-wide functional genomics screen reveals a novel mechanism of TNFalpha-induced nuclear accumulation of the HIF-1alpha transcription factor in cancer cells. PLoS ONE 2012, 7, e31270. [Google Scholar] [CrossRef] [PubMed]
- Hughes, B.G.; Harrison, P.M.; Hekimi, S. Estimating the occurrence of primary ubiquinone deficiency by analysis of large-scale sequencing data. Sci. Rep. 2017, 7, 17744. [Google Scholar] [CrossRef] [PubMed]
- Alcazar-Fabra, M.; Trevisson, E.; Brea-Calvo, G. Clinical syndromes associated with Coenzyme Q10 deficiency. Essays Biochem. 2018, 62, 377–398. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.Q.; Lai, W.S.; Stumpo, D.J.; Blackshear, P.J. Mouse Embryonic Fibroblast Cell Culture and Stimulation. Bio-protocol 2016, 6, e1859. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, G.; Kokocinski, F.; Lichter, P. AutoPrime: Selecting primers for expressed sequences. Genome Biol. 2004, 5, 11. [Google Scholar] [CrossRef]
- Richardson, A.; Fischer, K.E.; Speakman, J.R.; de Cabo, R.; Mitchell, S.J.; Peterson, C.A.; Rabinovitch, P.; Chiao, Y.A.; Taffet, G.; Miller, R.A.; et al. Measures of Healthspan as Indices of Aging in Mice-A Recommendation. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 427–430. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; van Putten, M. Assessing functional performance in the mdx mouse model. J. Vis. Exp. 2014, 85, e51303. [Google Scholar] [CrossRef]
- Calvino-Nunez, C.; Dominguez-del-Toro, E. Clonidine treatment delays postnatal motor development and blocks short-term memory in young mice. PLoS ONE 2014, 9, e114869. [Google Scholar] [CrossRef]
- Dornelles, A.; de Lima, M.N.; Grazziotin, M.; Presti-Torres, J.; Garcia, V.A.; Scalco, F.S.; Roesler, R.; Schroder, N. Adrenergic enhancement of consolidation of object recognition memory. Neurobiol. Learn. Mem. 2007, 88, 137–142. [Google Scholar] [CrossRef]
- Rodriguez-Aguilera, J.C.; Cortes, A.B.; Fernandez-Ayala, D.J.; Navas, P. Biochemical Assessment of Coenzyme Q10 Deficiency. J. Clin. Med. 2017, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Tekle, M.; Brismar, K.; Chojnacki, T.; Swiezewska, E.; Dallner, G. Polyisoprenoid epoxides stimulate the biosynthesis of coenzyme Q and inhibit cholesterol synthesis. J. Biol. Chem. 2008, 283, 14645–14653. [Google Scholar] [CrossRef] [PubMed]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Salabei, J.K.; Gibb, A.A.; Hill, B.G. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 2014, 9, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Madrigal-Matute, J.; Scheibye-Knudsen, M.; Fang, E.; Aon, M.; Gonzalez-Reyes, J.A.; Cortassa, S.; Kaushik, S.; Gonzalez-Freire, M.; Patel, B.; et al. Effects of Sex, Strain, and Energy Intake on Hallmarks of Aging in Mice. Cell Metab. 2016, 23, 1093–1112. [Google Scholar] [CrossRef]
- NCBI-GEO Database Repository. Available online: http://www.ncbi.nlm.nih.gov/geo/ (accessed on 1 August 2019).
- Mitchell, S.J.; Bernier, M.; Aon, M.A.; Cortassa, S.; Kim, E.Y.; Fang, E.F.; Palacios, H.H.; Ali, A.; Navas-Enamorado, I.; Di Francesco, A.; et al. Nicotinamide Improves Aspects of Healthspan, but Not Lifespan, in Mice. Cell Metab. 2018, 27, 667–676. [Google Scholar] [CrossRef]
- Fernandez-Ayala, D.J.; Guerra, I.; Jimenez-Gancedo, S.; Cascajo, M.V.; Gavilan, A.; Dimauro, S.; Hirano, M.; Briones, P.; Artuch, R.; De Cabo, R.; et al. Survival transcriptome in the coenzyme Q10 deficiency syndrome is acquired by epigenetic modifications: A modelling study for human coenzyme Q10 deficiencies. BMJ Open 2013, 3, e002524. [Google Scholar] [CrossRef]
- The National Heart, Lung, and Blood Institute (NHLBI) exome sequencing project exome variant server. Available online: http://evs.gs.washington.edu/EVS/ (accessed on 1 August 2019).
