Quantifying Cancer Epithelial-Mesenchymal Plasticity and its Association with Stemness and Immune Response

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
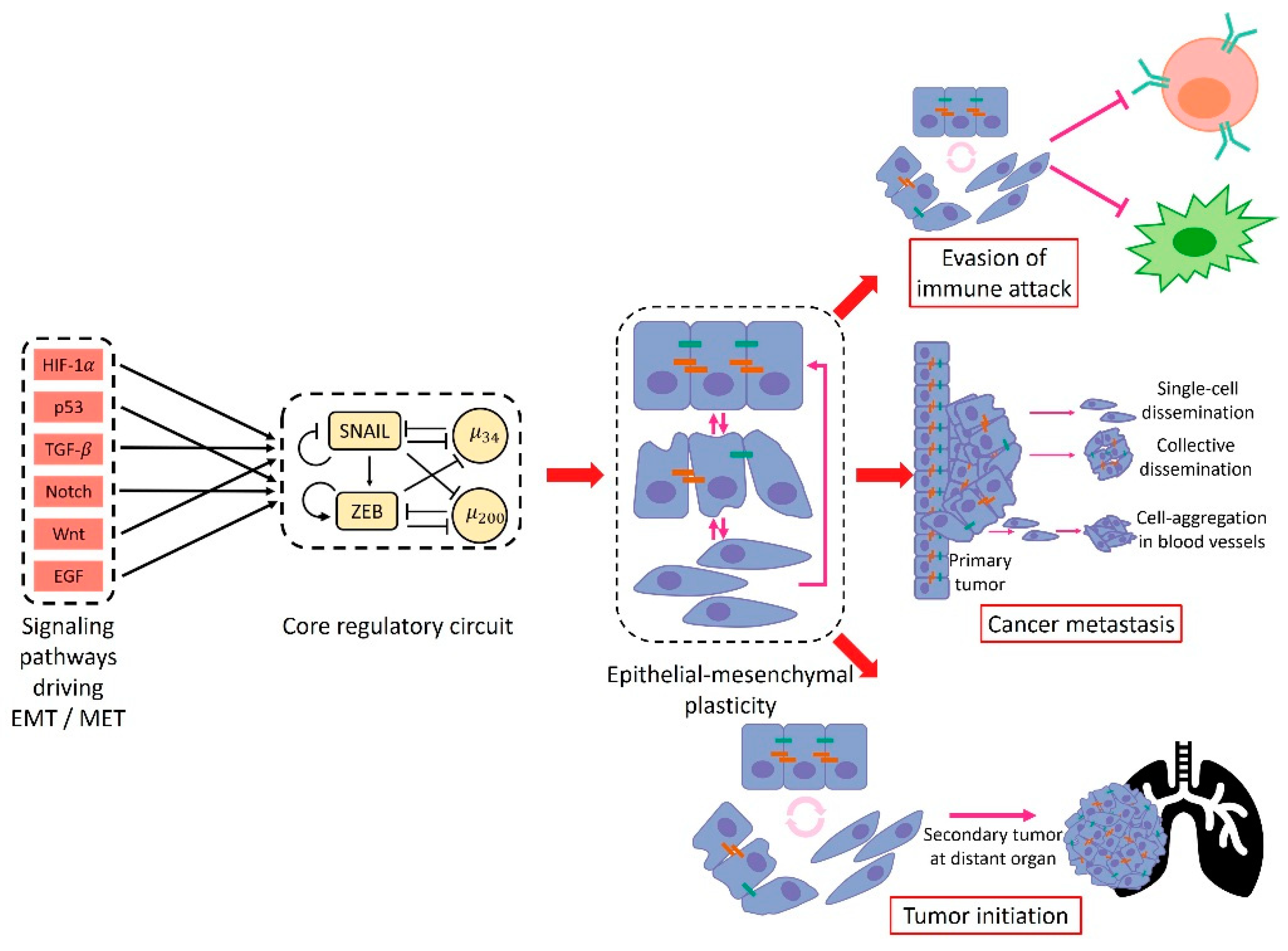
1. Introduction
2. Emergence of Hybrid Epithelial/Mesenchymal Phenotypes
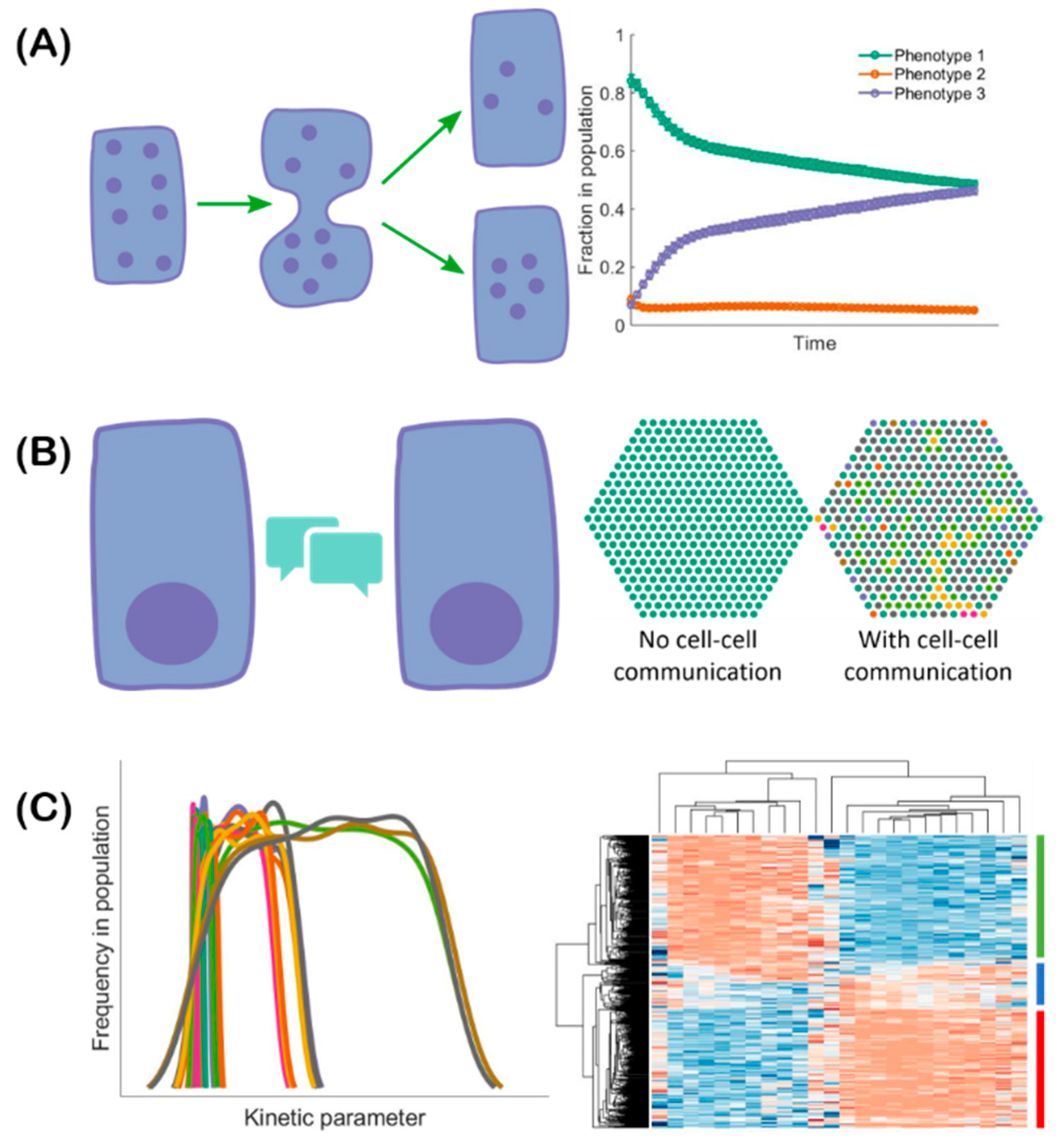
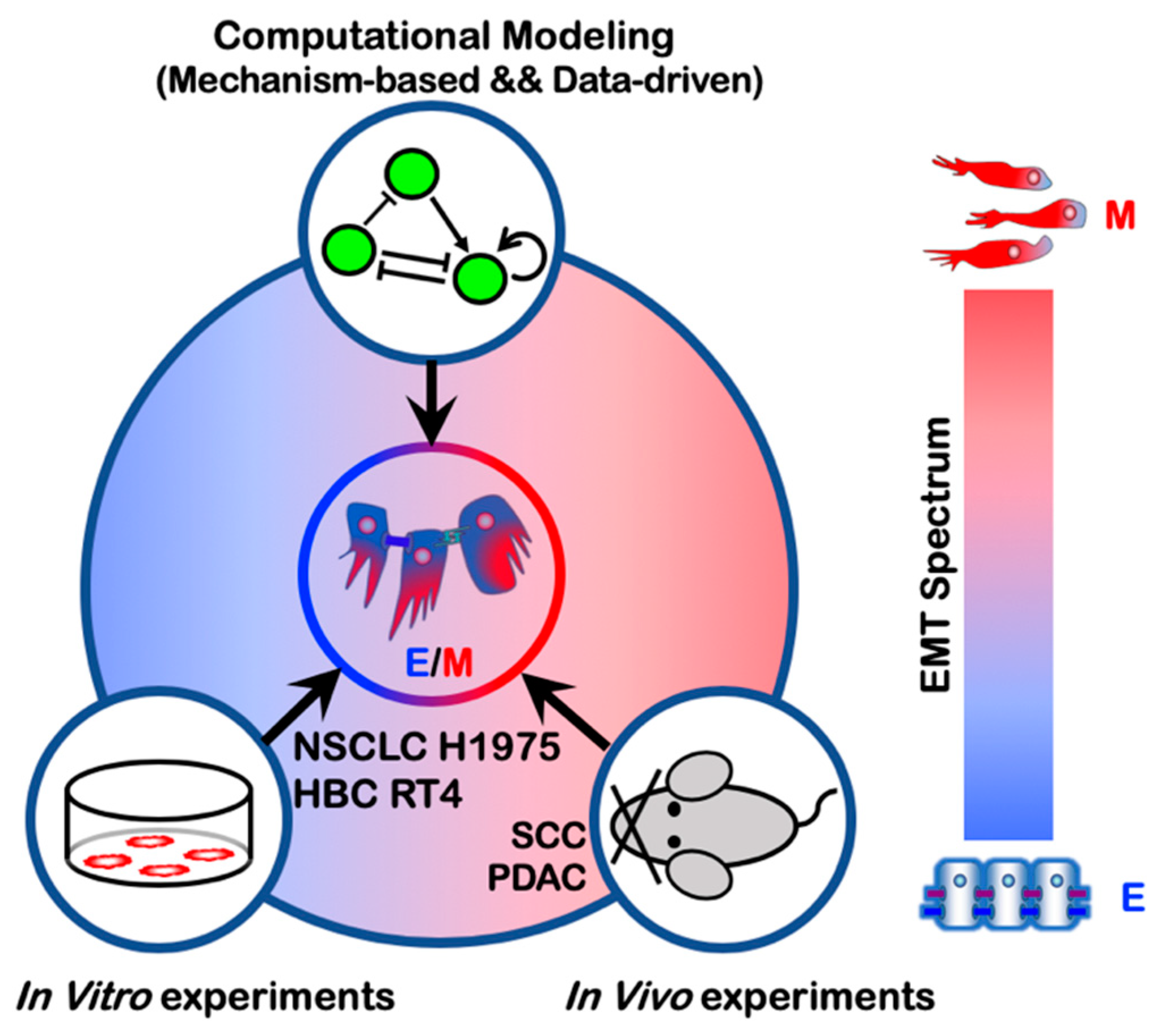
2.1. Hybrid E/M Phenotypes Are Predicted by Mathematical Modeling of EMT Regulation
2.2. In vitro characterization of hybrid E/M phenotypes
2.3. In Vivo Characterization of Hybrid E/M Phenotypes
3. EMT Regulation by Cell Cooperation
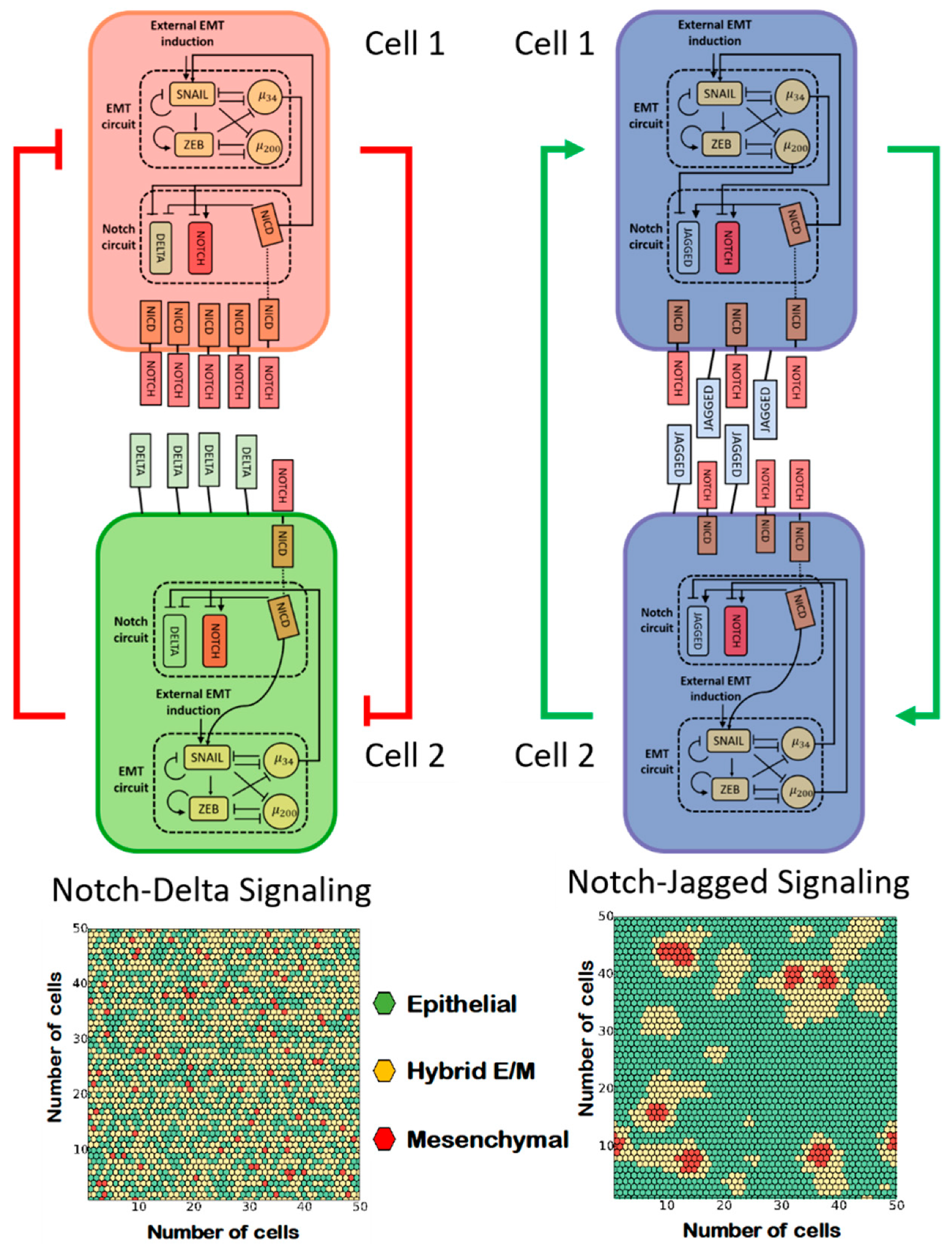
3.1. Notch Signaling
3.2. Interaction among Epithelial, Hybrid E/M, and Mesenchymal Cells
4. Mechanical Control of EMT by ECM
5. Quantification of the EMT Spectrum
6. EMT and CTC Clusters
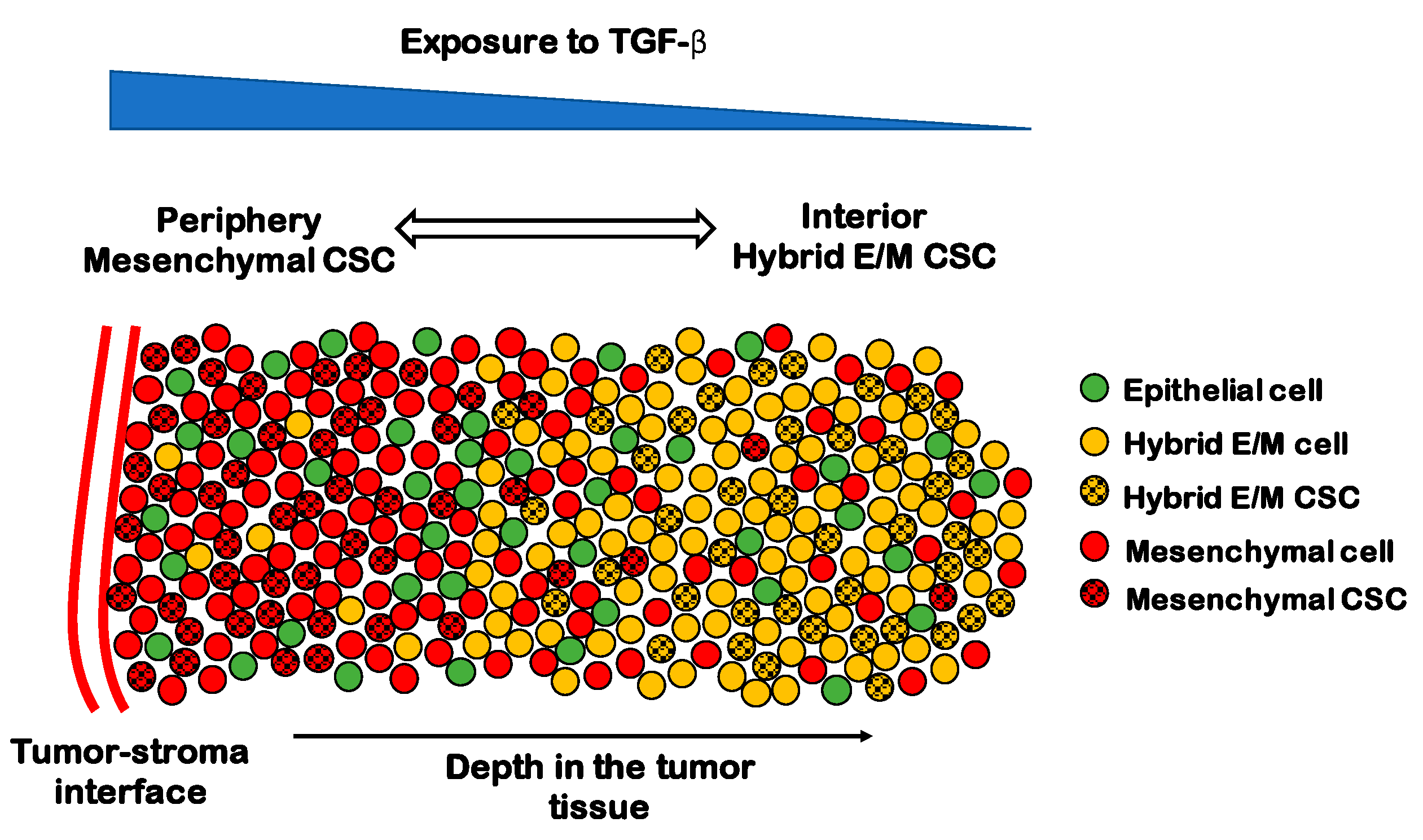
7. EMT and Stemness in Tumor Microenvironment
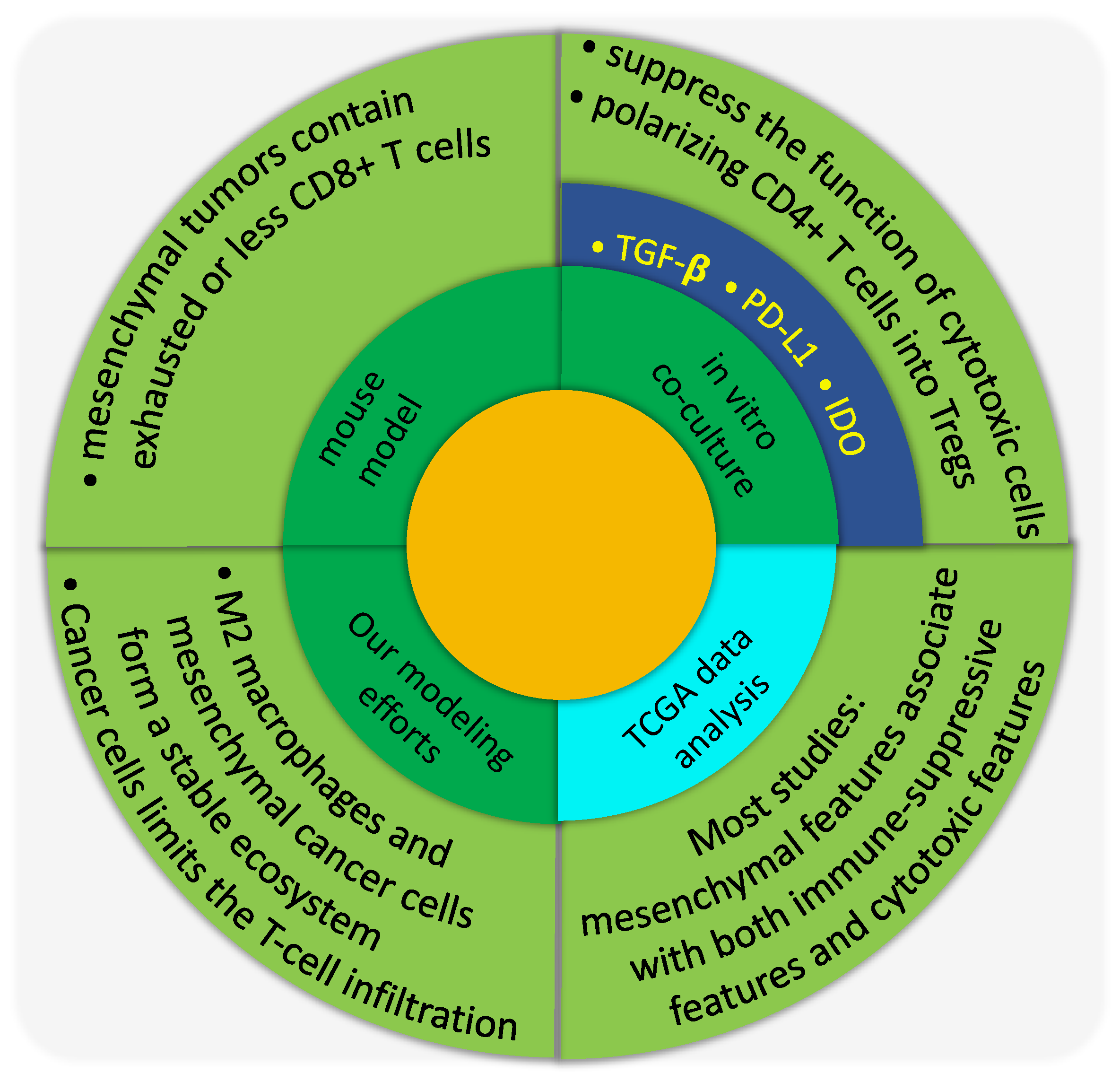
8. EMT and Immune Suppression
8.1. In Vitro Characterization of the Immune-Suppressing Role of Mesenchymal-Like Cancer Cells
8.2. In Vivo Characterization of the Immune-Suppressing Role of Mesenchymal-Like Cancer Cells
8.3. Gene Expression Data Analysis
8.4. Causal Relationship between EMT and Infiltration of Cancer-Killing Immune Cells
9. Hybrid E/M Phenotypes and Beyond
10. Conclusions and Future Vision
Acknowledgments
Conflicts of Interest
References
- Chaffer, C.L.; Weinberg, R.A. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef]
- Greenburg, G.; Hay, E.D. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Sargent, M.G.; Wilkinson, D.G.; Cooke, J. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science 1994, 264, 835–839. [Google Scholar] [CrossRef]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Liu Donaher, J.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2337–E2346. [Google Scholar] [CrossRef]
- Jolly, M.K.; Ward, C.; Eapen, M.S.; Myers, S.; Hallgren, O.; Levine, H.; Sohal, S.S. Epithelial-mesenchymal transition, a spectrum of states: Role in lung development, homeostasis, and disease. Dev. Dyn. 2018, 247, 346–358. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Keymeulen, A.V.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [PubMed]
- Fabisiewicz, A.; Grzybowska, E. CTC clusters in cancer progression and metastasis. Med. Oncol. 2017, 34, 12. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef]
- Cursons, J.; Pillman, K.A.; Scheer, K.G.; Gregory, P.A.; Foroutan, M.; Hediyeh-Zadeh, S.; Toubia, J.; Crampin, E.J.; Goodall, G.J.; Bracken, C.P.; et al. Combinatorial targeting by microRNAs co-ordinates post-transcriptional control of EMT. Cell Syst. 2018, 7, 77.e7–91.e7. [Google Scholar] [CrossRef]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion: Migrating cancer stem cells—An integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.-P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [PubMed]
- Datar, I.; Schalper, K.A. Epithelial–Mesenchymal Transition and Immune Evasion during Lung Cancer Progression: The Chicken or the Egg? Clin. Cancer Res. 2016, 22, 3422–3424. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.M.A.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Grainge, M.J.; Lee, A.H.S.; Ellis, I.O.; Green, A.R. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J. Clin. Oncol. 2011, 29, 1949–1955. [Google Scholar] [CrossRef]
- Carstens, J.L.; Correa de Sampaio, P.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Edin, S.; Wikberg, M.L.; Dahlin, A.M.; Rutegård, J.; Öberg, Å.; Oldenborg, P.-A.; Palmqvist, R. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PLoS ONE 2012, 7, e47045. [Google Scholar] [CrossRef]
- Ma, J.; Liu, L.; Che, G.; Yu, N.; Dai, F.; You, Z. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC Cancer 2010, 10, 112. [Google Scholar] [CrossRef]
- Ohri, C.M.; Shikotra, A.; Green, R.H.; Waller, D.A.; Bradding, P. Macrophages within NSCLC tumour islets are predominantly of a cytotoxic M1 phenotype associated with extended survival. Eur. Respir. J. 2009, 33, 118–126. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Grelet, S.; McShane, A.; Geslain, R.; Howe, P.H. Pleiotropic Roles of Non-Coding RNAs in TGF-β-Mediated Epithelial-Mesenchymal Transition and Their Functions in Tumor Progression. Cancers 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. MicroRNA-based regulation of epithelial–hybrid–mesenchymal fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18144–18149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.-J.; Zhang, H.; Teng, Y.; Li, R.; Bai, F.; Elankumaran, S.; Xing, J. TGF-β–induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci. Signal. 2014, 7, ra91. [Google Scholar] [CrossRef]
- Jia, D.; Jolly, M.K.; Tripathi, S.C.; Den Hollander, P.; Huang, B.; Lu, M.; Celiktas, M.; Ramirez-Peña, E.; Ben-Jacob, E.; Onuchic, J.N.; et al. Distinguishing mechanisms underlying EMT tristability. Cancer Converg. 2017, 1, 2. [Google Scholar] [CrossRef]
