Epithelial-Mesenchymal Plasticity in Organotropism Metastasis and Tumor Immune Escape



Abstract
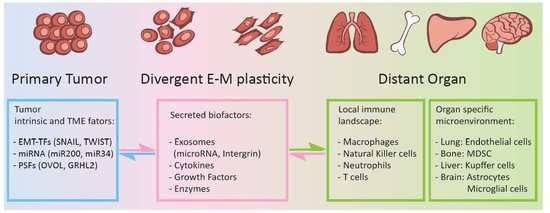
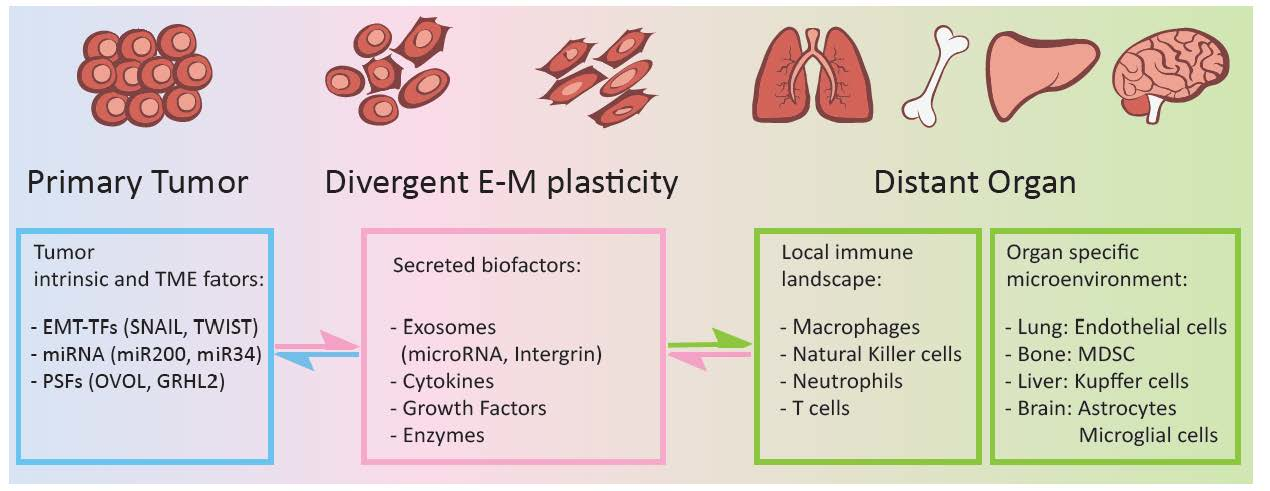{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Organotropic Metastasis
1.2. Heterogeneous EMT Phenotypes and the Activation/Regulation Complexity
2. Epithelial-Mesenchymal Plasticity and Cancer Organotropism Metastasis
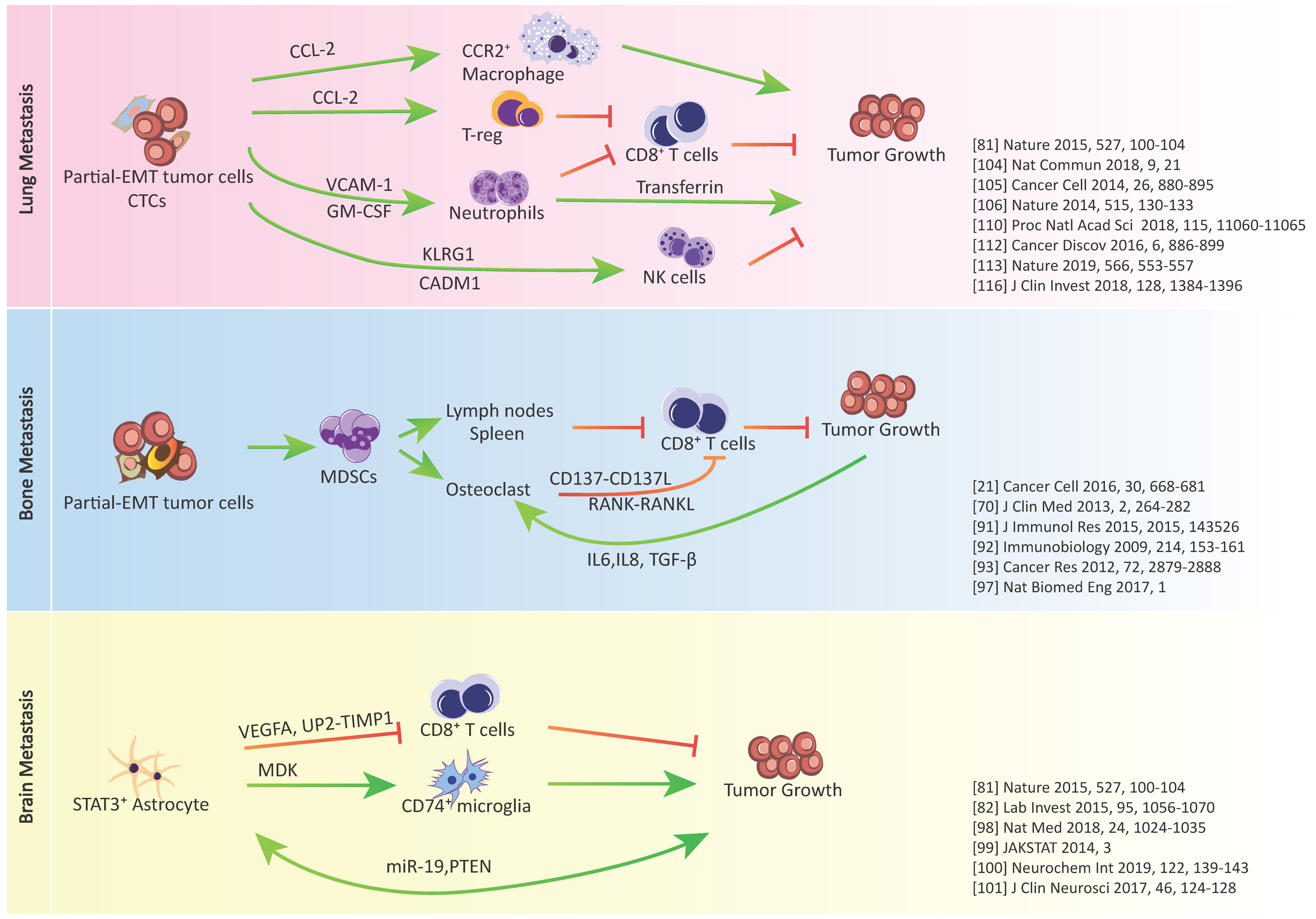
3. Epithelial-Mesenchymal Plasticity and Tumor Immune Escape in Metastatic Organs
3.1. Metastasis-Associated Macrophages
3.2. Metastasis-Associated Neutrophils
3.3. Metastasis-Associated Natural Killer (NK) Cells
3.4. Metastasis-Associated T Cells
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Turajlic, S.; Swanton, C. Metastasis as an evolutionary process. Science 2016, 352, 169–175. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar]
- Akhtar, M.; Haider, A.; Rashid, S.; Al-Nabet, A. Paget’s “Seed and Soil” theory of cancer metastasis: An idea whose time has come. Adv. Anat. Pathol. 2019, 26, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hoffmann, A.D.; Liu, H.; Liu, X. Organotropism: New insights into molecular mechanisms of breast cancer metastasis. NPJ Precis. Oncol. 2018, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, J.; Zhu, S.; Wu, J.; Chen, C.; Liu, Q.; Wei, W.; Zhang, Y.; Sun, S. Breast cancer subtypes predict the preferential site of distant metastases: A SEER based study. Oncotarget 2017, 8, 27990–27996. [Google Scholar] [CrossRef]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef]
- Obenauf, A.C.; Massague, J. Surviving at a distance: Organ specific metastasis. Trends Cancer 2015, 1, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Er, E.E.; Valiente, M.; Ganesh, K.; Zou, Y.; Agrawal, S.; Hu, J.; Griscom, B.; Rosenblum, M.; Boire, A.; Brogi, E.; et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat. Cell Biol. 2018, 20, 966–978. [Google Scholar] [CrossRef]
- Valiente, M.; Obenauf, A.C.; Jin, X.; Chen, Q.; Zhang, X.H.; Lee, D.J.; Chaft, J.E.; Kris, M.G.; Huse, J.T.; Brogi, E.; et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014, 156, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massague, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Kan, C.; Vargas, G.; Pape, F.L.; Clezardin, P. Cancer cell colonisation in the bone microenvironment. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef]
- Dong, X. Current strategies for brain drug delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Mojares, E.; Del Rio Hernandez, A. Role of extracellular matrix in development and cancer progression. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, K.; Szczylik, C.; Bielecka, Z.F.; Bartnik, E.; Porta, C.; Lian, F.; Czarnecka, A.M. The role of the cell–cell interactions in cancer progression. J. Cell Mol. Med. 2015, 19, 283–296. [Google Scholar] [CrossRef]
