The Role of Genetic Factors in Characterizing Extra-Intestinal Manifestations in Crohn’s Disease Patients: Are Bayesian Machine Learning Methods Improving Outcome Predictions?


 ,
,  , ,
, ,  , , , and
, , , and 
Abstract
1. Introduction
2. Material and Methods
2.1. BMLTs
2.2. Statistical Analysis
3. Results
4. Discussion
4.1. Study Limitations
4.2. Final Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burisch, J.; Jess, T.; Martinato, M.; Lakatos, P.L. The burden of inflammatory bowel disease in Europe. J. Crohn’s Colitis 2013, 7, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Shivananda, S.; Lennard-Jones, J.; Logan, R.; Fear, N.; Price, A.; Carpenter, L.; van Blankenstein, M. Incidence of inflammatory bowel disease across Europe: Is there a difference between north and south? Results of the European Collaborative Study on Inflammatory Bowel Disease (EC-IBD). Gut 1996, 39, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, L.; Pandur, T.; David, G.; Balogh, Z.; Kuronya, P.; Tollas, A.; Lakatos, P.L. Association of extraintestinal manifestations of inflammatory bowel disease in a province of western Hungary with disease phenotype: Results of a 25-year follow-up study. World J. Gastroenterol. 2003, 9, 2300–2307. [Google Scholar] [CrossRef] [PubMed]
- Vavricka, S.R.; Brun, L.; Ballabeni, P.; Pittet, V.; Prinz Vavricka, B.M.; Zeitz, J.; Rogler, G.; Schoepfer, A.M. Frequency and risk factors for extraintestinal manifestations in the Swiss inflammatory bowel disease cohort. Am. J. Gastroenterol. 2011, 106, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Orchard, T.R.; Chua, C.N.; Ahmad, T.; Cheng, H.; Welsh, K.I.; Jewell, D.P. Uveitis and erythema nodosum in inflammatory bowel disease: Clinical features and the role of HLA genes. Gastroenterology 2002, 123, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Farhi, D.; Cosnes, J.; Zizi, N.; Chosidow, O.; Seksik, P.; Beaugerie, L.; Aractingi, S.; Khosrotehrani, K. Significance of erythema nodosum and pyoderma gangrenosum in inflammatory bowel diseases: A cohort study of 2402 patients. Medicine 2008, 87, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Olsson, R.; Danielsson, A.; Järnerot, G.; Lindström, E.; Lööf, L.; Rolny, P.; Rydén, B.O.; Tysk, C.; Wallerstedt, S. Prevalence of primary sclerosing cholangitis in patients with ulcerative colitis. Gastroenterology 1991, 100, 1319–1323. [Google Scholar] [CrossRef]
- Danese, S.; Semeraro, S.; Papa, A.; Roberto, I.; Scaldaferri, F.; Fedeli, G.; Gasbarrini, G.; Gasbarrini, A. Extraintestinal manifestations in inflammatory bowel disease. World J. Gastroenterol. 2005, 11, 7227–7236. [Google Scholar] [CrossRef] [PubMed]
- Caprilli, R.; Gassull, M.A.; Escher, J.C.; Moser, G.; Munkholm, P.; Forbes, A.; Hommes, D.W.; Lochs, H.; Angelucci, E.; Cocco, A.; et al. European evidence based consensus on the diagnosis and management of Crohn’s disease: Special situations. Gut 2006, 55, i36–i58. [Google Scholar] [CrossRef] [PubMed]
- Stange, E.F.; Travis, S.P.L.; Vermeire, S.; Beglinger, C.; Kupcinskas, L.; Geboes, K.; Barakauskiene, A.; Villanacci, V.; Von Herbay, A.; Warren, B.F.; et al. European evidence based consensus on the diagnosis and management of Crohn’s disease: Definitions and diagnosis. Gut 2006, 55, i1–i15. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-B.; Lee, S.H.; Montgomery, G.W.; Wray, N.R.; Visscher, P.M.; Gearry, R.B.; Lawrance, I.C.; Andrews, J.M.; Bampton, P.; Mahy, G.; et al. Performance of risk prediction for inflammatory bowel disease based on genotyping platform and genomic risk score method. BMC Med. Genet. 2017, 18, 94. [Google Scholar] [CrossRef] [PubMed]