- The Genome Aggregation Database of Broad Institute of Harvard University and Massachusetts Institute of Technology. Available online: http://gnomad.broadinstitute.org (accessed on 1 August 2019).
- Newman, J.C.; Verdin, E. beta-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef]
- Yu, Q.; Zhou, B.; Zhang, Y.; Nguyen, E.T.; Du, J.; Glosson, N.L.; Kaplan, M.H. DNA methyltransferase 3a limits the expression of interleukin-13 in T helper 2 cells and allergic airway inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, 541–546. [Google Scholar] [CrossRef]
- Murphy, M.P.; O’Neill, L.A.J. Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 2018, 174, 780–784. [Google Scholar] [CrossRef]
- Teveroni, E.; Pellegrino, M.; Sacconi, S.; Calandra, P.; Cascino, I.; Farioli-Vecchioli, S.; Puma, A.; Garibaldi, M.; Morosetti, R.; Tasca, G.; et al. Estrogens enhance myoblast differentiation in facioscapulohumeral muscular dystrophy by antagonizing DUX4 activity. J. Clin. Investig. 2017, 127, 1531–1545. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Schneiderat, P.; Schoser, B.G.; Gempel, K.; Neuen-Jacob, E.; Ploger, H.; Muller-Hocker, J.; Pongratz, D.E.; Naini, A.; DiMauro, S.; et al. Coenzyme Q10 deficiency and isolated myopathy. Neurology 2006, 66, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Lalani, S.R.; Vladutiu, G.D.; Plunkett, K.; Lotze, T.E.; Adesina, A.M.; Scaglia, F. Isolated mitochondrial myopathy associated with muscle coenzyme Q10 deficiency. Arch. Neurol. 2005, 62, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef] [PubMed]
- Parrado-Fernandez, C.; Lopez-Lluch, G.; Rodriguez-Bies, E.; Santa-Cruz, S.; Navas, P.; Ramsey, J.J.; Villalba, J.M. Calorie restriction modifies ubiquinone and COQ transcript levels in mouse tissues. Free Radic. Biol. Med. 2011, 50, 1728–1736. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Alvarez, M.I.; Sebastian, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernandez, V.; Vasconcelos, N.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895. [Google Scholar] [CrossRef]
- Eisenberg-Bord, M.; Tsui, H.S.; Antunes, D.; Fernandez-Del-Rio, L.; Bradley, M.C.; Dunn, C.D.; Nguyen, T.P.T.; Rapaport, D.; Clarke, C.F.; Schuldiner, M. The Endoplasmic Reticulum-Mitochondria Encounter Structure Complex Coordinates Coenzyme Q Biosynthesis. Contact (Thousand Oaks) 2019, 2, 2515256418825409. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, P.; Matern, D.; Bennett, M.J. Fatty acid oxidation disorders. Annu. Rev. Physiol. 2002, 64, 477–502. [Google Scholar] [CrossRef]
- Schatz, U.A.; Ensenauer, R. The clinical manifestation of MCAD deficiency: Challenges towards adulthood in the screened population. J. Inherit. Metab. Dis. 2010, 33, 513–520. [Google Scholar] [CrossRef]
- Wicks, S.E.; Vandanmagsar, B.; Haynie, K.R.; Fuller, S.E.; Warfel, J.D.; Stephens, J.M.; Wang, M.; Han, X.; Zhang, J.; Noland, R.C.; et al. Impaired mitochondrial fat oxidation induces adaptive remodeling of muscle metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, E3300–E3309. [Google Scholar] [CrossRef]