- Blomberg, O.S.; Spagnuolo, L.; de Visser, K.E. Immune regulation of metastasis: Mechanistic insights and therapeutic opportunities. Dis. Model Mech. 2018, 11. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, X. Characteristics and significance of the pre-metastatic niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Giles, A.J.; Reid, C.M.; Evans, J.D.; Murgai, M.; Vicioso, Y.; Highfill, S.L.; Kasai, M.; Vahdat, L.; Mackall, C.L.; Lyden, D.; et al. Activation of hematopoietic stem/progenitor cells promotes immunosuppression within the pre-metastatic niche. Cancer Res. 2016, 76, 1335–1347. [Google Scholar] [CrossRef]
- Sceneay, J.; Parker, B.S.; Smyth, M.J.; Moller, A. Hypoxia-driven immunosuppression contributes to the pre-metastatic niche. Oncoimmunology 2013, 2, e22355. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Roche, J. The epithelial-to-mesenchymal transition in cancer. Cancers 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhu, X.; Cui, K.; Mancuso, J.; Federley, R.; Fischer, K.; Teng, G.; Mittal, V.; Gao, D.; Zhao, H.; et al. In vivo visualization and characterization of epithelial-mesenchymal transition in breast tumors. Cancer Res. 2016, 76, 2094–2104. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Segall, J.E. Intravital imaging of cell movement in tumours. Nat. Rev. Cancer 2003, 3, 921–930. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT subtype influences epithelial plasticity and mode of cell migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, J.J.; Rajasekaran, A.K. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006, 66, 8319–8326. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Lecharpentier, A.; Vielh, P.; Perez-Moreno, P.; Planchard, D.; Soria, J.C.; Farace, F. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br. J. Cancer 2011, 105, 1338–1341. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia-Blanco, M.A. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef]
- Lo, H.C.; Zhang, X.H. EMT in metastasis: Finding the right balance. Dev. Cell 2018, 45, 663–665. [Google Scholar] [CrossRef]
- Joosse, S.A.; Gorges, T.M.; Pantel, K. Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol. Med. 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef]
- Wingrove, E.; Liu, Z.Z.; Patel, K.D.; Arnal-Estape, A.; Cai, W.L.; Melnick, M.A.; Politi, K.; Monteiro, C.; Zhu, L.; Valiente, M.; et al. Transcriptomic hallmarks of tumor plasticity and stromal interactions in brain metastasis. Cell Rep. 2019, 27, 1277–1292. [Google Scholar] [CrossRef]
- Jeevan, D.S.; Cooper, J.B.; Braun, A.; Murali, R.; Jhanwar-Uniyal, M. Molecular pathways mediating metastases to the brain via epithelial-to-mesenchymal transition: Genes, proteins, and functional analysis. Anticancer Res. 2016, 36, 523–532. [Google Scholar] [PubMed]