- Kooperberg, C.; LeBlanc, M.; Obenchain, V. Risk prediction using genome-wide association studies. Genet. Epidemiol. 2010, 34, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Wang, W.; Bradfield, J.; Li, J.; Cardinale, C.; Frackelton, E.; Kim, C.; Mentch, F.; Van Steen, K.; Visscher, P.M.; et al. Large Sample Size, Wide Variant Spectrum, and Advanced Machine-Learning Technique Boost Risk Prediction for Inflammatory Bowel Disease. Am. J. Hum. Genet. 2013, 92, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Giachino, D.F.; Regazzoni, S.; Bardessono, M.; De Marchi, M.; Gregori, D.; Piedmont Study Group on the Genetics of IBD. Modeling the role of genetic factors in characterizing extra-intestinal manifestations in Crohn’s disease patients: Does this improve outcome predictions? Curr. Med. Res. Opin. 2007, 23, 1657–1665. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Lee, T.-W.; Sample, P.A.; Goldbaum, M.H.; Weinreb, R.N.; Sejnowski, T.J. Comparison of machine learning and traditional classifiers in glaucoma diagnosis. IEEE Trans. Biomed. Eng. 2002, 49, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Dreiseitl, S.; Ohno-Machado, L.; Kittler, H.; Vinterbo, S.; Billhardt, H.; Binder, M. A comparison of machine learning methods for the diagnosis of pigmented skin lesions. J. Biomed. Inf. 2001, 34, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Roy, J.; Stewart, W.F. Prediction modeling using EHR data: Challenges, strategies, and a comparison of machine learning approaches. Med. Care 2010, 48, S106–S113. [Google Scholar] [CrossRef]
- Barber, D. Bayesian Reasoning and Machine Learning. Available online: http://web4.cs.ucl.ac.uk/staff/D.Barber/textbook/090310.pdf (accessed on 17 June 2019).
- Hernández, B.; Raftery, A.E.; Pennington, S.R.; Parnell, A.C. Bayesian Additive Regression Trees using Bayesian model averaging. Stat. Comput. 2018, 28, 869–890. [Google Scholar] [CrossRef]
- Berchialla, P.; Scarinzi, C.; Snidero, S.; Gregori, D. Comparing models for quantitative risk assessment: An application to the European Registry of foreign body injuries in children. Stat. Methods Med. Res. 2016, 25, 1244–1259. [Google Scholar] [CrossRef]
- Palaniappan, S.; Awang, R. Intelligent Heart Disease Prediction System Using Data Mining Techniques. In Proceedings of the 2008 IEEE/ACS International Conference on Computer Systems and Applications, Doha, Qatar, 31 March–4 April 2008; IEEE Computer Society: Washington, DC, USA, 2008; pp. 108–115. [Google Scholar]
- Sesen, M.B.; Nicholson, A.E.; Banares-Alcantara, R.; Kadir, T.; Brady, M. Bayesian networks for clinical decision support in lung cancer care. PLoS ONE 2013, 8, e82349. [Google Scholar] [CrossRef]
- Wei, W.; Visweswaran, S.; Cooper, G.F. The application of naive Bayes model averaging to predict Alzheimer’s disease from genome-wide data. J. Am. Med. Inf. Assoc. 2011, 18, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Sparapani, R.A.; Logan, B.R.; McCulloch, R.E.; Laud, P.W. Nonparametric survival analysis using Bayesian Additive Regression Trees (BART). Stat. Med. 2016, 35, 2741–2753. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, P. Genome-wide prediction using Bayesian additive regression trees. Genet. Sel. Evol. 2016, 48, 42. [Google Scholar] [CrossRef] [PubMed]
- Menti, E.; Lanera, C.; Lorenzoni, G.; Giachino, D.F.; Marchi, M.D.; Gregori, D.; Berchialla, P.; Piedmont Study Group on the Genetics of IBD. Bayesian Machine Learning Techniques for revealing complex interactions among genetic and clinical factors in association with extra-intestinal Manifestations in IBD patients. AMIA Annu. Symp. Proc. 2016, 2016, 884–893. [Google Scholar] [PubMed]
