The “Usual Suspects”: Genes for Inflammation, Fibrosis, Regeneration, and Muscle Strength Modify Duchenne Muscular Dystrophy


Abstract
1. Introduction
2. Phenotype Variability in DMD
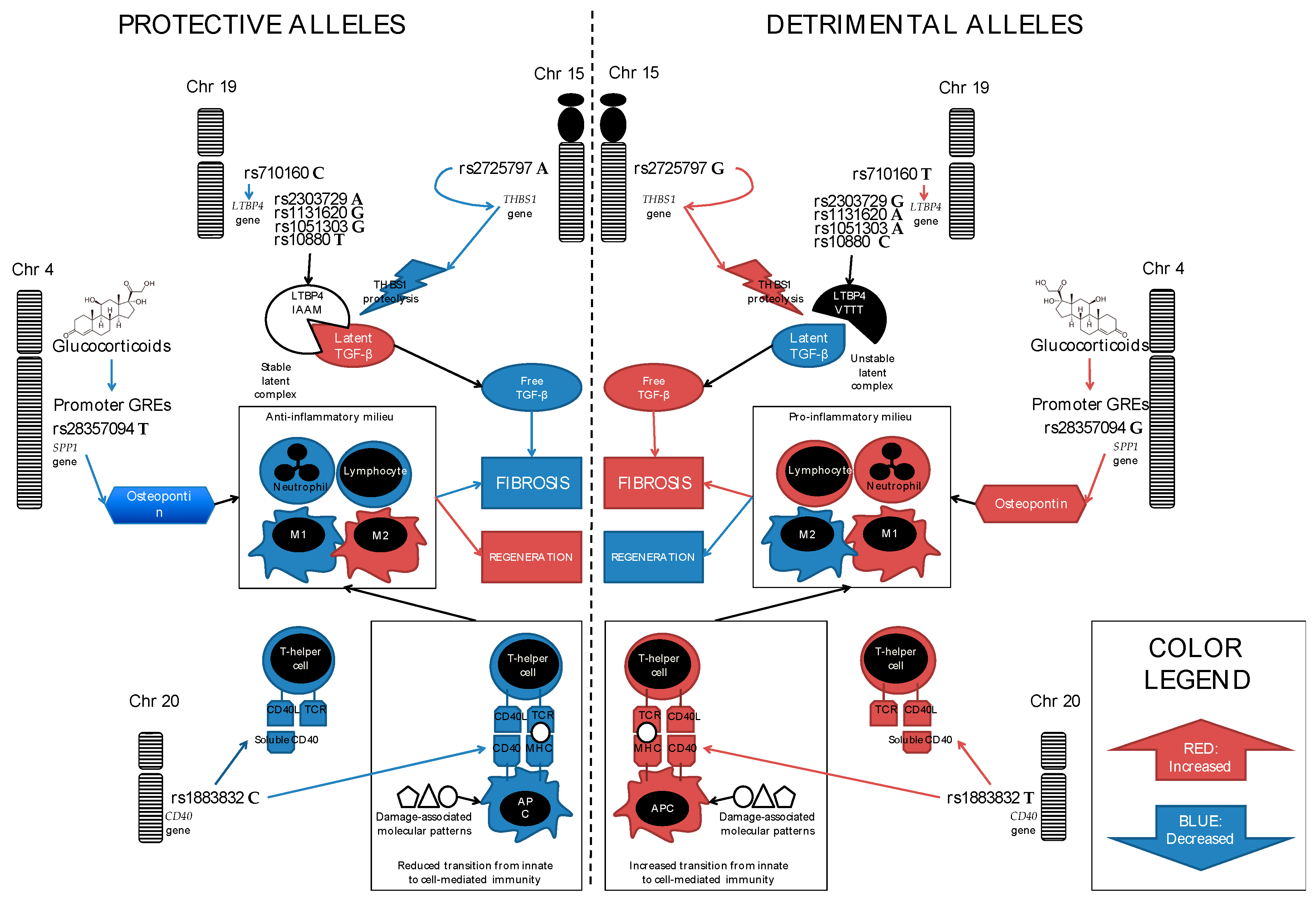
3. SPP1 (Secreted PhosphoProtein 1, Also Known as Osteopontin)
4. LTBP4 (Latent Transforming Growth Factor β Binding Protein 4)
5. CD40, a.k.a. TNFRSF5 (Tumor Necrosis Factor Receptor SuperFamily Member 5)
6. ACTN3 (Actinin-3)
7. THBS1 (Thrombospondin-1)
8. Alternative (Non-Skeletal Muscle) Phenotypes
9. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; de Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet. J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Bello, L.; Hoffman, E.P.; Pegoraro, E. Dystrophinopathies. In Muscular Dystrophy: Causes and Management; Nova Science Publihser, Inc.: Hauppage, NY, USA, 2013; pp. 67–96. [Google Scholar]
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Nigro, G.; Comi, L.I.; Limongelli, F.M.; Giugliano, M.A.; Politano, L.; Petretta, V.; Passamano, L.; Stefanelli, S. Prospective study of X-linked progressive muscular dystrophy in Campania. Muscle Nerve. 1983, 6, 253–262. [Google Scholar] [CrossRef]
- Angelini, C.; Fanin, M.; Pegoraro, E.; Freda, M.P.; Cadaldini, M.; Martinello, F. Clinical-molecular correlation in 104 mild X-linked muscular dystrophy patients: Characterization of sub-clinical phenotypes. Neuromuscul. Disord. 1994, 4, 349–358. [Google Scholar] [CrossRef]
- Comi, G.P.; Prelle, A.; Bresolin, N.; Moggio, M.; Bardoni, A.; Gallanti, A.; Vita, G.; Toscano, A.; Ferro, M.T.; Bordoni, A. Clinical variability in Becker muscular dystrophy. Genetic, biochemical and immunohistochemical correlates. Brain 1994, 117, 1–14. [Google Scholar] [CrossRef]
- Bello, L.; Campadello, P.; Barp, A.; Fanin, M.; Semplicini, C.; Sorarù, G.; Caumo, L.; Calore, C.; Angelini, C.; Pegoraro, E. Functional changes in Becker muscular dystrophy: Implications for clinical trials in dystrophinopathies. Sci. Rep. 2016, 6, 32439. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Henricson, E.K.; Abresch, R.T.; Duong, T.; Joyce, N.C.; Hu, F.; Clemens, P.R.; Hoffman, E.P.; Cnaan, A.; Gordish-Dressman, H.; et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet 2018, 391, 451–461. [Google Scholar] [CrossRef]
- Humbertclaude, V.; Hamroun, D.; Bezzou, K.; Bérard, C.; Boespflug-Tanguy, O.; Bommelaer, C.; Campana-Salort, E.; Cances, C.; Chabrol, B.; Commare, M.-C.; et al. Motor and respiratory heterogeneity in Duchenne patients: Implication for clinical trials. Eur. J. Paediatr. Neurol. 2012, 16, 149–160. [Google Scholar] [CrossRef]
- Jimenez, C.; Moreno; Eagle, M.; Mayhew, A.; James, M.; Straub, V.; Bushby, K. Impact of three decades of improvement in standards of care for Duchenne muscular dystrophy. Neuromus. Dis. 2015, 25, S201–S202. [Google Scholar] [CrossRef]
- Bello, L.; Gordish-Dressman, H.; Morgenroth, L.P.; Henricson, E.K.; Duong, T.; Hoffman, E.P.; Cnaan, A.; McDonald, C.M. CINRG investigators prednisone/prednisolone and deflazacort regimens in the CINRG Duchenne natural history study. Neurology 2015, 85, 1048–1055. [Google Scholar] [CrossRef]
- Barp, A.; Bello, L.; Politano, L.; Melacini, P.; Calore, C.; Polo, A.; Vianello, S.; Sorarù, G.; Semplicini, C.; Pantic, B.; et al. Genetic modifiers of Duchenne muscular dystrophy and dilated cardiomyopathy. PLoS ONE 2015, 10, e0141240. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Van Deutekom, J.C.T.; Fokkema, I.F.; Van Ommen, G.-J.B.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006, 34, 135–144. [Google Scholar] [CrossRef]
- Muntoni, F.; Gobbi, P.; Sewry, C.; Sherratt, T.; Taylor, J.; Sandhu, S.K.; Abbs, S.; Roberts, R.; Hodgson, S.V.; Bobrow, M. Deletions in the 5’ region of dystrophin and resulting phenotypes. J. Med. Genet. 1994, 31, 843–847. [Google Scholar] [CrossRef]