- Somarelli, J.A.; Schaeffer, D.; Marengo, M.S.; Bepler, T.; Rouse, D.; Ware, K.E.; Hish, A.J.; Zhao, Y.; Buckley, A.F.; Epstein, J.I.; et al. Distinct routes to metastasis: Plasticity-dependent and plasticity-independent pathways. Oncogene 2016, 35, 4302–4311. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; von der Ohe, J.; Lehnert, H.; Ungefroren, H.; Hass, R. Cancer stem cell niche models and contribution by mesenchymal stroma/stem cells. Mol. Cancer 2017, 16, 28. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Battistelli, C.; Cicchini, C.; Santangelo, L.; Tramontano, A.; Grassi, L.; Gonzalez, F.J.; de Nonno, V.; Grassi, G.; Amicone, L.; Tripodi, M. The Snail repressor recruits EZH2 to specific genomic sites through the enrollment of the lncRNA HOTAIR in epithelial-to-mesenchymal transition. Oncogene 2017, 36, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Battistelli, C.; Tripodi, M.; Cicchini, C. Targeting of polycombs to DNA in EMT. Oncotarget 2017, 8, 57936–57937. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.L.; Shepard, C.R.; Wells, A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer 2010, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tang, Y.L.; Liang, X.H. EMT: A new vision of hypoxia promoting cancer progression. Cancer Biol. Ther. 2011, 11, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.P.; Harishankar, M.K.; Pillai, A.A.; Devi, A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018, 80, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.J.; Xing, J. Signal transduction pathways of EMT induced by TGF-beta, SHH, and WNT and their crosstalks. J. Clin. Med. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Jia, D.; Boareto, M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Coupling the modules of EMT and stemness: A tunable ‘stemness window’ model. Oncotarget 2015, 6, 25161–25174. [Google Scholar] [CrossRef]
- Garg, M. Epithelial plasticity and cancer stem cells: Major mechanisms of cancer pathogenesis and therapy resistance. World J. Stem Cells 2017, 9, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Reichert, M.; Bakir, B.; Moreira, L.; Pitarresi, J.R.; Feldmann, K.; Simon, L.; Suzuki, K.; Maddipati, R.; Rhim, A.D.; Schlitter, A.M.; et al. Regulation of epithelial plasticity determines metastatic organotropism in pancreatic cancer. Dev. Cell 2018, 45, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.T.; Yang, M.H. Revisiting epithelial-mesenchymal transition in cancer metastasis: The connection between epithelial plasticity and stemness. Mol. Oncol. 2017, 11, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Chen, L.; Ma, Z.; Du, Z.; Zhao, Z.; Hu, Z.; Li, Q. Isolation and phenotypic characterization of colorectal cancer stem cells with organ-specific metastatic potential. Gastroenterology 2013, 145, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, M.; Nosrati, R.; Salmaninejad, A.; Dehghani, S.; Shahryari, A.; Saberi, A. Organ-specific metastasis of breast cancer: Molecular and cellular mechanisms underlying lung metastasis. Cell Oncol. 2018, 41, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Zhu, X.; Kong, R.; Zhao, Z.; Sheng, J.; Wang, J.; Xu, X.; Liu, J.; Cui, K.; Zhang, X.H.; et al. Targeting brain-adaptive cancer stem cells prohibits brain metastatic colonization of triple-negative breast cancer. Cancer Res. 2018, 78, 2052–2064. [Google Scholar] [CrossRef]