- Langley, P.; Iba, W.; Thompson, K. An Analysis of Bayesian Classifiers. In Proceedings of the Tenth National Conference on Artificial Intelligence, San Jose, CA, USA, 12–16 July 1992; AAAI Press: San Jose, CA, USA, 1992; pp. 223–228. [Google Scholar]
- Sordo, M.; Zeng, Q. On Sample Size and Classification Accuracy: A Performance Comparison. In Proceedings of the Biological and Medical Data Analysis, Thessaloniki, Greece, 7–8 December 2006; Springer: Berlin/Heidelberg, Germany, 2005; pp. 193–201. [Google Scholar]
- Domingos, P.; Pazzani, M. On the Optimality of the Simple Bayesian Classifier under Zero-One Loss. Mach. Learn. 1997, 29, 103–130. [Google Scholar] [CrossRef]
- Jensen, F.V. Introduction to Bayesian Networks, 1st ed.; Springer-Verlag New York, Inc.: Secaucus, NJ, USA, 1996; ISBN 978-0-387-91502-9. [Google Scholar]
- Chipman, H.A.; George, E.I.; McCulloch, R.E. BART: Bayesian additive regression trees. Ann. Appl. Stat. 2010, 4, 266–298. [Google Scholar] [CrossRef]
- Zhang, H. The Optimality of Naïve Bayes. In Proceedings of the FLAIRS2004 Conference, Miami Beach, FL, USA, 12–14 May 2004. [Google Scholar]
- Dos Santos, E.B.; Ebecken, N.F.F.; Hruschka, E.R.; Elkamel, A.; Madhuranthakam, C.M.R. Bayesian Classifiers Applied to the Tennessee Eastman Process. Risk Anal. 2014, 34, 485–497. [Google Scholar] [CrossRef]
- Roos, T.; Wettig, H.; Grünwald, P.; Myllymäki, P.; Tirri, H. On Discriminative Bayesian Network Classifiers and Logistic Regression. Mach. Learn 2005, 59, 267–296. [Google Scholar] [CrossRef]
- Sherif, F.F.; Zayed, N.; Fakhr, M. Discovering Alzheimer Genetic Biomarkers Using Bayesian Networks. Adv. Bioinform. 2015, 2015, 639367. [Google Scholar] [CrossRef]
- Avila, R.; Horn, B.; Moriarty, E.; Hodson, R.; Moltchanova, E. Evaluating statistical model performance in water quality prediction. J. Environ. Manag. 2018, 206, 910–919. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, S.; Li, R.; Shahabi, H. Performance evaluation of the GIS-based data mining techniques of best-first decision tree, random forest, and naïve Bayes tree for landslide susceptibility modeling. Sci. Total Environ. 2018, 644, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Frizzell, J.D.; Liang, L.; Schulte, P.J.; Yancy, C.W.; Heidenreich, P.A.; Hernandez, A.F.; Bhatt, D.L.; Fonarow, G.C.; Laskey, W.K. Prediction of 30-Day All-Cause Readmissions in Patients Hospitalized for Heart Failure: Comparison of Machine Learning and Other Statistical Approaches. JAMA Cardiol. 2017, 2, 204–209. [Google Scholar] [CrossRef]
- Montazeri, M.; Montazeri, M.; Montazeri, M.; Beigzadeh, A. Machine learning models in breast cancer survival prediction. Technol. Health Care 2016, 24, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, L.; Ren, J.; Ahlgren, N.A.; Fuhrman, J.A.; Sun, F. Prediction of virus-host infectious association by supervised learning methods. BMC Bioinform. 2017, 18, 60. [Google Scholar] [CrossRef] [PubMed]
- Efron, B. Estimating the Error Rate of a Prediction Rule: Improvement on Cross-Validation. J. Am. Stat. Assoc. 1983, 78, 316–331. [Google Scholar] [CrossRef]
- Harrell, F., Jr. Regression Modeling Strategies with Applications to Linear Models, Logistic and Ordinal Regression, and Survival Analysis, 2nd ed.; Springer: New York, NY, USA, 2015. [Google Scholar]
- Gelman, A.; Rubin, D.B. Avoiding Model Selection in Bayesian Social Research. Sociol. Methodol. 1995, 25, 165–173. [Google Scholar] [CrossRef]
- Claeskens, G.; Hjort, N.L. Model Selection and Model Averaging by Gerda Claeskens. Available online: /core/books/model-selection-and-model-averaging/E6F1EC77279D1223423BB64FC3A12C37 (accessed on 13 August 2018).