- Winnard, A.V.; Mendell, J.R.; Prior, T.W.; Florence, J.; Burghes, A.H. Frameshift deletions of exons 3-7 and revertant fibers in Duchenne muscular dystrophy: Mechanisms of dystrophin production. Am. J. Hum. Genet. 1995, 56, 158–166. [Google Scholar]
- Gualandi, F.; Rimessi, P.; Trabanelli, C.; Spitali, P.; Neri, M.; Patarnello, T.; Angelini, C.; Yau, S.C.; Abbs, S.; Muntoni, F.; et al. Intronic breakpoint definition and transcription analysis in DMD/BMD patients with deletion/duplication at the 5’ mutation hot spot of the dystrophin gene. Gene 2006, 370, 26–33. [Google Scholar] [CrossRef]
- Bello, L.; Morgenroth, L.P.; Gordish-Dressman, H.; Hoffman, E.P.; McDonald, C.M.; Cirak, S. CINRG investigators DMD genotypes and loss of ambulation in the CINRG Duchenne natural history study. Neurology 2016, 87, 401–409. [Google Scholar] [CrossRef]
- Flanigan, K.M.; Dunn, D.M.; von Niederhausern, A.; Soltanzadeh, P.; Howard, M.T.; Sampson, J.B.; Swoboda, K.J.; Bromberg, M.B.; Mendell, J.R.; Taylor, L.E.; et al. Nonsense mutation-associated Becker muscular dystrophy: Interplay between exon definition and splicing regulatory elements within the DMD gene. Hum. Mutat. 2011, 32, 299–308. [Google Scholar] [CrossRef]
- Dwianingsih, E.K.; Malueka, R.G.; Nishida, A.; Itoh, K.; Lee, T.; Yagi, M.; Iijima, K.; Takeshima, Y.; Matsuo, M. A novel splicing silencer generated by DMD exon 45 deletion junction could explain upstream exon 44 skipping that modifies dystrophinopathy. J. Hum. Genet. 2014, 59, 423–429. [Google Scholar] [CrossRef]
- Wang, D.-N.; Wang, Z.-Q.; Yan, L.; He, J.; Lin, M.-T.; Chen, W.-J.; Wang, N. Clinical and mutational characteristics of Duchenne muscular dystrophy patients based on a comprehensive database in South China. Neuromuscul. Disord. 2017, 27, 715–722. [Google Scholar] [CrossRef]
- van den Bergen, J.C.; Ginjaar, H.B.; Niks, E.H.; Aartsma-Rus, A.; Verschuuren, J.J.G.M. Prolonged ambulation in Duchenne patients with a mutation amenable to exon 44 skipping. J. Neuromuscul. Dis. 2014, 1, 91–94. [Google Scholar]
- Pane, M.; Mazzone, E.S.; Sormani, M.P.; Messina, S.; Vita, G.L.; Fanelli, L.; Berardinelli, A.; Torrente, Y.; D’Amico, A.; Lanzillotta, V.; et al. 6 minute walk test in Duchenne MD patients with different mutations: 12 month changes. PLoS ONE 2014, 9, e83400. [Google Scholar] [CrossRef]
- Hufton, M.; Roper, H. Variations in Duchenne muscular dystrophy course in a multi-ethnic UK population: Potential influence of socio-economic factors. Dev. Med. Child Neurol. 2017, 59, 837–842. [Google Scholar] [CrossRef]
- Fanin, M.; Danieli, G.A.; Cadaldini, M.; Miorin, M.; Vitiello, L.; Angelini, C. Dystrophin-positive fibers in duchenne dystrophy: Origin and correlation to clinical course. Muscle Nerve. 1995, 18, 1115–1120. [Google Scholar] [CrossRef]
- Partridge, T.; Lu, Q.-L. Recent Advances in Skeletal Muscle Differentiation; Research Signpost: Fort P.O., Kerala, India, 2008; pp. 93–107. [Google Scholar]
- Pegoraro, E.; Hoffman, E.; Piva, L.; Gavassini, B.; Cagnin, S.; Ermani, M.; Bello, L.; Soraru, G.; Pacchioni, B.; Bonifati, M.; et al. SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology 2011, 76, 219–226. [Google Scholar] [CrossRef]
- Nelson, S.F.; Griggs, R.C. Predicting the severity of Duchenne muscular dystrophy: Implications for treatment. Neurology 2011, 76, 208–209. [Google Scholar] [CrossRef]
- Castello, L.M.; Raineri, D.; Salmi, L.; Clemente, N.; Vaschetto, R.; Quaglia, M.; Garzaro, M.; Gentilli, S.; Navalesi, P.; Cantaluppi, V.; et al. Osteopontin at the crossroads of inflammation and tumor progression. Mediators Inflamm. 2017, 2017, 1–22. [Google Scholar] [CrossRef]
- Hao, C.; Cui, Y.; Owen, S.; Li, W.; Cheng, S.; Jiang, W.G. Human osteopontin: Potential clinical applications in cancer (Review). Int. J. Mol. Med. 2017, 39, 1327–1337. [Google Scholar] [CrossRef]