- Stankic, M.; Pavlovic, S.; Chin, Y.; Brogi, E.; Padua, D.; Norton, L.; Massague, J.; Benezra, R. TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013, 5, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.M.; Panzilius, E.; Bartsch, H.S.; Irmler, M.; Beckers, J.; Kari, V.; Linnemann, J.R.; Dragoi, D.; Hirschi, B.; Kloos, U.J.; et al. Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Rep. 2015, 10, 131–139. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and functional characterization of epithelial-mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.; Li, Z.Y.; Liu, Y.; Beyer, I.; Persson, J.; Sova, P.; Moller, T.; Pesonen, S.; Hemminki, A.; Hamerlik, P.; et al. Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLoS ONE 2011, 6, e16186. [Google Scholar] [CrossRef]
- Beerling, E.; Seinstra, D.; de Wit, E.; Kester, L.; van der Velden, D.; Maynard, C.; Schafer, R.; van Diest, P.; Voest, E.; van Oudenaarden, A.; et al. Plasticity between epithelial and mesenchymal states unlinks EMT from metastasis-enhancing stem cell capacity. Cell Rep. 2016, 14, 2281–2288. [Google Scholar] [CrossRef]
- Perdigao-Henriques, R.; Petrocca, F.; Altschuler, G.; Thomas, M.P.; Le, M.T.; Tan, S.M.; Hide, W.; Lieberman, J. miR-200 promotes the mesenchymal to epithelial transition by suppressing multiple members of the Zeb2 and Snail1 transcriptional repressor complexes. Oncogene 2016, 35, 158–172. [Google Scholar] [CrossRef]
- Bockhorn, J.; Dalton, R.; Nwachukwu, C.; Huang, S.; Prat, A.; Yee, K.; Chang, Y.F.; Huo, D.; Wen, Y.; Swanson, K.E.; et al. MicroRNA-30c inhibits human breast tumour chemotherapy resistance by regulating TWF1 and IL-11. Nat. Commun. 2013, 4, 1393. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H.; Xing, F.; Pandey, P.R.; Sharma, S.; Watabe, M.; Pai, S.K.; Mo, Y.Y.; Iiizumi-Gairani, M.; Hirota, S.; Liu, Y.; et al. miR-7 suppresses brain metastasis of breast cancer stem-like cells by modulating KLF4. Cancer Res. 2013, 73, 1434–1444. [Google Scholar] [CrossRef] [PubMed]
- Demirkan, B. The roles of epithelial-to-mesenchymal transition (EMT) and mesenchymal-to-epithelial transition (MET) in breast cancer bone metastasis: Potential targets for prevention and treatment. J. Clin. Med. 2013, 2, 264–282. [Google Scholar] [CrossRef]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef] [PubMed]
- Ell, B.; Qiu, Q.; Wei, Y.; Mercatali, L.; Ibrahim, T.; Amadori, D.; Kang, Y. The microRNA-23b/27b/24 cluster promotes breast cancer lung metastasis by targeting metastasis-suppressive gene prosaposin. J. Biol. Chem. 2014, 289, 21888–21895. [Google Scholar] [CrossRef]
- Ma, F.; Li, W.; Liu, C.; Li, W.; Yu, H.; Lei, B.; Ren, Y.; Li, Z.; Pang, D.; Qian, C. MiR-23a promotes TGF-beta1-induced EMT and tumor metastasis in breast cancer cells by directly targeting CDH1 and activating Wnt/beta-catenin signaling. Oncotarget 2017, 8, 69538–69550. [Google Scholar]
- Jiang, G.; Shi, W.; Fang, H.; Zhang, X. miR27a promotes human breast cancer cell migration by inducing EMT in a FBXW7dependent manner. Mol. Med. Rep. 2018, 18, 5417–5426. [Google Scholar]