- Scutari, M.; Nagarajan, R. Identifying significant edges in graphical models of molecular networks. Artif. Intell. Med. 2013, 57, 207–217. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R foundation for statistical computing: Vienna, Austria, 2018. [Google Scholar]
- Scutari, M. Learning Bayesian Networks with the bnlearn R Package. J. Stat. Softw. 2010, 35. [Google Scholar] [CrossRef]
- Kapelner, A.; Bleich, J. bartMachine: Machine Learning with Bayesian Additive Regression Trees. J. Stat. Softw. 2016, 70. [Google Scholar] [CrossRef]
- Napolioni, V.; Predazzi, I.M. Age- and gender-specific association between ADA (22G>A) and TNF-α (-308G>A) genetic polymorphisms. Tissue Antigens 2010, 76, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Altman, D.G.; Royston, P. The cost of dichotomising continuous variables. BMJ 2006, 332, 1080. [Google Scholar] [CrossRef] [PubMed]
- Dawson, N.V.; Weiss, R. Dichotomizing continuous variables in statistical analysis: A practice to avoid. Med. Decis. Mak. 2012, 32, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, V.; Mannino, F.; Zhang, R. Consequences of dichotomization. Pharm. Stat. 2009, 8, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Royston, P.; Altman, D.G.; Sauerbrei, W. Dichotomizing continuous predictors in multiple regression: A bad idea. Stat. Med. 2006, 25, 127–141. [Google Scholar] [CrossRef] [PubMed]



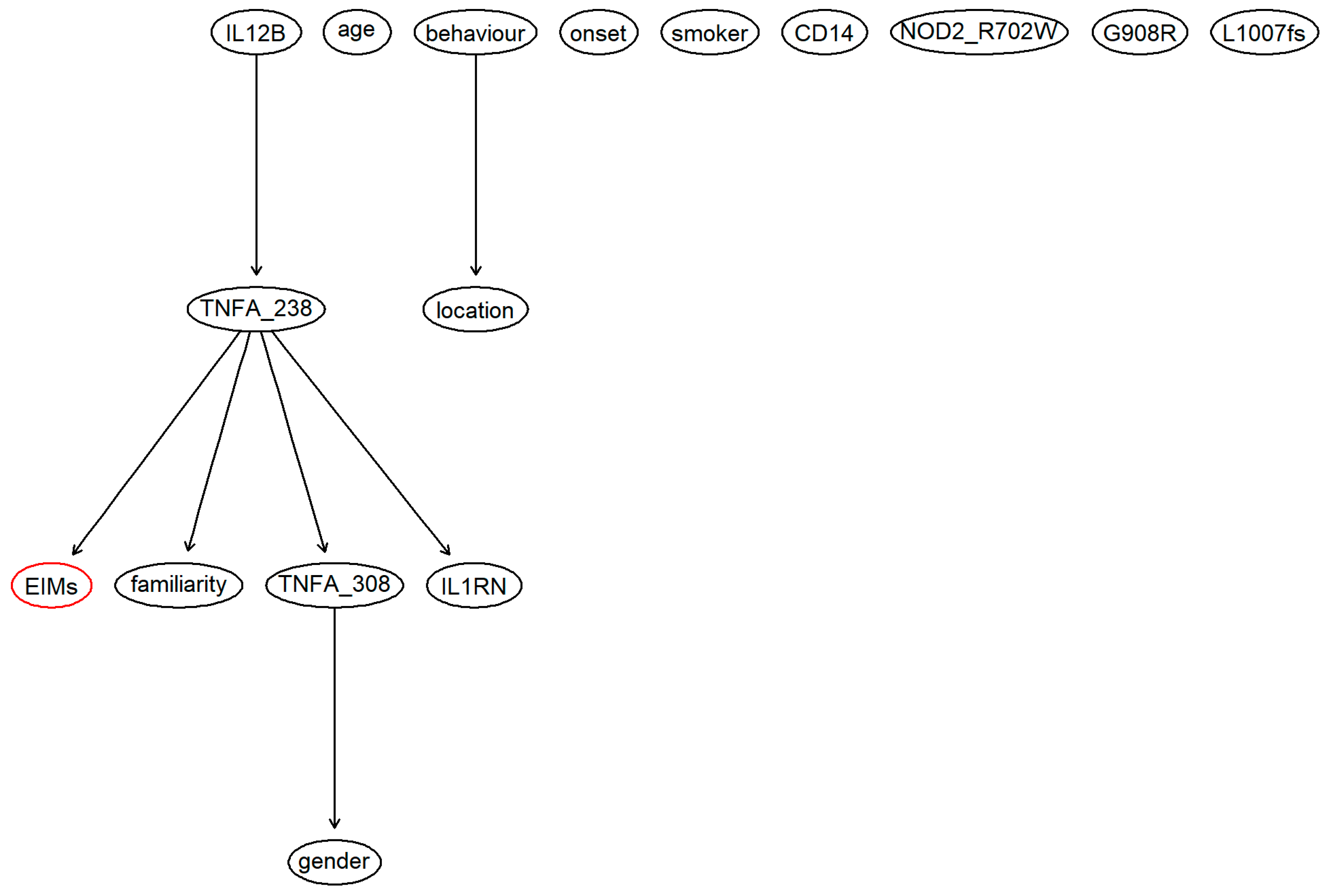{kind=link}
{kind=link}
| Variable (n*) | EIM = No, n (%) N = 77 | EIM = Yes, n (%) N = 75 | Combined, n (%) N = 152 | |
|---|---|---|---|---|
| Onset (147) | Medical | 70 (90.9) | 63 (84.0) | 133 (87.5) |
| Surgical | 7 (9.1) | 7 (9.3) | 14 (9.2) | |
| Behavior (108) | B1 | 25 (32.5) | 25 (33.3) | 50 (32.9) |
| B2 | 15 (19.5) | 20 (26.7) | 35 (23.0) | |
| B3 | 9 (11.7) | 14 (18.7) | 23 (15.1) | |
| Location (109) | L1 | 14 (18.2) | 14 (18.7) | 28 (18.4) |
| L2 | 11 (14.3) | 14 (18.7) | 25 (16.4) | |
| L3 | 21 (27.3) | 27 (36.0) | 48 (31.6) | |
| L4 | 4 (5.2) | 4 (5.3) | 8 (5.3) | |
| Age (146) | A1 | 53 (68.8) | 51 (68.0) | 104 (68.4) |
| A2 | 21 (27.3) | 21 (28.0) | 42 (27.6) | |
| Gender (152) | M | 46 (59.7) | 34 (45.3) | 80 (52.6) |
| F | 31 (40.3) | 41 (54.7) | 72 (47.4) | |