- Rittling, S.R.; Singh, R. Osteopontin in immune-mediated diseases. J. Dental Res. 2015, 94, 1638–1645. [Google Scholar] [CrossRef]
- Many, G.M.; Yokosaki, Y.; Uaesoontrachoon, K.; Nghiem, P.P.; Bello, L.; Dadgar, S.; Yin, Y.; Damsker, J.M.; Cohen, H.B.; Kornegay, J.N.; et al. OPN-a induces muscle inflammation by increasing recruitment and activation of pro-inflammatory macrophages. Exp. Physiol. 2016, 101, 1285–1300. [Google Scholar] [CrossRef]
- Hirata, A.; Masuda, S.; Tamura, T.; Kai, K.; Ojima, K.; Fukase, A.; Motoyoshi, K.; Kamakura, K.; Miyagoe-Suzuki, Y.; Takeda, S. Expression profiling of cytokines and related genes in regenerating skeletal muscle after cardiotoxin injection: A role for osteopontin. Am. J. Pathol. 2003, 163, 203–215. [Google Scholar] [CrossRef]
- Vetrone, S.A.; Montecino-Rodriguez, E.; Kudryashova, E.; Kramerova, I.; Hoffman, E.P.; Liu, S.D.; Miceli, M.C.; Spencer, M.J. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-β. J. Clin. Investig. 2009, 119, 1583–1594. [Google Scholar] [CrossRef]
- Chen, Y.-W.; Nagaraju, K.; Bakay, M.; McIntyre, O.; Rawat, R.; Shi, R.; Hoffman, E.P. Early onset of inflammation and later involvement of TGF in Duchenne muscular dystrophy. Neurology 2005, 65, 826–834. [Google Scholar] [CrossRef]
- Capote, J.; Kramerova, I.; Martinez, L.; Vetrone, S.; Barton, E.R.; Sweeney, H.L.; Miceli, M.C.; Spencer, M.J. Osteopontin ablation ameliorates muscular dystrophy by shifting macrophages to a pro-regenerative phenotype. J. Cell Biol. 2016, 213, 275–288. [Google Scholar] [CrossRef]
- Pagel, C.N.; Wasgewatte Wijesinghe, D.K.; Taghavi Esfandouni, N.; Mackie, E.J. Osteopontin, inflammation and myogenesis: Influencing regeneration, fibrosis and size of skeletal muscle. J. Cell Commun. Signal. 2014, 8, 95–103. [Google Scholar] [CrossRef]
- Zanotti, S.; Gibertini, S.; Di Blasi, C.; Cappelletti, C.; Bernasconi, P.; Mantegazza, R.; Morandi, L.; Mora, M. Osteopontin is highly expressed in severely dystrophic muscle and seems to play a role in muscle regeneration and fibrosis. Histopathology 2011, 59, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Uaesoontrachoon, K.; Wasgewatte Wijesinghe, D.K.; Mackie, E.J.; Pagel, C.N. Osteopontin deficiency delays inflammatory infiltration and the onset of muscle regeneration in a mouse model of muscle injury. Dis. Model. Mech. 2013, 6, 197–205. [Google Scholar] [CrossRef]
- Uaesoontrachoon, K.; Yoo, H.-J.; Tudor, E.M.; Pike, R.N.; Mackie, E.J.; Pagel, C.N. Osteopontin and skeletal muscle myoblasts: Association with muscle regeneration and regulation of myoblast function in vitro. Int. J. Biochem. Cell Biol. 2008, 40, 2303–2314. [Google Scholar] [CrossRef]
- Giacopelli, F.; Marciano, R.; Pistorio, A.; Catarsi, P.; Canini, S.; Karsenty, G.; Ravazzolo, R. Polymorphisms in the osteopontin promoter affect its transcriptional activity. Physiol. Genom. 2004, 20, 87–96. [Google Scholar] [CrossRef]
- Piva, L.; Gavassini, B.F.; Bello, L.; Fanin, M.; Soraru, G.; Barp, A.; Ermani, M.; Angelini, C.; Hoffman, E.P.; Pegoraro, E. TGFBR2 but not SPP1 genotype modulates osteopontin expression in Duchenne muscular dystrophy muscle. J. Pathol. 2012, 228, 251–259. [Google Scholar] [CrossRef]
- Bello, L.; Piva, L.; Barp, A.; Taglia, A.; Picillo, E.; Vasco, G.; Pane, M.; Previtali, S.C.; Torrente, Y.; Gazzerro, E.; et al. Importance of SPP1 genotype as a covariate in clinical trials in Duchenne muscular dystrophy. Neurology 2012, 79, 159–162. [Google Scholar] [CrossRef]
- van den Bergen, J.C.; Hiller, M.; Böhringer, S.; Vijfhuizen, L.; Ginjaar, H.B.; Chaouch, A.; Bushby, K.; Straub, V.; Scoto, M.; Cirak, S.; et al. Validation of genetic modifiers for Duchenne muscular dystrophy: A multicentre study assessing SPP1 and LTBP4 variants. J. Neurol. Neurosurg. Psychiatr. 2015, 86, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M.; Ceco, E.; Lamar, K.-M.; Kaminoh, Y.; Dunn, D.M.; Mendell, J.R.; King, W.M.; Pestronk, A.; Florence, J.M.; Mathews, K.D.; et al. LTBP4 genotype predicts age of ambulatory loss in Duchenne muscular dystrophy. Ann. Neurol. 2013, 73, 481–488. [Google Scholar] [CrossRef]