- Schrijver, W.A.; van Diest, P.J.; Dutch Distant Breast Cancer Metastases Consortium; Moelans, C.B. Unravelling site-specific breast cancer metastasis: A microRNA expression profiling study. Oncotarget 2017, 8, 3111–3123. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, D.; Liang, S.; Wang, J.; Liu, C.; Nie, C.; Shan, Z.; Wang, L.; Fan, Q.; Wang, F. miR-106b promotes cell invasion and metastasis via PTEN mediated EMT in ESCC. Oncol. Lett. 2018, 15, 4619–4626. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hu, Y.; Dai, L.; Wang, Y.; Zhou, J.; Wang, W.; Di, W.; Qiu, L. MicroRNA-7 inhibits tumor metastasis and reverses epithelial-mesenchymal transition through AKT/ERK1/2 inactivation by targeting EGFR in epithelial ovarian cancer. PLoS ONE 2014, 9, e96718. [Google Scholar] [CrossRef] [PubMed]
- Mody, H.R.; Hung, S.W.; Pathak, R.K.; Griffin, J.; Cruz-Monserrate, Z.; Govindarajan, R. miR-202 diminishes TGFbeta receptors and attenuates TGFbeta1-induced EMT in pancreatic cancer. Mol. Cancer Res. 2017, 15, 1029–1039. [Google Scholar] [CrossRef]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, S.; Yan, W.; Yang, J.; Qin, Y.J.; Lin, X.L.; Xie, R.Y.; Wang, S.C.; Jin, W.; Gao, F.; et al. MicroRNA-19 triggers epithelial-mesenchymal transition of lung cancer cells accompanied by growth inhibition. Lab. Investig. 2015, 95, 1056–1070. [Google Scholar] [CrossRef]
- Rana, S.; Malinowska, K.; Zoller, M. Exosomal tumor microRNA modulates premetastatic organ cells. Neoplasia 2013, 15, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tao, X.; Li, C.B.; Wang, C.M. MicroRNA-494 acts as a tumor suppressor in pancreatic cancer, inhibiting epithelial-mesenchymal transition, migration and invasion by binding to SDC1. Int. J. Oncol. 2018, 53, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Liu, Z.; Wang, Y.; Wang, L.; Yao, B.; Li, Q.; Wang, C.; Tu, K.; Liu, Q. MiR-542-3p inhibits metastasis and epithelial-mesenchymal transition of hepatocellular carcinoma by targeting UBE3C. Biomed. Pharmacother 2017, 93, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, L.; Roato, I. The impact of immune system in regulating bone metastasis formation by osteotropic tumors. J. Immunol. Res. 2015, 2015, 143526. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumar, R.; Lee, H.W. CD137L- and RANKL-mediated reverse signals inhibit osteoclastogenesis and T lymphocyte proliferation. Immunobiology 2009, 214, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Palafox, M.; Ferrer, I.; Pellegrini, P.; Vila, S.; Hernandez-Ortega, S.; Urruticoechea, A.; Climent, F.; Soler, M.T.; Munoz, P.; Vinals, F.; et al. RANK induces epithelial-mesenchymal transition and stemness in human mammary epithelial cells and promotes tumorigenesis and metastasis. Cancer Res. 2012, 72, 2879–2888. [Google Scholar] [CrossRef]
- Capietto, A.H.; Kim, S.; Sanford, D.E.; Linehan, D.C.; Hikida, M.; Kumosaki, T.; Novack, D.V.; Faccio, R. Down-regulation of PLCgamma2-beta-catenin pathway promotes activation and expansion of myeloid-derived suppressor cells in cancer. J. Exp. Med. 2013, 210, 2257–2271. [Google Scholar] [CrossRef]