- Steinway, S.N.; Zañudo, J.G.T.; Michel, P.J.; Feith, D.J.; Loughran, T.P.; Albert, R. Combinatorial interventions inhibit TGFβ-driven epithelial-to-mesenchymal transition and support hybrid cellular phenotypes. NPJ Syst. Biol. Appl. 2015, 1, 15014. [Google Scholar] [CrossRef]
- Huang, B.; Lu, M.; Jia, D.; Ben-Jacob, E.; Levine, H.; Onuchic, J.N. Interrogating the topological robustness of gene regulatory circuits by randomization. PLoS Comput. Biol. 2017, 13, e1005456. [Google Scholar] [CrossRef]
- Jolly, M.K.; Preca, B.-T.; Tripathi, S.C.; Jia, D.; George, J.T.; Hanash, S.M.; Brabletz, T.; Stemmler, M.P.; Maurer, J.; Levine, H. Interconnected feedback loops among ESRP1, HAS2, and CD44 regulate epithelial-mesenchymal plasticity in cancer. APL Bioeng. 2018, 2, 031908. [Google Scholar] [CrossRef]
- Font-Clos, F.; Zapperi, S.; Porta, C.A.M.L. Topography of epithelial–mesenchymal plasticity. Proc. Natl. Acad. Sci. USA 2018, 115, 5902–5907. [Google Scholar] [CrossRef]
- Li, C.; Hong, T.; Nie, Q. Quantifying the landscape and kinetic paths for epithelial–mesenchymal transition from a core circuit. Phys. Chem. Chem. Phys. 2016, 18, 17949–17956. [Google Scholar] [CrossRef]
- Biswas, K.; Jolly, M.K.; Ghosh, A. Stability and mean residence times for hybrid epithelial/mesenchymal phenotype. Phys. Biol. 2019, 16, 025003. [Google Scholar] [CrossRef]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef]
- Jia, D.; Jolly, M.K.; Boareto, M.; Parsana, P.; Mooney, S.M.; Pienta, K.J.; Levine, H.; Ben-Jacob, E. OVOL guides the epithelial-hybrid-mesenchymal transition. Oncotarget 2015, 6, 15436–15448. [Google Scholar] [CrossRef]
- Bocci, F.; Jolly, M.K.; Tripathi, S.C.; Aguilar, M.; Hanash, S.M.; Levine, H.; Onuchic, J.N. Numb prevents a complete epithelial–mesenchymal transition by modulating Notch signalling. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef]
- Gould, R.; Bassen, D.M.; Chakrabarti, A.; Varner, J.D.; Butcher, J. Population Heterogeneity in the Epithelial to Mesenchymal Transition Is Controlled by NFAT and Phosphorylated Sp1. PLoS Comput. Biol. 2016, 12, e1005251. [Google Scholar] [CrossRef]
- Biddle, A.; Gammon, L.; Liang, X.; Costea, D.E.; Mackenzie, I.C. Phenotypic Plasticity Determines Cancer Stem Cell Therapeutic Resistance in Oral Squamous Cell Carcinoma. EBioMedicine 2016, 4, 138–145. [Google Scholar] [CrossRef]
- Kohar, V.; Lu, M. Role of noise and parametric variation in the dynamics of gene regulatory circuits. NPJ Syst. Biol. Appl. 2018, 4, 40. [Google Scholar] [CrossRef]
- Huang, R.Y.-J.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.C.; Chung, V.Y.; Chu, Y.-S.; Matsumura, N.; Lai, H.-C.; Lee, Y.F.; et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef]
- Varankar, S.S.; Kamble, S.C.; Mali, A.M.; More, M.M.; Abraham, A.; Kumar, B.; Pansare, K.J.; Narayanan, N.J.; Sen, A.; Dhake, R.D.; et al. Functional Balance between TCF21-Slug defines phenotypic plasticity and sub-classes in high-grade serous ovarian cancer. bioRxiv 2018, 307934. [Google Scholar]
- Schliekelman, M.J.; Taguchi, A.; Zhu, J.; Dai, X.; Rodriguez, J.; Celiktas, M.; Zhang, Q.; Chin, A.; Wong, C.H.; Wang, H.; et al. Molecular portraits of epithelial, mesenchymal, and hybrid States in lung adenocarcinoma and their relevance to survival. Cancer Res. 2015, 75, 1789–1800. [Google Scholar] [CrossRef]
- Jia, D.; George, J.T.; Tripathi, S.C.; Kundnani, D.L.; Lu, M.; Hanash, S.M.; Onuchic, J.N.; Jolly, M.K.; Levine, H. Testing the gene expression classification of the EMT spectrum. Phys. Biol. 2019, 16, 025002. [Google Scholar] [CrossRef]
- Jolly, M.K.; Tripathi, S.C.; Somarelli, J.A.; Hanash, S.M.; Levine, H. Epithelial/mesenchymal plasticity: How have quantitative mathematical models helped improve our understanding? Mol. Oncol. 2017, 11, 739–754. [Google Scholar] [CrossRef]
- Bocci, F.; Tripathi, S.C.; Mercedes, S.V.; George, J.T.; Casabar, J.; Wong, P.K.; Hanash, S.; Levine, H.; Onuchic, J.N.; Jolly, M.K. NRF2 activates a partial Epithelial-Mesenchymal Transition and is maximally present in a hybrid Epithelial/Mesenchymal phenotype. bioRxiv 2018, 390237. [Google Scholar]
- Fustaino, V.; Presutti, D.; Colombo, T.; Cardinali, B.; Papoff, G.; Brandi, R.; Bertolazzi, P.; Felici, G.; Ruberti, G. Characterization of epithelial-mesenchymal transition intermediate/hybrid phenotypes associated to resistance to EGFR inhibitors in non-small cell lung cancer cell lines. Oncotarget 2017, 8, 103340–103363. [Google Scholar] [CrossRef]
- Grosse-Wilde, A.; d’Hérouël, A.F.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; del Sol, A.; Walters, K.-A.; Huang, S. Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef]
- Grigore, A.D.; Jolly, M.K.; Jia, D.; Farach-Carson, M.C.; Levine, H. Tumor Budding: The Name is EMT. Partial EMT. J. Clin. Med. 2016, 5, 51. [Google Scholar] [CrossRef]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2019, 194, 161–184. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]