- Bello, L.; Kesari, A.; Gordish-Dressman, H.; Cnaan, A.; Morgenroth, L.P.; Punetha, J.; Duong, T.; Henricson, E.K.; Pegoraro, E.; McDonald, C.M.; et al. Genetic modifiers of ambulation in the cooperative international neuromuscular research group Duchenne natural history study: Ambulation in CINRG-DNHS. Ann. Neurol. 2015, 77, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yamamoto, S.; Hijiya, N.; Benveniste, E.N.; Gladson, C.L. Transcriptional regulation of the human osteopontin promoter: functional analysis and DNA-protein interactions. Oncogene 2000, 19, 5801–5809. [Google Scholar] [CrossRef][Green Version]
- Newton, R.; Holden, N.S. Separating transrepression and transactivation: A distressing divorce for the glucocorticoid receptor? Mol. Pharmacol. 2007, 72, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Gordish-Dressman, H.; McLane, V.D.; Devaney, J.M.; Thompson, P.D.; Visich, P.; Gordon, P.M.; Pescatello, L.S.; Zoeller, R.F.; Moyna, N.M.; et al. Alterations in osteopontin modify muscle size in females in both humans and mice. Med. Sci. Sports Exerc. 2013, 45, 1060–1068. [Google Scholar] [CrossRef]
- Barfield, W.L.; Uaesoontrachoon, K.; Wu, C.-S.; Lin, S.; Chen, Y.; Wang, P.C.; Kanaan, Y.; Bond, V.; Hoffman, E.P. Eccentric muscle challenge shows osteopontin polymorphism modulation of muscle damage. Hum. Mol. Genet. 2014, 23, 4043–4050. [Google Scholar] [CrossRef]
- Vianello, S.; Pantic, B.; Fusto, A.; Bello, L.; Galletta, E.; Borgia, D.; Gavassini, B.F.; Semplicini, C.; Sorarù, G.; Vitiello, L.; et al. SPP1 genotype and glucocorticoid treatment modify osteopontin expression in Duchenne muscular dystrophy cells. Hum. Mol. Genet. 2017, 26, 3342–3351. [Google Scholar] [CrossRef]
- Gimba, E.R.; Tilli, T.M. Human osteopontin splicing isoforms: Known roles, potential clinical applications and activated signaling pathways. Cancer Lett. 2013, 331, 11–17. [Google Scholar] [CrossRef]
- Kornegay, J.N.; Childers, M.K.; Bogan, D.J.; Bogan, J.R.; Nghiem, P.; Wang, J.; Fan, Z.; Howard, J.F.; Schatzberg, S.J.; Dow, J.L.; et al. The paradox of muscle hypertrophy in muscular dystrophy. Phys. Med. Rehabil. Clin. N. Am. 2012, 23, 149–172. [Google Scholar] [CrossRef]
- Nghiem, P.P.; Kornegay, J.N.; Uaesoontrachoon, K.; Bello, L.; Yin, Y.; Kesari, A.; Mittal, P.; Schatzberg, S.J.; Many, G.M.; Lee, N.H.; et al. Osteopontin is linked with AKT, FoxO1, and myostatin in skeletal muscle cells. Muscle Nerve. 2017, 56, 1119–1127. [Google Scholar] [CrossRef]
- Bonifati, D.M.; Witchel, S.F.; Ermani, M.; Hoffman, E.P.; Angelini, C.; Pegoraro, E. The glucocorticoid receptor N363S polymorphism and steroid response in Duchenne dystrophy. J. Neurol. Neurosurg. Psychiatry 2006, 77, 1177–1179. [Google Scholar] [CrossRef]
- Heydemann, A.; Ceco, E.; Lim, J.E.; Hadhazy, M.; Ryder, P.; Moran, J.L.; Beier, D.R.; Palmer, A.A.; McNally, E.M. Latent TGF-beta-binding protein 4 modifies muscular dystrophy in mice. J. Clin. Investig. 2009, 119, 3703–3712. [Google Scholar] [CrossRef] [PubMed]
- Giltay, R.; Kostka, G.; Timpl, R. Sequence and expression of a novel member (LTBP-4) of the family of latent transforming growth factor-beta binding proteins. FEBS Lett. 1997, 411, 164–168. [Google Scholar] [CrossRef]
- Saharinen, J.; Taipale, J.; Monni, O.; Keski-Oja, J. Identification and characterization of a new latent transforming growth factor-beta-binding protein, LTBP-4. J. Biol. Chem. 1998, 273, 18459–18469. [Google Scholar] [CrossRef]
- Saharinen, J.; Keski-Oja, J. Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol. Biol. Cell 2000, 11, 2691–2704. [Google Scholar] [CrossRef]