- Gao, D.; Joshi, N.; Choi, H.; Ryu, S.; Hahn, M.; Catena, R.; Sadik, H.; Argani, P.; Wagner, P.; Vahdat, L.T.; et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res. 2012, 72, 1384–1394. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular vesicles in cancer: Cell-to-cell mediators of metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef]
- Aguado, B.A.; Bushnell, G.G.; Rao, S.S.; Jeruss, J.S.; Shea, L.D. Engineering the pre-metastatic niche. Nat. Biomed. Eng. 2017, 1. [Google Scholar] [CrossRef]
- Priego, N.; Zhu, L.; Monteiro, C.; Mulders, M.; Wasilewski, D.; Bindeman, W.; Doglio, L.; Martinez, L.; Martinez-Saez, E.; Ramon, Y.C.S.; et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat. Med. 2018, 24, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Balanis, N.; Carlin, C.R.; Schiemann, W.P. STAT3 and epithelial-mesenchymal transitions in carcinomas. JAKSTAT 2014, 3, e28975. [Google Scholar] [CrossRef]
- Iser, I.C.; Lenz, G.; Wink, M.R. EMT-like process in glioblastomas and reactive astrocytes. Neurochem. Int. 2019, 122, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Nagaishi, M.; Nakata, S.; Ono, Y.; Hirata, K.; Tanaka, Y.; Suzuki, K.; Yokoo, H.; Hyodo, A. Tumoral and stromal expression of Slug, ZEB1, and ZEB2 in brain metastasis. J. Clin. Neurosci. 2017, 46, 124–128. [Google Scholar] [CrossRef]
- Gonzalez, H.; Robles, I.; Werb, Z. Innate and acquired immune surveillance in the postdissemination phase of metastasis. FEBS J. 2018, 285, 654–664. [Google Scholar] [CrossRef]
- Piao, Y.J.; Kim, H.S.; Hwang, E.H.; Woo, J.; Zhang, M.; Moon, W.K. Breast cancer cell-derived exosomes and macrophage polarization are associated with lymph node metastasis. Oncotarget 2018, 9, 7398–7410. [Google Scholar] [CrossRef]
- Linde, N.; Casanova-Acebes, M.; Sosa, M.S.; Mortha, A.; Rahman, A.; Farias, E.; Harper, K.; Tardio, E.; Reyes Torres, I.; Jones, J.; et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 2018, 9, 21. [Google Scholar] [CrossRef]
- Srivastava, K.; Hu, J.; Korn, C.; Savant, S.; Teichert, M.; Kapel, S.S.; Jugold, M.; Besemfelder, E.; Thomas, M.; Pasparakis, M.; et al. Postsurgical adjuvant tumor therapy by combining anti-angiopoietin-2 and metronomic chemotherapy limits metastatic growth. Cancer Cell 2014, 26, 880–895. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, L.; Coissieux, M.M.; Wyckoff, J.; Mertz, K.D.; Varga, Z.; Junt, T.; Bentires-Alj, M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014, 515, 130–133. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.J.; Izumi, K. Androgen receptor, CCL2, and epithelial-mesenchymal transition: A dangerous affair in the tumor microenvironment. Oncoimmunology 2014, 3, e27871. [Google Scholar] [CrossRef]
- Tuting, T.; de Visser, K.E. How neutrophils promote metastasis. Science 2016, 352, 145–146. [Google Scholar] [CrossRef]