| Smoker (146) | No | 42 (54.5) | 36 (48.0) | 78 (51.3) |
| Yes | 19 (24.7) | 26 (34.7) | 45 (29.6) | |
| Ex | 11 (14.3) | 12 (16.0) | 23 (15.1) | |
| Family History (139) | No | 58 (75.3) | 57 (76.0) | 115 (75.7) |
| Yes | 11 (14.3) | 13 (17.3) | 24 (15.8) | |
| NOD2:R702W (152) | RR | 63 (81.8) | 64 (85.3) | 127 (83.6) |
| RW | 11 (14.3) | 9 (12.0) | 20 (13.2) | |
| WW | 3 (3.9) | 2 (2.7) | 5 (3.3) | |
| G908R (152) | GG | 73 (94.8) | 67 (89.3) | 140(92.1) |
| GR | 4 (5.2) | 8 (10.7) | 12 (7.9) | |
| L1007fs (152) | LL | 71 (92.2) | 65 (86.7) | 136 (89.5) |
| L/insC | 5 (6.5) | 8 (10.7) | 13 (8.6) | |
| insC/insC | 1 (1.3) | 2 (2.7) | 3 (2.0) | |
| CD14 (152) | CC | 20 (26.0) | 20 (26.7) | 40 (26.3) |
| TC | 39 (50.6) | 36 (48.0) | 75 (49.3) | |
| TT | 18 (23.4) | 19 (25.3) | 37 (24.3) | |
| TNF-308 (72) | GG | 35 (45.5) | 18 (24.0) | 53 (34.9) |
| GA | 9 (11.7) | 4 (5.3) | 13 (8.6) | |
| AA | 5 (6.5) | 1 (1.3) | 6 (3.9) | |
| TNF -238 (72) | GG | 49 (63.6) | 23 (30.7) | 72 (47.4) |
| IL12B (72) | AA | 17 (22.1) | 11 (14.7) | 28 (18.4) |
| AC | 24 (31.2) | 10 (13.3) | 34 (22.4) | |
| CC | 8 (10.4) | 2 (2.7) | 10 (6.6) | |
| IL1RN (72) | ILRN*1 | 29 (37.7) | 12 (16.0) | 41 (27.0) |
| ILRN*1/ILRN* | 15 (19.5) | 7 (9.3) | 22 (14.5) | |
| ILRN*2 | 3 (3.9) | 3 (4.0) | 6 (3.9) | |
| ILRN*1/ILRN* | 1(1.3) | 1 (1.3) | 2 (1.3) | |
| ILRN*2/ILRN* | 1 (1.3) | 0 | 1 (0.7) |
| Learning algorithm | MCR |
|---|---|
| Model without genetic variables | |
| Grow-Shrink (GS) | 0.57 |
| Incremental Association Markov-Blanket (IAMB) | 0.61 |
| Fast Incremental Association Markov-Blanket (Fast-IAMB) | 0.61 |
| Interleaved Incremental Association Markov-Blanket (Inter-IAMB) | 0.59 |
| Hill-Climbing (HC) | 0.57 |
| Tabu-Search (TS) | 0.61 |
| Max-Min Hill-Climbing (MMHC) | 0.53 |
| Restricted Maximization (RSMAX2) | 0.60 |
| Max-Min Parents and Children (MMPC) | 0.55 |
| Hiton Parents and Children (SI-HITON-PC) | 0.51 |
| Chow‒Liu (CL) | 0.56 |
| ARACNE | 0.58 |
| Model with genetic variables | |
| Grow-Shrink (GS) | 0.57 |
| Incremental Association Markov-Blanket (IAMB) | 0.62 |
| Fast Incremental Association Markov-Blanket (Fast-IAMB) | 0.61 |
| Interleaved Incremental Association Markov-Blanket (Inter-IAMB) | 0.59 |