- Chen, Y.; Ali, T.; Todorovic, V.; O’leary, J.M.; Kristina Downing, A.; Rifkin, D.B. Amino acid requirements for formation of the TGF-beta-latent TGF-beta binding protein complexes. J. Mol. Biol. 2005, 345, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Koli, K.; Saharinen, J.; Hyytiäinen, M.; Penttinen, C.; Keski-Oja, J. Latency, activation, and binding proteins of TGF-beta. Microsc. Res. Tech. 2001, 52, 354–362. [Google Scholar] [CrossRef]
- Lamar, K.-M.; Miller, T.; Dellefave-Castillo, L.; McNally, E.M. Genotype-specific interaction of latent TGFβ binding protein 4 with TGFβ. PLoS ONE 2016, 11, e0150358. [Google Scholar] [CrossRef]
- Weiss, R.B.; Vieland, V.J.; Dunn, D.M.; Kaminoh, Y.; Flanigan, K.M. United dystrophinopathy project long-range genomic regulators of THBS1 and LTBP4 modify disease severity in Duchenne muscular dystrophy. Ann. Neurol. 2018, 84, 234–245. [Google Scholar] [CrossRef]
- Lamar, K.-M.; Bogdanovich, S.; Gardner, B.B.; Gao, Q.Q.; Miller, T.; Earley, J.U.; Hadhazy, M.; Vo, A.H.; Wren, L.; Molkentin, J.D.; et al. Overexpression of latent TGFβ binding protein 4 in muscle ameliorates muscular dystrophy through myostatin and TGFβ. PLoS Genet. 2016, 12, e1006019. [Google Scholar] [CrossRef] [PubMed]
- Ceco, E.; Bogdanovich, S.; Gardner, B.; Miller, T.; DeJesus, A.; Earley, J.U.; Hadhazy, M.; Smith, L.R.; Barton, E.R.; Molkentin, J.D.; et al. Targeting latent TGFβ release in muscular dystrophy. Sci. Transl. Med. 2014, 6, 259. [Google Scholar] [CrossRef] [PubMed]
- Bello, L.; Flanigan, K.M.; Weiss, R.B.; Spitali, P.; Aartsma-Rus, A.; Muntoni, F.; Zaharieva, I.; Ferlini, A.; Mercuri, E.; Tuffery-Giraud, S.; et al. Association study of exon variants in the NF-κB and TGFβ pathways identifies CD40 as a modifier of Duchenne muscular dystrophy. Am. J. Hum. Genet. 2016, 99, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, E.M.; Concepcion, E.; Oashi, T.; Tomer, Y. A Graves’ disease-associated Kozak sequence single-nucleotide polymorphism enhances the efficiency of CD40 gene translation: A case for translational pathophysiology. Endocrinology 2005, 146, 2684–2691. [Google Scholar] [CrossRef]
- Gandhi, K.S.; McKay, F.C.; Cox, M.; Riveros, C.; Armstrong, N.; Heard, R.N.; Vucic, S.; Williams, D.W.; Stankovich, J.; Brown, M.; et al. The multiple sclerosis whole blood mRNA transcriptome and genetic associations indicate dysregulation of specific T cell pathways in pathogenesis. Hum. Mol. Genet. 2010, 19, 2134–2143. [Google Scholar] [CrossRef]
- Japan Kawasaki Disease Genome Consortium; US Kawasaki Disease Genetics Consortium; Onouchi, Y.; Ozaki, K.; Burns, J.C.; Shimizu, C.; Terai, M.; Hamada, H.; Honda, T.; Suzuki, H.; et al. A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat. Genet. 2012, 44, 517–521. [Google Scholar] [CrossRef]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015, 7, 299. [Google Scholar] [CrossRef]
- Gussoni, E.; Pavlath, G.K.; Miller, R.G.; Panzara, M.A.; Powell, M.; Blau, H.M.; Steinman, L. Specific T cell receptor gene rearrangements at the site of muscle degeneration in Duchenne muscular dystrophy. J. Immunol. 1994, 153, 4798–4805. [Google Scholar]
- Morrison, J.; Lu, Q.L.; Pastoret, C.; Partridge, T.; Bou-Gharios, G. T-cell-dependent fibrosis in the mdx dystrophic mouse. Lab. Investig. 2000, 80, 881–891. [Google Scholar] [CrossRef]
- Morrison, J.; Palmer, D.B.; Cobbold, S.; Partridge, T.; Bou-Gharios, G. Effects of T-lymphocyte depletion on muscle fibrosis in the mdx mouse. Am. J. Pathol. 2005, 166, 1701–1710. [Google Scholar] [CrossRef]
- Cascabulho, C.M.; Corrêa, C.B.; Cotta-de-Almeida, V.; Henriques-Pons, A. Defective T-lymphocyte migration to muscles in dystrophin-deficient mice. Am. J. Pathol. 2012, 181, 593–604. [Google Scholar] [CrossRef]