- Liang, W.; Li, Q.; Ferrara, N. Metastatic growth instructed by neutrophil-derived transferrin. Proc. Natl. Acad. Sci. USA 2018, 115, 11060–11065. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Z.; Chen, Y.; Lv, Z.; Ding, X.; Wang, R.; Xiao, H.; Hou, C.; Shen, B.; Feng, J.; et al. An epithelial-to-mesenchymal transition-inducing potential of granulocyte macrophage colony-stimulating factor in colon cancer. Sci. Rep. 2017, 7, 8265. [Google Scholar] [CrossRef] [PubMed]
- Waghray, M.; Yalamanchili, M.; Dziubinski, M.; Zeinali, M.; Erkkinen, M.; Yang, H.; Schradle, K.A.; Urs, S.; Pasca Di Magliano, M.; Welling, T.H.; et al. GM-CSF mediates mesenchymal-epithelial cross-talk in pancreatic cancer. Cancer Discov. 2016, 6, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Wang, P.C.; Weng, C.C.; Hou, Y.S.; Jian, S.F.; Fang, K.T.; Hou, M.F.; Cheng, K.H. Activation of VCAM-1 and its associated molecule CD44 leads to increased malignant potential of breast cancer cells. Int. J. Mol. Sci. 2014, 15, 3560–3579. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of metastasis by NK cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef]
- Chockley, P.J.; Chen, J.; Chen, G.; Beer, D.G.; Standiford, T.J.; Keshamouni, V.G. Epithelial-mesenchymal transition leads to NK cell-mediated metastasis-specific immunosurveillance in lung cancer. J. Clin. Investig. 2018, 128, 1384–1396. [Google Scholar] [CrossRef]
- Speiser, D.E.; Ho, P.C.; Verdeil, G. Regulatory circuits of T cell function in cancer. Nat. Rev. Immunol. 2016, 16, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, A.S.; Ren, H.; Sutor, S.; Sarangi, V.; Nair, A.; Davila, J.; Elsbernd, L.R.; Udell, J.B.; Dronca, R.S.; Park, S.; et al. Contraction of T cell richness in lung cancer brain metastases. Sci. Rep. 2018, 8, 2171. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.B.; Gettinger, S.N.; Mahajan, A.; Chiang, A.C.; Herbst, R.S.; Sznol, M.; Tsiouris, A.J.; Cohen, J.; Vortmeyer, A.; Jilaveanu, L.; et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: Early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 976–983. [Google Scholar] [CrossRef]
- Kamath, S.D.; Kumthekar, P.U. Immune checkpoint inhibitors for the treatment of central nervous system (CNS) metastatic disease. Front. Oncol. 2018, 8, 414. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nan, X.; Wang, J.; Liu, H.N.; Wong, S.T.C.; Zhao, H. Epithelial-Mesenchymal Plasticity in Organotropism Metastasis and Tumor Immune Escape. J. Clin. Med. 2019, 8, 747. https://doi.org/10.3390/jcm8050747
Nan X, Wang J, Liu HN, Wong STC, Zhao H. Epithelial-Mesenchymal Plasticity in Organotropism Metastasis and Tumor Immune Escape. Journal of Clinical Medicine. 2019; 8(5):747. https://doi.org/10.3390/jcm8050747
Chicago/Turabian StyleNan, Xiang, Jiang Wang, Haowen Nikola Liu, Stephen T.C. Wong, and Hong Zhao. 2019. "Epithelial-Mesenchymal Plasticity in Organotropism Metastasis and Tumor Immune Escape" Journal of Clinical Medicine 8, no. 5: 747. https://doi.org/10.3390/jcm8050747
APA StyleNan, X., Wang, J., Liu, H. N., Wong, S. T. C., & Zhao, H. (2019). Epithelial-Mesenchymal Plasticity in Organotropism Metastasis and Tumor Immune Escape. Journal of Clinical Medicine, 8(5), 747. https://doi.org/10.3390/jcm8050747