| Hill-Climbing (HC) | 0.34 |
| Tabu-Search (TS) | 0.34 |
| Max-Min Hill-Climbing (MMHC) | 0.53 |
| Restricted Maximization (RSMAX2) | 0.60 |
| Max-Min Parents and Children (MMPC) | 0.56 |
| Hiton Parents and Children (SI-HITON-PC) | 0.51 |
| Chow‒Liu (CL) | 0.57 |
| ARACNE | 0.58 |
| MCR | Sensitivity | Specificity | PPV | NPV | AUC | Somer’s D | |
|---|---|---|---|---|---|---|---|
| Model without genetic variables | |||||||
| LR | 0.46 | _ | _ | 0.77 | 0.52 | 0.72 | 0.45 |
| GAM | 0.44 | _ | _ | 0.81 | 0.53 | 0.72 | 0.45 |
| PPR | 0.36 | _ | _ | 0.98 | 0.58 | 0.82 | 0.64 |
| LDA | 0.49 | _ | _ | 0.98 | 0.52 | 0.70 | 0.40 |
| QDA | 0.49 | _ | _ | 0.72 | 0.52 | 0.67 | 0.34 |
| ANN | 0.38 | _ | _ | 0.94 | 0.57 | 0.79 | 0.58 |
| NB | 0.34 | 0.45 | 0.81 | 0.68 | 0.65 | 0.71 | 0.42 |
| BN | 0.50 | 1.00 | 0.00 | 0.51 | 0.49 | 0.50 | 0.00 |
| BART | 0.32 | 0.64 | 0.68 | 0.67 | 0.69 | 0.76 | 0.51 |
| Model with genetic variables | |||||||
| LR | 0.39 | _ | _ | 0.89 | 0.56 | 0.77 | 0.53 |
| GAM | 0.37 | _ | _ | 0.90 | 0.57 | 0.77 | 0.54 |
| PPR | 0.30 | _ | _ | 0.99 | 0.62 | 0.94 | 0.87 |
| LDA | 0.38 | _ | _ | 0.99 | 0.57 | 0.77 | 0.53 |
| QDA | 0.22 | _ | _ | 0.74 | 0.52 | 0.88 | 0.75 |
| ANN | 0.33 | _ | _ | 0.92 | 0.60 | 0.87 | 0.73 |
| NB | 0.33 | 0.65 | 0.69 | 0.69 | 0.66 | 0.75 | 0.51 |
| BN | 0.34 | 0.64 | 0.69 | 0.68 | 0.65 | 0.67 | 0.33 |
| BART | 0.32 | 0.66 | 0.69 | 0.67 | 0.69 | 0.78 | 0.56 |
| ID | EIMs | IL12B | TNFA-308 | TNFA-238 | IL1RN | NB | BN | BART |
|---|---|---|---|---|---|---|---|---|
| 63 | NO | AC | GG | GG | ILRN*1 | 0.06 | 0.36 | 0.26 |
| 64 | NO | AA | GG | GG | ILRN1/ILRN3 | 0.13 | 0.36 | 0.29 |
| 65 | NO | AC | GG | GG | ILRN*1 | 0.15 | 0.36 | 0.48 |
| 66 | NO | AA | GG | GG | ILRN*1 | 0.22 | 0.36 | 0.48 |
| 67 | NO | AC | GG | GG | ILRN*1 | 0.02 | 0.36 | 0.35 |
| 68 | NO | AC | GG | GG | ILRN*1 | 0.03 | 0.36 | 0.31 |
| 69 | NO | AA | GA | GG | ILRN1/ILRN2 | 0.09 | 0.36 | 0.32 |
| 70 | NO | AA | AA | GG | ILRN*1 | 0.04 | 0.36 | 0.37 |
| 71 | YES | AA | GA | GG | ILRN*1 | 0.02 | 0.36 | 0.28 |
| 72 | YES | AA | GG | GG | ILRN*2 | 0.28 | 0.36 | 0.36 |
| 73 | YES | Missing | Missing | Missing | Missing | 0.91 | 0.65 | 0.65 |
| 74 | NO | Missing | Missing | Missing | Missing | 0.69 | 0.65 | 0.61 |