- Farini, A.; Meregalli, M.; Belicchi, M.; Battistelli, M.; Parolini, D.; D’Antona, G.; Gavina, M.; Ottoboni, L.; Constantin, G.; Bottinelli, R.; et al. T and B lymphocyte depletion has a marked effect on the fibrosis of dystrophic skeletal muscles in the scid/mdx mouse. J. Pathol. 2007, 213, 229–238. [Google Scholar] [CrossRef]
- Villalta, S.A.; Rosenthal, W.; Martinez, L.; Kaur, A.; Sparwasser, T.; Tidball, J.G.; Margeta, M.; Spencer, M.J.; Bluestone, J.A. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci. Transl. Med. 2014, 6, 258ra142. [Google Scholar]
- Kissel, J.T.; Burrow, K.L.; Rammohan, K.W.; Mendell, J.R. Mononuclear cell analysis of muscle biopsies in prednisone-treated and untreated Duchenne muscular dystrophy. CIDD Study Group. Neurology 1991, 41, 667–672. [Google Scholar] [CrossRef]
- MacArthur, D.G.; North, K.N. ACTN3: A genetic influence on muscle function and athletic performance. Exerc. Sport Sci. Rev. 2007, 35, 30–34. [Google Scholar] [CrossRef]
- North, K.N.; Yang, N.; Wattanasirichaigoon, D.; Mills, M.; Easteal, S.; Beggs, A.H. A common nonsense mutation results in alpha-actinin-3 deficiency in the general population. Nat. Genet. 1999, 21, 353–354. [Google Scholar] [CrossRef]
- Schiaffino, S. Knockout of human muscle genes revealed by large scale whole-exome studies. Mol. Genet. Metab. 2018, 123, 411–415. [Google Scholar] [CrossRef]
- Moran, C.N.; Yang, N.; Bailey, M.E.S.; Tsiokanos, A.; Jamurtas, A.; MacArthur, D.G.; North, K.; Pitsiladis, Y.P.; Wilson, R.H. Association analysis of the ACTN3 R577X polymorphism and complex quantitative body composition and performance phenotypes in adolescent Greeks. Eur. J. Hum. Genet. 2007, 15, 88–93. [Google Scholar] [CrossRef]
- Yang, N.; MacArthur, D.G.; Gulbin, J.P.; Hahn, A.G.; Beggs, A.H.; Easteal, S.; North, K. ACTN3 genotype is associated with human elite athletic performance. Am. J. Hum. Genet. 2003, 73, 627–631. [Google Scholar] [CrossRef]
- Eynon, N.; Duarte, J.A.; Oliveira, J.; Sagiv, M.; Yamin, C.; Meckel, Y.; Sagiv, M.; Goldhammer, E. ACTN3 R577X polymorphism and Israeli top-level athletes. Int. J. Sports Med. 2009, 30, 695–698. [Google Scholar] [CrossRef]
- Papadimitriou, I.D.; Lockey, S.J.; Voisin, S.; Herbert, A.J.; Garton, F.; Houweling, P.J.; Cieszczyk, P.; Maciejewska-Skrendo, A.; Sawczuk, M.; Massidda, M.; et al. No association between ACTN3 R577X and ACE I/D polymorphisms and endurance running times in 698 Caucasian athletes. BMC Genomics 2018, 19, 13. [Google Scholar] [CrossRef]
- MacArthur, D.G.; Seto, J.T.; Chan, S.; Quinlan, K.G.R.; Raftery, J.M.; Turner, N.; Nicholson, M.D.; Kee, A.J.; Hardeman, E.C.; Gunning, P.W.; et al. An Actn3 knockout mouse provides mechanistic insights into the association between alpha-actinin-3 deficiency and human athletic performance. Hum. Mol. Genet. 2008, 17, 1076–1086. [Google Scholar] [CrossRef]
- Seto, J.T.; Quinlan, K.G.R.; Lek, M.; Zheng, X.F.; Garton, F.; MacArthur, D.G.; Hogarth, M.W.; Houweling, P.J.; Gregorevic, P.; Turner, N.; et al. ACTN3 genotype influences muscle performance through the regulation of calcineurin signaling. J. Clin. Investig. 2013, 123, 4255–4263. [Google Scholar] [CrossRef]
- Hogarth, M.W.; Houweling, P.J.; Thomas, K.C.; Gordish-Dressman, H.; Bello, L.; Cooperative International Neuromuscular Research Group (CINRG); Pegoraro, E.; Hoffman, E.P.; Head, S.I.; North, K.N. Evidence for ACTN3 as a genetic modifier of Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14143. [Google Scholar] [CrossRef]
- Webster, C.; Silberstein, L.; Hays, A.P.; Blau, H.M. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell 1988, 52, 503–513. [Google Scholar] [CrossRef]
- Javierre, B.M.; Burren, O.S.; Wilder, S.P.; Kreuzhuber, R.; Hill, S.M.; Sewitz, S.; Cairns, J.; Wingett, S.W.; Várnai, C.; Thiecke, M.J.; et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 2016, 167, 1369–1384. [Google Scholar] [CrossRef]