- Losick, R.; Desplan, C. Stochasticity and Cell Fate. Science 2008, 320, 65–68. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial Persistence as a Phenotypic Switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef]
- Huh, D.; Paulsson, J. Non-genetic heterogeneity from stochastic partitioning at cell division. Nat. Genet. 2011, 43, 95–100. [Google Scholar] [CrossRef]
- Huh, D.; Paulsson, J. Random partitioning of molecules at cell division. Proc. Natl. Acad. Sci. USA 2011, 108, 15004–15009. [Google Scholar] [CrossRef]
- Tripathi, S.; Levine, H.; Jolly, M.K. A Mechanism for Epithelial-Mesenchymal Heterogeneity in a Population of Cancer Cells. bioRxiv 2019, 592691. [Google Scholar]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells During Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef]
- Boareto, M.; Jolly, M.K.; Goldman, A.; Pietilä, M.; Mani, S.A.; Sengupta, S.; Ben-Jacob, E.; Levine, H.; Onuchic, J.N. Notch-Jagged signalling can give rise to clusters of cells exhibiting a hybrid epithelial/mesenchymal phenotype. J. R. Soc. Interface 2016, 13. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681.e4–695.e4. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611.e24–1624.e24. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Bastian, C.; Liu, D.; Ell, B.; Aiello, N.M.; Wei, Y.; Zamalloa, J.; Blanco, A.M.; Hang, X.; Kunisky, D.; et al. Hysteresis control of epithelial-mesenchymal transition dynamics conveys a distinct program with enhanced metastatic ability. Nat. Commun. 2018, 9, 5005. [Google Scholar] [CrossRef]
- Siebel, C.; Lendahl, U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef]
- Li, D.; Masiero, M.; Banham, A.H.; Harris, A.L. The notch ligand JAGGED1 as a target for anti-tumor therapy. Front. Oncol. 2014, 4, 254. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, S.; Bajdak, K.; Meidhof, S.; Burk, U.; Niedermann, G.; Firat, E.; Wellner, U.; Dimmler, A.; Faller, G.; Schubert, J.; et al. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011, 30, 770–782. [Google Scholar] [CrossRef] [PubMed]
- de Antonellis, P.; Medaglia, C.; Cusanelli, E.; Andolfo, I.; Liguori, L.; Vita, G.D.; Carotenuto, M.; Bello, A.; Formiggini, F.; Galeone, A.; et al. MiR-34a Targeting of Notch Ligand Delta-Like 1 Impairs CD15+/CD133+ Tumor-Propagating Cells and Supports Neural Differentiation in Medulloblastoma. PLoS ONE 2011, 6, e24584. [Google Scholar] [CrossRef]
- Bu, P.; Chen, K.-Y.; Chen, J.H.; Wang, L.; Walters, J.; Shin, Y.J.; Goerger, J.P.; Sun, J.; Witherspoon, M.; Rakhilin, N.; et al. A microRNA miR-34a-regulated bimodal switch targets Notch in colon cancer stem cells. Cell Stem Cell 2013, 12, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Bocci, F.; Jolly, M.K.; Levine, H.; Onuchic, J.N. Quantitative Characteristic of ncRNA Regulation in Gene Regulatory Networks. Methods Mol. Biol. 2019, 1912, 341–366. [Google Scholar]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef]
- Niessen, K.; Fu, Y.; Chang, L.; Hoodless, P.A.; McFadden, D.; Karsan, A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 2008, 182, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.R.; Monk, N.A.; Maini, P.K.; Lewis, J.H. Pattern formation by lateral inhibition with feedback: A mathematical model of delta-notch intercellular signalling. J. Theor. Biol. 1996, 183, 429–446. [Google Scholar] [CrossRef]
- Shaya, O.; Sprinzak, D. From Notch signaling to fine-grained patterning: Modeling meets experiments. Curr. Opin. Genet. Dev. 2011, 21, 732–739. [Google Scholar] [CrossRef]
- Boareto, M.; Jolly, M.K.; Lu, M.; Onuchic, J.N.; Clementi, C.; Ben-Jacob, E. Jagged–Delta asymmetry in Notch signaling can give rise to a Sender/Receiver hybrid phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E402–E409. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Debeb, B.G.; Aceto, N.; Farach-Carson, M.C.; Woodward, W.A.; Levine, H. Inflammatory breast cancer: A model for investigating cluster-based dissemination. NPJ Breast Cancer 2017, 3, 21. [Google Scholar] [CrossRef]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Song, Y.; Sheng, X.; Ren, F.; Xiong, K.; Chen, L.; Zhang, H.; Liu, D.; Lengner, C.J.; et al. Numb and Numbl act to determine mammary myoepithelial cell fate, maintain epithelial identity, and support lactogenesis. FASEB J. 2016, 30, 3474–3488. [Google Scholar] [CrossRef]
- Flores, A.N.; McDermott, N.; Meunier, A.; Marignol, L. NUMB inhibition of NOTCH signalling as a therapeutic target in prostate cancer. Nat. Rev. Urol. 2014, 11, 499–507. [Google Scholar] [CrossRef]
- Santosh, A.B.R.; Jones, T.J. The Epithelial-Mesenchymal Interactions: Insights into Physiological and Pathological Aspects of Oral Tissues. Oncol. Rev. 2014, 8, 239. [Google Scholar]
- Tsuji, T.; Ibaragi, S.; Shima, K.; Hu, M.G.; Katsurano, M.; Sasaki, A.; Hu, G. Epithelial-mesenchymal transition induced by growth suppressor p12CDK2-AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res. 2008, 68, 10377–10386. [Google Scholar] [CrossRef]