| 75 | YES | Missing | Missing | Missing | Missing | 0.98 | 0.65 | 0.70 |
| 76 | YES | Missing | Missing | Missing | Missing | 0.95 | 0.65 | 0.65 |
| 77 | YES | Missing | Missing | Missing | Missing | 0.97 | 0.65 | 0.64 |
| 78 | YES | Missing | Missing | Missing | Missing | 0.95 | 0.65 | 0.68 |
| 79 | YES | Missing | Missing | Missing | Missing | 0.98 | 0.65 | 0.70 |
| 80 | YES | Missing | Missing | Missing | Missing | 0.88 | 0.65 | 0.68 |
| 81 | YES | Missing | Missing | Missing | Missing | 0.93 | 0.65 | 0.68 |
| 82 | YES | Missing | Missing | Missing | Missing | 0.98 | 0.65 | 0.75 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bottigliengo, D.; Berchialla, P.; Lanera, C.; Azzolina, D.; Lorenzoni, G.; Martinato, M.; Giachino, D.; Baldi, I.; Gregori, D. The Role of Genetic Factors in Characterizing Extra-Intestinal Manifestations in Crohn’s Disease Patients: Are Bayesian Machine Learning Methods Improving Outcome Predictions? J. Clin. Med. 2019, 8, 865. https://doi.org/10.3390/jcm8060865
Bottigliengo D, Berchialla P, Lanera C, Azzolina D, Lorenzoni G, Martinato M, Giachino D, Baldi I, Gregori D. The Role of Genetic Factors in Characterizing Extra-Intestinal Manifestations in Crohn’s Disease Patients: Are Bayesian Machine Learning Methods Improving Outcome Predictions? Journal of Clinical Medicine. 2019; 8(6):865. https://doi.org/10.3390/jcm8060865
Chicago/Turabian StyleBottigliengo, Daniele, Paola Berchialla, Corrado Lanera, Danila Azzolina, Giulia Lorenzoni, Matteo Martinato, Daniela Giachino, Ileana Baldi, and Dario Gregori. 2019. "The Role of Genetic Factors in Characterizing Extra-Intestinal Manifestations in Crohn’s Disease Patients: Are Bayesian Machine Learning Methods Improving Outcome Predictions?" Journal of Clinical Medicine 8, no. 6: 865. https://doi.org/10.3390/jcm8060865
APA StyleBottigliengo, D., Berchialla, P., Lanera, C., Azzolina, D., Lorenzoni, G., Martinato, M., Giachino, D., Baldi, I., & Gregori, D. (2019). The Role of Genetic Factors in Characterizing Extra-Intestinal Manifestations in Crohn’s Disease Patients: Are Bayesian Machine Learning Methods Improving Outcome Predictions? Journal of Clinical Medicine, 8(6), 865. https://doi.org/10.3390/jcm8060865