- Crawford, S.E.; Stellmach, V.; Murphy-Ullrich, J.E.; Ribeiro, S.M.; Lawler, J.; Hynes, R.O.; Boivin, G.P.; Bouck, N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 1998, 93, 1159–1170. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018, 17, 347–361. [Google Scholar] [CrossRef]
- Rouger, K.; Le Cunff, M.; Steenman, M.; Potier, M.-C.; Gibelin, N.; Dechesne, C.A.; Leger, J.J. Global/temporal gene expression in diaphragm and hindlimb muscles of dystrophin-deficient (mdx) mice. Am. J. Physiol. Cell Physiol. 2002, 283, C773–C784. [Google Scholar] [CrossRef]
- Porter, J.D.; Merriam, A.P.; Leahy, P.; Gong, B.; Feuerman, J.; Cheng, G.; Khanna, S. Temporal gene expression profiling of dystrophin-deficient (mdx) mouse diaphragm identifies conserved and muscle group-specific mechanisms in the pathogenesis of muscular dystrophy. Hum. Mol. Genet. 2004, 13, 257–269. [Google Scholar] [CrossRef]
- McDonald, C.M.; Gordish-Dressman, H.; Henricson, E.K.; Duong, T.; Joyce, N.C.; Jhawar, S.; Leinonen, M.; Hsu, F.; Connolly, A.M.; Cnaan, A.; et al. Longitudinal pulmonary function testing outcome measures in Duchenne muscular dystrophy: Long-term natural history with and without glucocorticoids. Neuromuscul. Disord. 2018, 28, 897–909. [Google Scholar] [CrossRef]
- LoMauro, A.; Romei, M.; Gandossini, S.; Pascuzzo, R.; Vantini, S.; D’Angelo, M.G.; Aliverti, A. Evolution of respiratory function in Duchenne muscular dystrophy from childhood to adulthood. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef]
- Spurney, C.F. Cardiomyopathy of Duchenne muscular dystrophy: Current understanding and future directions. Muscle Nerve. 2011, 44, 8–19. [Google Scholar] [CrossRef]
- Kaspar, R.W.; Allen, H.D.; Ray, W.C.; Alvarez, C.E.; Kissel, J.T.; Pestronk, A.; Weiss, R.B.; Flanigan, K.M.; Mendell, J.R.; Montanaro, F. Analysis of dystrophin deletion mutations predicts age of cardiomyopathy onset in becker muscular dystrophy. Circ. Cardiovasc. Genet. 2009, 2, 544–551. [Google Scholar] [CrossRef]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Markham, L.W.; Kinnett, K.; Wong, B.L.; Woodrow Benson, D.; Cripe, L.H. Corticosteroid treatment retards development of ventricular dysfunction in Duchenne muscular dystrophy. Neuromuscul. Disord. 2008, 18, 365–370. [Google Scholar] [CrossRef]
- Silversides, C.K.; Webb, G.D.; Harris, V.A.; Biggar, D.W. Effects of deflazacort on left ventricular function in patients with Duchenne muscular dystrophy. Am. J. Cardiol. 2003, 91, 769–772. [Google Scholar] [CrossRef]
- Barber, B.J.; Andrews, J.G.; Lu, Z.; West, N.A.; Meaney, F.J.; Price, E.T.; Gray, A.; Sheehan, D.W.; Pandya, S.; Yang, M.; et al. Oral corticosteroids and onset of cardiomyopathy in Duchenne muscular dystrophy. J. Pediatr. 2013, 163, 1080–1084. [Google Scholar] [CrossRef]
- Ashwath, M.L.; Jacobs, I.B.; Crowe, C.A.; Ashwath, R.C.; Super, D.M.; Bahler, R.C. Left ventricular dysfunction in Duchenne muscular dystrophy and genotype. Am. J. Cardiol. 2014, 114, 284–289. [Google Scholar] [CrossRef]
- Spurney, C.; Shimizu, R.; Morgenroth, L.P.; Kolski, H.; Gordish-Dressman, H.; Clemens, P.R.; CINRG Investigators. Cooperative international neuromuscular research group Duchenne natural history study demonstrates insufficient diagnosis and treatment of cardiomyopathy in Duchenne muscular dystrophy. Muscle Nerve. 2014, 50, 250–256. [Google Scholar] [CrossRef]
- Yue, Y.; Meng, K.; Pu, Y.; Zhang, X. Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res. Clin. Pract. 2017, 133, 124–130. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.-J.; et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Lijnen, P.J.; Petrov, V.V.; Fagard, R.H. Association between transforming growth factor-beta and hypertension. Am. J. Hypertens. 2003, 16, 604–611. [Google Scholar] [CrossRef]
- Cohn, R.D.; Dubowitz, V. Duchenne muscular dystrophy: Ringo to the rescue? Neuromuscul. Disord. 2016, 26, 5–6. [Google Scholar] [CrossRef]