- Neelakantan, D.; Zhou, H.; Oliphant, M.U.J.; Zhang, X.; Simon, L.M.; Henke, D.M.; Shaw, C.A.; Wu, M.-F.; Hilsenbeck, S.G.; White, L.D.; et al. EMT cells increase breast cancer metastasis via paracrine GLI activation in neighbouring tumour cells. Nat. Commun. 2017, 8, 15773. [Google Scholar] [CrossRef]
- Grosse-Wilde, A.; Kuestner, R.E.; Skelton, S.M.; MacIntosh, E.; d’Hérouël, A.F.; Ertaylan, G.; Del Sol, A.; Skupin, A.; Huang, S. Loss of inter-cellular cooperation by complete epithelial-mesenchymal transition supports favorable outcomes in basal breast cancer patients. Oncotarget 2018, 9, 20018–20033. [Google Scholar] [CrossRef]
- Campbell, K.; Casanova, J. A role for E-cadherin in ensuring cohesive migration of a heterogeneous population of non-epithelial cells. Nat. Commun. 2015, 6, 7998. [Google Scholar] [CrossRef]
- Kröger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Meca-Cortés, O.; Mateo, F.; Martínez de Paz, A.; Rubio, N.; Arnal-Estapé, A.; Ell, B.J.; Bermudo, R.; Díaz, A.; Guerra-Rebollo, M.; et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J. Clin. Investig. 2012, 122, 1849–1868. [Google Scholar] [CrossRef]
- Ocaña, O.H.; Córcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef]
- Tarin, D.; Thompson, E.W.; Newgreen, D.F. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005, 65, 5996–6000; discussion 6000–6001. [Google Scholar] [CrossRef]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial–mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Ewald, A.J.; Brenot, A.; Duong, M.; Chan, B.S.; Werb, Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev. Cell 2008, 14, 570–581. [Google Scholar] [CrossRef]
- Theveneau, E.; Mayor, R. Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol. 2012, 366, 34–54. [Google Scholar] [CrossRef]
- Kai, K.; Iwamoto, T.; Zhang, D.; Shen, L.; Takahashi, Y.; Rao, A.; Thompson, A.; Sen, S.; Ueno, N.T. CSF-1/CSF-1R axis is associated with epithelial/mesenchymal hybrid phenotype in epithelial-like inflammatory breast cancer. Sci. Rep. 2018, 8, 9427. [Google Scholar] [CrossRef]
- Tiwari, P.; Mrigwani, A.; Kaur, H.; Kaila, P.; Kumar, R.; Guptasarma, P. Structural-Mechanical and Biochemical Functions of Classical Cadherins at Cellular Junctions: A Review and Some Hypotheses. Adv. Exp. Med. Biol. 2018, 1112, 107–138. [Google Scholar]
- Knútsdóttir, H.; Pálsson, E.; Edelstein-Keshet, L. Mathematical model of macrophage-facilitated breast cancer cells invasion. J. Theor. Biol. 2014, 357, 184–199. [Google Scholar] [CrossRef]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef]
- Matte, B.F.; Kumar, A.; Placone, J.K.; Zanella, V.G.; Martins, M.D.; Engler, A.J.; Lamers, M.L. Matrix stiffness mechanically conditions EMT and migratory behavior of oral squamous cell carcinoma. J. Cell Sci. 2019, 132, jcs224360. [Google Scholar] [CrossRef]
- Razinia, Z.; Castagnino, P.; Xu, T.; Vázquez-Salgado, A.; Puré, E.; Assoian, R.K. Stiffness-dependent motility and proliferation uncoupled by deletion of CD44. Sci. Rep. 2017, 7, 16499. [Google Scholar] [CrossRef]
- Gjorevski, N.; Boghaert, E.; Nelson, C.M. Regulation of Epithelial-Mesenchymal Transition by Transmission of Mechanical Stress through Epithelial Tissues. Cancer Microenviron. 2011, 5, 29–38. [Google Scholar] [CrossRef]
- Leight, J.L.; Wozniak, M.A.; Chen, S.; Lynch, M.L.; Chen, C.S. Matrix rigidity regulates a switch between TGF-β1-induced apoptosis and epithelial-mesenchymal transition. Mol. Biol. Cell 2012, 23, 781–791. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Ondeck, M.G.; Kumar, A.; Placone, J.K.; Plunkett, C.M.; Matte, B.F.; Wong, K.C.; Fattet, L.; Yang, J.; Engler, A.J. Dynamically stiffened matrix promotes malignant transformation of mammary epithelial cells via collective mechanical signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 3502–3507. [Google Scholar] [CrossRef]
- Tang, X.; Kuhlenschmidt, T.B.; Zhou, J.; Bell, P.; Wang, F.; Kuhlenschmidt, M.S.; Saif, T.A. Mechanical Force Affects Expression of an In Vitro Metastasis-Like Phenotype in HCT-8 Cells. Biophys. J. 2010, 99, 2460–2469. [Google Scholar] [CrossRef]
- Cox, T.R.; Bird, D.; Baker, A.-M.; Barker, H.E.; Ho, M.W.-Y.; Lang, G.; Erler, J.T. LOX-Mediated Collagen Crosslinking Is Responsible for Fibrosis-Enhanced Metastasis. Cancer Res. 2013, 73, 1721–1732. [Google Scholar] [CrossRef]
- Peng, D.H.; Ungewiss, C.; Tong, P.; Byers, L.A.; Wang, J.; Canales, J.R.; Villalobos, P.A.; Uraoka, N.; Mino, B.; Behrens, C.; et al. ZEB1 induces LOXL2-mediated collagen stabilization and deposition in the extracellular matrix to drive lung cancer invasion and metastasis. Oncogene 2017, 36, 1925–1938. [Google Scholar] [CrossRef]