- Zhao, L.; Pascual, F.; Bacudio, L.; Suchanek, A.L.; Young, P.A.; Li, L.O.; Martin, S.A.; Camporez, J.P.; Perry, R.J.; Shulman, G.I.; et al. Defective fatty acid oxidation in mice with muscle-specific acyl-CoA synthetase 1 deficiency increases amino acid use and impairs muscle function. J. Biol. Chem. 2019, 294, 8819–8833. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Wicks, S.E.; Vandanmagsar, B.; Mendoza, T.M.; Bayless, D.S.; Salbaum, J.M.; Dearth, S.P.; Campagna, S.R.; Mynatt, R.L.; Noland, R.C. Extensive metabolic remodeling after limiting mitochondrial lipid burden is consistent with an improved metabolic health profile. J. Biol. Chem. 2019, 294, 12313–12327. [Google Scholar] [CrossRef] [PubMed]
- Li, L.O.; Grevengoed, T.J.; Paul, D.S.; Ilkayeva, O.; Koves, T.R.; Pascual, F.; Newgard, C.B.; Muoio, D.M.; Coleman, R.A. Compartmentalized acyl-CoA metabolism in skeletal muscle regulates systemic glucose homeostasis. Diabetes 2015, 64, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Kozawa, S.; Honda, A.; Kajiwara, N.; Takemoto, Y.; Nagase, T.; Nikami, H.; Okano, Y.; Nakashima, S.; Shimozawa, N. Induction of peroxisomal lipid metabolism in mice fed a high-fat diet. Mol. Med. Rep. 2011, 4, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Fonseca, L.; Schäefer, J.; Navas-Enamorado, I.; Santos-Ocaña, C.; Hernández-Camacho, J.D.; Guerra, I.; Cascajo, M.V.; Sánchez-Cuesta, A.; Horvath, Z.; Siendones, E.; et al. ADCK2 Haploinsufficiency Reduces Mitochondrial Lipid Oxidation and Causes Myopathy Associated with CoQ Deficiency. J. Clin. Med. 2019, 8, 1374. https://doi.org/10.3390/jcm8091374
Vázquez-Fonseca L, Schäefer J, Navas-Enamorado I, Santos-Ocaña C, Hernández-Camacho JD, Guerra I, Cascajo MV, Sánchez-Cuesta A, Horvath Z, Siendones E, et al. ADCK2 Haploinsufficiency Reduces Mitochondrial Lipid Oxidation and Causes Myopathy Associated with CoQ Deficiency. Journal of Clinical Medicine. 2019; 8(9):1374. https://doi.org/10.3390/jcm8091374
Chicago/Turabian StyleVázquez-Fonseca, Luis, Jochen Schäefer, Ignacio Navas-Enamorado, Carlos Santos-Ocaña, Juan D. Hernández-Camacho, Ignacio Guerra, María V. Cascajo, Ana Sánchez-Cuesta, Zoltan Horvath, Emilio Siendones, and et al. 2019. "ADCK2 Haploinsufficiency Reduces Mitochondrial Lipid Oxidation and Causes Myopathy Associated with CoQ Deficiency" Journal of Clinical Medicine 8, no. 9: 1374. https://doi.org/10.3390/jcm8091374
APA StyleVázquez-Fonseca, L., Schäefer, J., Navas-Enamorado, I., Santos-Ocaña, C., Hernández-Camacho, J. D., Guerra, I., Cascajo, M. V., Sánchez-Cuesta, A., Horvath, Z., Siendones, E., Jou, C., Casado, M., Gutierrez-Rios, P., Brea-Calvo, G., López-Lluch, G., Fernández-Ayala, D. J. M., Cortés, A. B., Rodríguez-Aguilera, J. C., Matté, C., ... Navas, P. (2019). ADCK2 Haploinsufficiency Reduces Mitochondrial Lipid Oxidation and Causes Myopathy Associated with CoQ Deficiency. Journal of Clinical Medicine, 8(9), 1374. https://doi.org/10.3390/jcm8091374