- Zatz, M.; Pavanello, R.C.M.; Lazar, M.; Yamamoto, G.L.; Lourenço, N.C.V.; Cerqueira, A.; Nogueira, L.; Vainzof, M. Milder course in Duchenne patients with nonsense mutations and no muscle dystrophin. Neuromuscul. Disord. 2014, 24, 986–989. [Google Scholar] [CrossRef]


{kind=link}
| Locus | Protein Product | rs# | Alleles | Chr Position (GRCh38.p12) | Inheritance Model | SNP Effect | Minor Allele Effect on Protein Product | Minor Allele Effect on DMD Severity | Studied DMD Sub-Phenotypes | Outcome Measures | Positive Association Studies (n of DMD Participants) | Negative Association Studies (n of DMD Participants) | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Discovery | Validation | Discovery | Validation | ||||||||||||
| SPP1 | Secreted PhosphoProtein 1, also known as osteopontin | rs28357094 | T>G | 4:87975645 | Dominant | Transcriptional (promoter) | Reduced expression, increased steroid responsiveness | Detrimental | Skeletal muscle strength | MRC strength | 80 | - | - | - | 43 |
| Grip strength | 156 | - | - | - | 27 | ||||||||||
| NSAA (1-year change) | 80 | - | - | - | |||||||||||
| 6MWT (1-year change) | 80 | - | - | - | |||||||||||
| LoA | 106 | 279 | - | 254; 336 | 27, 44, 45, 46 | ||||||||||
| Dilated cardiomyopathy | Onset (age at LVEF < 50% or LVEDV > 70 mL/m2) | - | - | 178 | - | 13 | |||||||||
| LTBP4 | Latent Transforming growth factor β Binding Protein 4 | rs2303729, rs1131620, rs1051303, rs10880 | G>A, A>G, A>G, C>T | 19:40605163, 19:40611963, 19:40612150, 19:40622404 | Recessive | Coding haplotype (VTTT/IAAM) | Resistance to proteolysis, increased TGF-β binding avidity | Protective | Skeletal muscle strength | LoA | 254 | 274; 265 | - | 137 | 13, 44, 45, 46 |
| Dilated cardiomyopathy | Onset (age at LVEF < 50% or LVEDV > 70 mL/m2) | 178 | - | - | - | 13 | |||||||||
| rs710160 | T>C | 19:40581585 | Recessive | Upstream regulatory | Reduced expression | Protective | Skeletal muscle strength | LoA | 253 | - | - | - | 63 | ||
| CD40 | Tumor Necrosis Factor Receptor SuperFamily member 5 (TNFRSF5) | rs1883832 | C>T | 20:46118343 | Additive/ dominant | 5’-UTR (Kozak sequence) | Reduced expression | Detrimental | Skeletal muscle strength | LoA | 109 | 660 | - | - | 66 |
| THBS1 | Thrombospondin-1 | rs2725797 | G>A | 15:38817032 | Recessive | Long-range regulator | Reduced expression | Protective | Skeletal muscle strength | LoA | 253 | - | - | - | 63 |
| ACTN3 | α-actinin-3 | rs1815739 | C>T | 11:66560624 | Additive/ genotypic | Nonsense | Complete defect | Detrimental (worse in heterozygotes?) | Skeletal muscle strength | Grip strength | 59 | - | - | - | 87 |
| Detrimental (worse in heterozygotes?) | QMT strength | 61 | - | - | - | ||||||||||
| Detrimental (worse in heterozygotes?) | 10 m run/walk speed | 61 | - | - | - | ||||||||||
| Detrimental (worse in heterozygotes?) | LoA | 266 | 102 | - | - | ||||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bello, L.; Pegoraro, E. The “Usual Suspects”: Genes for Inflammation, Fibrosis, Regeneration, and Muscle Strength Modify Duchenne Muscular Dystrophy. J. Clin. Med. 2019, 8, 649. https://doi.org/10.3390/jcm8050649
Bello L, Pegoraro E. The “Usual Suspects”: Genes for Inflammation, Fibrosis, Regeneration, and Muscle Strength Modify Duchenne Muscular Dystrophy. Journal of Clinical Medicine. 2019; 8(5):649. https://doi.org/10.3390/jcm8050649
Chicago/Turabian StyleBello, Luca, and Elena Pegoraro. 2019. "The “Usual Suspects”: Genes for Inflammation, Fibrosis, Regeneration, and Muscle Strength Modify Duchenne Muscular Dystrophy" Journal of Clinical Medicine 8, no. 5: 649. https://doi.org/10.3390/jcm8050649
APA StyleBello, L., & Pegoraro, E. (2019). The “Usual Suspects”: Genes for Inflammation, Fibrosis, Regeneration, and Muscle Strength Modify Duchenne Muscular Dystrophy. Journal of Clinical Medicine, 8(5), 649. https://doi.org/10.3390/jcm8050649