- Sun, M.; Spill, F.; Zaman, M.H. A Computational Model of YAP/TAZ Mechanosensing. Biophys. J. 2016, 110, 2540–2550. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Somarelli, J.A.; Schaeffer, D.; Marengo, M.S.; Bepler, T.; Rouse, D.; Ware, K.E.; Hish, A.J.; Zhao, Y.; Buckley, A.F.; Epstein, J.I.; et al. Distinct routes to metastasis: Plasticity-dependent and plasticity-independent pathways. Oncogene 2016, 35, 4302–4311. [Google Scholar] [CrossRef]
- Leggett, S.E.; Sim, J.Y.; Rubins, J.E.; Neronha, Z.J.; Williams, E.K.; Wong, I.Y. Morphological single cell profiling of the epithelial-mesenchymal transition. Integr. Biol. (Camb.) 2016, 8, 1133–1144. [Google Scholar] [CrossRef]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival Outcomes in Cancer Patients Predicted by a Partial EMT Gene Expression Scoring Metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.-J.; Thiery, J.P. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef]
- Liu, X.; Taftaf, R.; Kawaguchi, M.; Chang, Y.-F.; Chen, W.; Entenberg, D.; Zhang, Y.; Gerratana, L.; Huang, S.; Patel, D.B.; et al. Homophilic CD44 interactions mediate tumor cell aggregation and polyclonal metastasis in patient-derived breast cancer models. Cancer Discov. 2018, 9, 96–113. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Khalil, A.A.; Ilina, O.; Gritsenko, P.G.; Bult, P.; Span, P.N.; Friedl, P. Collective invasion in ductal and lobular breast cancer associates with distant metastasis. Clin. Exp. Metastasis 2017, 34, 421–429. [Google Scholar] [CrossRef]
- Li, C.-F.; Chen, J.-Y.; Ho, Y.-H.; Hsu, W.-H.; Wu, L.-C.; Lan, H.-Y.; Hsu, D.S.-S.; Tai, S.-K.; Chang, Y.-C.; Yang, M.-H. Snail-induced claudin-11 prompts collective migration for tumour progression. Nat. Cell Biol. 2019, 21, 251–262. [Google Scholar] [CrossRef]
- Bocci, F.; Jolly, M.K.; Onuchic, J.N. A biophysical model uncovers the size distribution of migrating cell clusters across cancer types. bioRxiv 2019, 563049. [Google Scholar]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Sarioglu, A.F.; Aceto, N.; Kojic, N.; Donaldson, M.C.; Zeinali, M.; Hamza, B.; Engstrom, A.; Zhu, H.; Sundaresan, T.K.; Miyamoto, D.T.; et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat. Methods 2015, 12, 685–691. [Google Scholar] [CrossRef]
- Leone, K.; Poggiana, C.; Zamarchi, R. The Interplay between Circulating Tumor Cells and the Immune System: From Immune Escape to Cancer Immunotherapy. Diagnostics (Basel) 2018, 8, 59. [Google Scholar] [CrossRef]
- Mierke, C.T. Physics of Cancer; IOP Publishing: Bristol, UK, 2015; The Role of Macrophages during Cancer Cell Transendothelial Migration. [Google Scholar]
- Nielsen, S.R.; Schmid, M.C. Macrophages as Key Drivers of Cancer Progression and Metastasis. Mediators Inflamm. 2017, 2017, 9624760. [Google Scholar] [CrossRef]
- Hojo, N.; Huisken, A.L.; Wang, H.; Chirshev, E.; Kim, N.S.; Nguyen, S.M.; Campos, H.; Glackin, C.A.; Ioffe, Y.J.; Unternaehrer, J.J. Snail knockdown reverses stemness and inhibits tumour growth in ovarian cancer. Sci. Rep. 2018, 8, 8704. [Google Scholar] [CrossRef]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef]
- Patsialou, A.; Wang, Y.; Lin, J.; Whitney, K.; Goswami, S.; Kenny, P.A.; Condeelis, J.S. Selective gene-expression profiling of migratory tumor cells in vivo predicts clinical outcome in breast cancer patients. Breast Cancer Res. 2012, 14, R139. [Google Scholar] [CrossRef]
- Liao, T.-T.; Hsu, W.-H.; Ho, C.-H.; Hwang, W.-L.; Lan, H.-Y.; Lo, T.; Chang, C.-C.; Tai, S.-K.; Yang, M.-H. let-7 Modulates Chromatin Configuration and Target Gene Repression through Regulation of the ARID3B Complex. Cell Rep. 2016, 14, 520–533. [Google Scholar] [CrossRef]
- Jolly, M.K.; Jia, D.; Boareto, M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Coupling the modules of EMT and stemness: A tunable ‘stemness window’ model. Oncotarget 2015, 6, 25161–25174. [Google Scholar] [CrossRef]
- Strauss, R.; Li, Z.-Y.; Liu, Y.; Beyer, I.; Persson, J.; Sova, P.; Möller, T.; Pesonen, S.; Hemminki, A.; Hamerlik, P.; et al. Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLoS ONE 2011, 6, e16186. [Google Scholar] [CrossRef]
- Bocci, F.; Jolly, M.K.; George, J.T.; Levine, H.; Onuchic, J.N. A mechanism-based computational model to capture the interconnections among epithelial-mesenchymal transition, cancer stem cells and Notch-Jagged signaling. Oncotarget 2018, 9, 29906–29920. [Google Scholar] [CrossRef]
- Bocci, F.; Gearhart-Serna, L.; Boareto, M.; Ribeiro, M.; Ben-Jacob, E.; Devi, G.R.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2019, 116, 148–157. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Varga, J.; Greten, F.R. Cell plasticity in epithelial homeostasis and tumorigenesis. Nat. Cell Biol. 2017, 19, 1133–1141. [Google Scholar] [CrossRef]
- Bocci, F.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Deciphering the dynamics of Epithelial-Mesenchymal Transition and Cancer Stem Cells in tumor progression. Curr. Stem Cell Rep. 2019, 5, 11–21. [Google Scholar] [CrossRef]
- Nie, Q. Stem cells: A window of opportunity in low-dimensional EMT space. Oncotarget 2018, 9, 31790–31791. [Google Scholar] [CrossRef]
- Yamamoto, M.; Taguchi, Y.; Ito-Kureha, T.; Semba, K.; Yamaguchi, N.; Inoue, J. NF-κB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat. Commun. 2013, 4, 2299. [Google Scholar] [CrossRef]
- Sikandar, S.S.; Pate, K.T.; Anderson, S.; Dizon, D.; Edwards, R.A.; Waterman, M.L.; Lipkin, S.M. NOTCH Signaling Is Required for Formation and Self-Renewal of Tumor-Initiating Cells and for Repression of Secretory Cell Differentiation in Colon Cancer. Cancer Res. 2010, 70, 1469–1478. [Google Scholar] [CrossRef]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Colacino, J.A.; Azizi, E.; Brooks, M.D.; Harouaka, R.; Fouladdel, S.; McDermott, S.P.; Lee, M.; Hill, D.; Madden, J.; Boerner, J.; et al. Heterogeneity of Human Breast Stem and Progenitor Cells as Revealed by Transcriptional Profiling. Stem Cell Rep. 2018, 10, 1596–1609. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, X.; Peng, Y.; Wu, M.; Zhang, P.; Xie, R.; Wu, Y.; Yan, Q.; Liu, S.; Wang, J. HIF-1α Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS ONE 2015, 10, e0129603. [Google Scholar] [CrossRef]
- Guo, J.; Wang, B.; Fu, Z.; Wei, J.; Lu, W. Hypoxic Microenvironment Induces EMT and Upgrades Stem-Like Properties of Gastric Cancer Cells. Technol. Cancer Res. Treat. 2016, 15, 60–68. [Google Scholar] [CrossRef]
- Salnikov, A.V.; Liu, L.; Platen, M.; Gladkich, J.; Salnikova, O.; Ryschich, E.; Mattern, J.; Moldenhauer, G.; Werner, J.; Schemmer, P.; et al. Hypoxia Induces EMT in Low and Highly Aggressive Pancreatic Tumor Cells but Only Cells with Cancer Stem Cell Characteristics Acquire Pronounced Migratory Potential. PLoS ONE 2012, 7, e46391. [Google Scholar] [CrossRef]
- Jolly, M.K.; Kulkarni, P.; Weninger, K.; Orban, J.; Levine, H. Phenotypic Plasticity, Bet-Hedging, and Androgen Independence in Prostate Cancer: Role of Non-Genetic Heterogeneity. Front. Oncol. 2018, 8, 50. [Google Scholar] [CrossRef]
- Goldman, A.; Majumder, B.; Dhawan, A.; Ravi, S.; Goldman, D.; Kohandel, M.; Majumder, P.K.; Sengupta, S. Temporally sequenced anticancer drugs overcome adaptive resistance by targeting a vulnerable chemotherapy-induced phenotypic transition. Nat. Commun. 2015, 6, 6139. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Bjerkvig, R.; Tysnes, B.B.; Aboody, K.S.; Najbauer, J.; Terzis, A.J.A. Opinion: The origin of the cancer stem cell: Current controversies and new insights. Nat. Rev. Cancer 2005, 5, 899–904. [Google Scholar] [CrossRef]
- Yang, G.; Quan, Y.; Wang, W.; Fu, Q.; Wu, J.; Mei, T.; Li, J.; Tang, Y.; Luo, C.; Ouyang, Q.; et al. Dynamic equilibrium between cancer stem cells and non-stem cancer cells in human SW620 and MCF-7 cancer cell populations. Br. J. Cancer 2012, 106, 1512–1519. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef]
- Avgustinova, A.; Benitah, S.A. Epigenetic control of adult stem cell function. Nat. Rev. Mol. Cell Biol. 2016, 17, 643–658. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Norton, K.-A.; Jin, K.; Popel, A.S. Modeling triple-negative breast cancer heterogeneity: Effects of stromal macrophages, fibroblasts and tumor vasculature. J. Theor. Biol. 2018, 452, 56–68. [Google Scholar] [CrossRef]
- Mahlbacher, G.E.; Reihmer, K.C.; Frieboes, H.B. Mathematical modeling of tumor-immune cell interactions. J. Theor. Biol. 2019, 469, 47–60. [Google Scholar] [CrossRef]
- Mahlbacher, G.; Curtis, L.T.; Lowengrub, J.; Frieboes, H.B. Mathematical modeling of tumor-associated macrophage interactions with the cancer microenvironment. J. ImmunoTher. Cancer 2018, 6, 10. [Google Scholar] [CrossRef]
- Robertson-Tessi, M.; El-Kareh, A.; Goriely, A. A mathematical model of tumor–immune interactions. J. Theor. Biol. 2012, 294, 56–73. [Google Scholar] [CrossRef]
- Louzoun, Y.; Xue, C.; Lesinski, G.B.; Friedman, A. A mathematical model for pancreatic cancer growth and treatments. J. Theor. Biol. 2014, 351, 74–82. [Google Scholar] [CrossRef]
- den Breems, N.Y.; Eftimie, R. The re-polarisation of M2 and M1 macrophages and its role on cancer outcomes. J. Theor. Biol. 2016, 390, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Ma, B.; Shao, H.; Clark, A.M.; Wells, A. Macrophage phenotypic subtypes diametrically regulate epithelial-mesenchymal plasticity in breast cancer cells. BMC Cancer 2016, 16, 419. [Google Scholar] [CrossRef]
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Mönkkönen, J.; Kellokumpu-Lehtinen, P.-L.; Lauttia, S.; Tynninen, O.; Joensuu, H.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015, 17, 101. [Google Scholar] [CrossRef]
- Weigert, A.; Tzieply, N.; von Knethen, A.; Johann, A.M.; Schmidt, H.; Geisslinger, G.; Brüne, B. Tumor Cell Apoptosis Polarizes Macrophages—Role of Sphingosine-1-Phosphate. Mol. Biol. Cell 2007, 18, 3810–3819. [Google Scholar] [CrossRef]
- Yuan, A.; Hsiao, Y.-J.; Chen, H.-Y.; Chen, H.-W.; Ho, C.-C.; Chen, Y.-Y.; Liu, Y.-C.; Hong, T.-H.; Yu, S.-L.; Chen, J.J.W.; et al. Opposite Effects of M1 and M2 Macrophage Subtypes on Lung Cancer Progression. Sci. Rep. 2015, 5, 14273. [Google Scholar] [CrossRef] [PubMed]
- Hollmén, M.; Roudnicky, F.; Karaman, S.; Detmar, M. Characterization of macrophage-cancer cell crosstalk in estrogen receptor positive and triple-negative breast cancer. Sci. Rep. 2015, 5, 9188. [Google Scholar] [CrossRef]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef]
- Li, X.; Jolly, M.K.; George, J.T.; Pienta, K.J.; Levine, H. Computational Modeling of the Crosstalk Between Macrophage Polarization and Tumor Cell Plasticity in the Tumor Microenvironment. Front. Oncol. 2019, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef]
- Ao, M.; Franco, O.E.; Park, D.; Raman, D.; Williams, K.; Hayward, S.W. Cross-talk between paracrine-acting cytokine and chemokine pathways promotes malignancy in benign human prostatic epithelium. Cancer Res. 2007, 67, 4244–4253. [Google Scholar] [CrossRef]
- Yu, Y.; Xiao, C.-H.; Tan, L.-D.; Wang, Q.-S.; Li, X.-Q.; Feng, Y.-M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef]
- Liu, Z.; Kuang, W.; Zhou, Q.; Zhang, Y. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int. J. Mol. Med. 2018, 42, 3395–3403. [Google Scholar] [CrossRef]
- Tu, E.; Chia, P.Z.C.; Chen, W. TGFβ in T cell biology and tumor immunity: Angel or devil? Cytokine Growth Factor Rev. 2014, 25, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Joffroy, C.M.; Buck, M.B.; Stope, M.B.; Popp, S.L.; Pfizenmaier, K.; Knabbe, C. Antiestrogens induce transforming growth factor beta-mediated immunosuppression in breast cancer. Cancer Res. 2010, 70, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massagué, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef]
- Chockley, P.J.; Keshamouni, V.G. Immunological Consequences of Epithelial-Mesenchymal Transition in Tumor Progression. J. Immunol. 2016, 197, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Standiford, T.J.; Kuick, R.; Bhan, U.; Chen, J.; Newstead, M.; Keshamouni, V.G. TGF-β-induced IRAK-M expression in tumor-associated macrophages regulates lung tumor growth. Oncogene 2011, 30, 2475–2484. [Google Scholar] [CrossRef]
- Chen, X.-H.; Liu, Z.-C.; Zhang, G.; Wei, W.; Wang, X.-X.; Wang, H.; Ke, H.-P.; Zhang, F.; Wang, H.-S.; Cai, S.-H.; et al. TGF-β and EGF induced HLA-I downregulation is associated with epithelial-mesenchymal transition (EMT) through upregulation of snail in prostate cancer cells. Mol. Immunol. 2015, 65, 34–42. [Google Scholar] [CrossRef]
- Tripathi, S.C.; Peters, H.L.; Taguchi, A.; Katayama, H.; Wang, H.; Momin, A.; Jolly, M.K.; Celiktas, M.; Rodriguez-Canales, J.; Liu, H.; et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc. Natl. Acad. Sci. USA 2016, 113, E1555–E1564. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Hasmim, M.; Noman, M.Z.; Messai, Y.; Bordereaux, D.; Gros, G.; Baud, V.; Chouaib, S. Cutting Edge: Hypoxia-Induced Nanog Favors the Intratumoral Infiltration of Regulatory T Cells and Macrophages via Direct Regulation of TGF-β1. J. Immunol. 2013, 191, 5802–5806. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.-Y.; Chen, W.; Bai, X.-L.; Xu, X.-Y.; Zhang, Q.; Xia, X.-F.; Sun, X.; Li, G.-G.; Hu, Q.-D.; Fu, Q.-H.; et al. Hypoxia-Induced Epithelial-to-Mesenchymal Transition in Hepatocellular Carcinoma Induces an Immunosuppressive Tumor Microenvironment to Promote Metastasis. Cancer Res. 2016, 76, 818–830. [Google Scholar] [CrossRef]
- Ricciardi, M.; Zanotto, M.; Malpeli, G.; Bassi, G.; Perbellini, O.; Chilosi, M.; Bifari, F.; Krampera, M. Epithelial-to-mesenchymal transition (EMT) induced by inflammatory priming elicits mesenchymal stromal cell-like immune-modulatory properties in cancer cells. Br. J. Cancer 2015, 112, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Kudo-Saito, C.; Shirako, H.; Ohike, M.; Tsukamoto, N.; Kawakami, Y. CCL2 is critical for immunosuppression to promote cancer metastasis. Clin. Exp. Metastasis 2013, 30, 393–405. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Bourcy, M.; Lesage, J.; Leroi, N.; Syne, L.; Blacher, S.; Hubert, P.; Erpicum, C.; Foidart, J.-M.; Delvenne, P.; et al. Soluble factors regulated by epithelial-mesenchymal transition mediate tumour angiogenesis and myeloid cell recruitment. J. Pathol. 2015, 236, 491–504. [Google Scholar] [CrossRef]
- Dominguez, C.; David, J.M.; Palena, C. Epithelial-mesenchymal transition and inflammation at the site of the primary tumor. Semin. Cancer Biol. 2017, 47, 177–184. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Diao, L.; Cuentas, E.R.P.; Denning, W.L.; Chen, L.; Fan, Y.H.; Byers, L.A.; Wang, J.; Papadimitrakopoulou, V.A.; Behrens, C.; et al. Epithelial–Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin. Cancer Res. 2016, 22, 3630–3642. [Google Scholar] [CrossRef]
- Kardos, J.; Chai, S.; Mose, L.E.; Selitsky, S.R.; Krishnan, B.; Saito, R.; Iglesia, M.D.; Milowsky, M.I.; Parker, J.S.; Kim, W.Y.; et al. Claudin-low bladder tumors are immune infiltrated and actively immune suppressed. JCI Insight 2016, 1, e85902. [Google Scholar] [CrossRef]
- Yao, J.; Caballero, O.L.; Huang, Y.; Lin, C.; Rimoldi, D.; Behren, A.; Cebon, J.S.; Hung, M.-C.; Weinstein, J.N.; Strausberg, R.L.; et al. Altered Expression and Splicing of ESRP1 in Malignant Melanoma Correlates with Epithelial-Mesenchymal Status and Tumor-Associated Immune Cytolytic Activity. Cancer Immunol. Res. 2016, 4, 552–561. [Google Scholar] [CrossRef]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef]
- Ueno, T.; Tsuchikawa, T.; Hatanaka, K.C.; Hatanaka, Y.; Mitsuhashi, T.; Nakanishi, Y.; Noji, T.; Nakamura, T.; Okamura, K.; Matsuno, Y.; et al. Prognostic impact of programmed cell death ligand 1 (PD-L1) expression and its association with epithelial-mesenchymal transition in extrahepatic cholangiocarcinoma. Oncotarget 2018, 9, 20034–20047. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Choi, W.; Porten, S.; Kim, S.; Willis, D.; Plimack, E.R.; Hoffman-Censits, J.; Roth, B.; Cheng, T.; Tran, M.; Lee, I.-L.; et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014, 25, 152–165. [Google Scholar] [CrossRef]
- Teng, M.W.L.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef]
- Sweis, R.F.; Spranger, S.; Bao, R.; Paner, G.P.; Stadler, W.M.; Steinberg, G.; Gajewski, T.F. Molecular Drivers of the Non-T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol. Res. 2016, 4, 563–568. [Google Scholar] [CrossRef]
- Seo, A.N.; Lee, H.J.; Kim, E.J.; Kim, H.J.; Jang, M.H.; Lee, H.E.; Kim, Y.J.; Kim, J.H.; Park, S.Y. Tumour-infiltrating CD8+ lymphocytes as an independent predictive factor for pathological complete response to primary systemic therapy in breast cancer. Br. J. Cancer 2013, 109, 2705–2713. [Google Scholar] [CrossRef]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178.e7–193.e7. [Google Scholar] [CrossRef]
- Boareto, M.; Jolly, M.K.; Ben-Jacob, E.; Onuchic, J.N. Jagged mediates differences in normal and tumor angiogenesis by affecting tip-stalk fate decision. Proc. Natl. Acad. Sci. USA 2015, 112, E3836–E3844. [Google Scholar] [CrossRef]
- Yu, L.; Lu, M.; Jia, D.; Ma, J.; Ben-Jacob, E.; Levine, H.; Kaipparettu, B.A.; Onuchic, J.N. Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 2017, 77, 1564–1574. [Google Scholar] [CrossRef]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Park, J.; Jung, K.; Levine, H.; Kaipparettu, B. Elucidating the metabolic plasticity of cancer: Mitochondrial reprogramming and hybrid metabolic states. Cells 2018, 7, 21. [Google Scholar] [CrossRef]
- Sone, M.; Morone, N.; Nakamura, T.; Tanaka, A.; Okita, K.; Woltjen, K.; Nakagawa, M.; Heuser, J.E.; Yamada, Y.; Yamanaka, S.; et al. Hybrid Cellular Metabolism Coordinated by Zic3 and Esrrb Synergistically Enhances Induction of Naive Pluripotency. Cell Metab. 2017, 25, 1103.e6–1117.e6. [Google Scholar] [CrossRef]
- Pacella, I.; Procaccini, C.; Focaccetti, C.; Miacci, S.; Timperi, E.; Faicchia, D.; Severa, M.; Rizzo, F.; Coccia, E.M.; Bonacina, F.; et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc. Natl. Acad. Sci. USA 2018, 115, E6546–E6555. [Google Scholar] [CrossRef]
- Huang, S. Hybrid T-Helper Cells: Stabilizing the Moderate Center in a Polarized System. PLoS Biol. 2013, 11, e1001632. [Google Scholar] [CrossRef]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef]
- Peterson, V.M.; Zhang, K.X.; Kumar, N.; Wong, J.; Li, L.; Wilson, D.C.; Moore, R.; McClanahan, T.K.; Sadekova, S.; Klappenbach, J.A. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 2017, 35, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Karreman, M.A.; Hyenne, V.; Schwab, Y.; Goetz, J.G. Intravital correlative microscopy: Imaging life at the nanoscale. Trends Cell Biol. 2016, 26, 848–863. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, D.; Li, X.; Bocci, F.; Tripathi, S.; Deng, Y.; Jolly, M.K.; Onuchic, J.N.; Levine, H. Quantifying Cancer Epithelial-Mesenchymal Plasticity and its Association with Stemness and Immune Response. J. Clin. Med. 2019, 8, 725. https://doi.org/10.3390/jcm8050725
Jia D, Li X, Bocci F, Tripathi S, Deng Y, Jolly MK, Onuchic JN, Levine H. Quantifying Cancer Epithelial-Mesenchymal Plasticity and its Association with Stemness and Immune Response. Journal of Clinical Medicine. 2019; 8(5):725. https://doi.org/10.3390/jcm8050725
Chicago/Turabian StyleJia, Dongya, Xuefei Li, Federico Bocci, Shubham Tripathi, Youyuan Deng, Mohit Kumar Jolly, José N. Onuchic, and Herbert Levine. 2019. "Quantifying Cancer Epithelial-Mesenchymal Plasticity and its Association with Stemness and Immune Response" Journal of Clinical Medicine 8, no. 5: 725. https://doi.org/10.3390/jcm8050725
APA StyleJia, D., Li, X., Bocci, F., Tripathi, S., Deng, Y., Jolly, M. K., Onuchic, J. N., & Levine, H. (2019). Quantifying Cancer Epithelial-Mesenchymal Plasticity and its Association with Stemness and Immune Response. Journal of Clinical Medicine, 8(5), 725. https://doi.org/10.3390/jcm8050725