Control of Invasion by Epithelial-to-Mesenchymal Transition Programs during Metastasis

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. EMT Program Regulation and Function
2.1. Mechanism EMT Program Activation
2.2. Suppression of Epithelial Traits
2.3. Induction of Mesenchymal Features
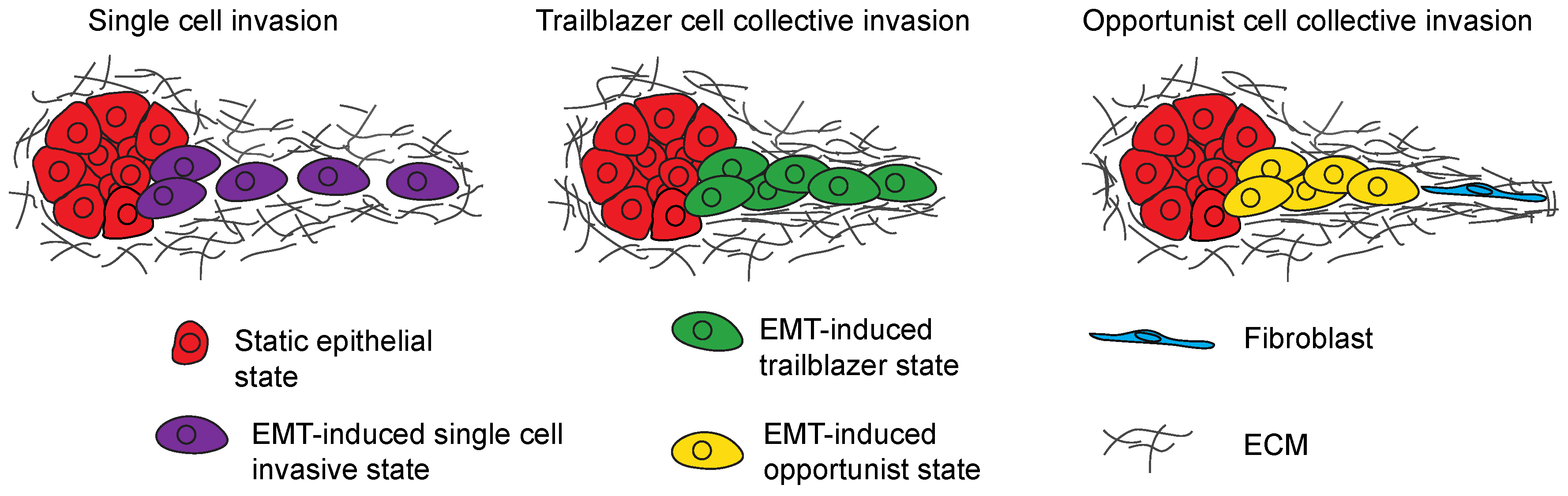
3. EMT Program Heterogeneity Confers Distinct Invasive Phenotypes
3.1. Regulation of Single-Cell Invasion
3.2. Collective Invasion Is the Predominant Mode of Tumor Cell Invasion
3.3. Trailblazer-Type Collective Invasion
3.4. Opportunistic-Type Collective Invasion
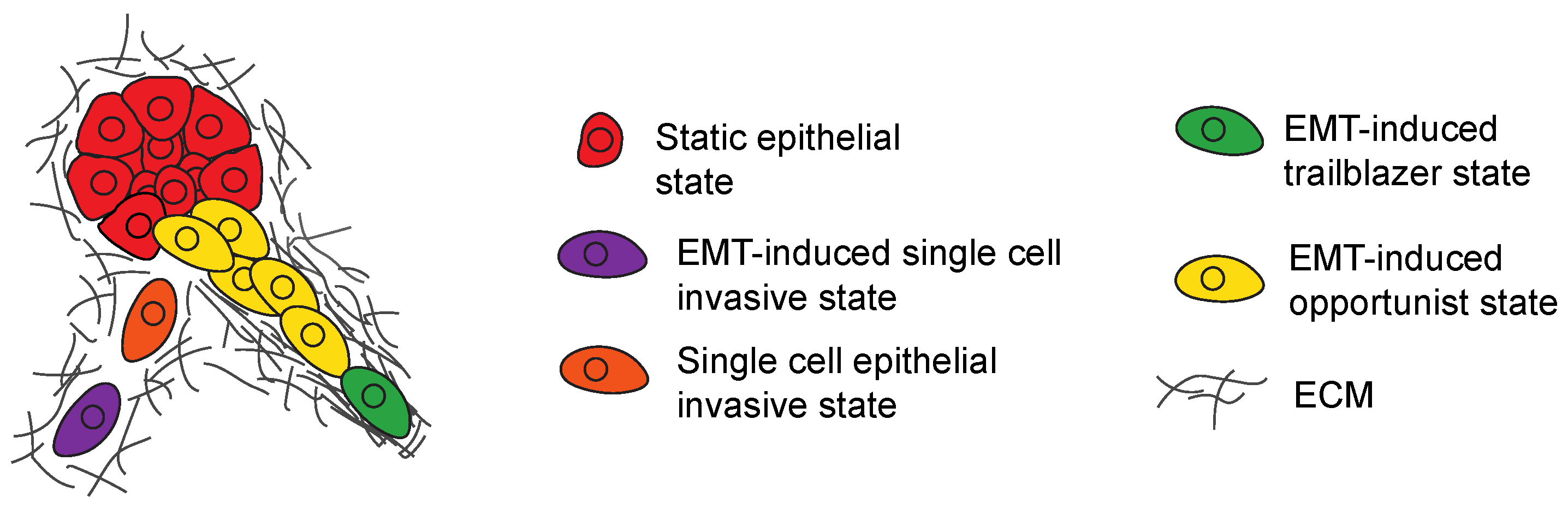
3.5. Regulation of Hybrid EMT States That Promote Opportunistic Collective Invasion
4. EMT-Induced Cells Can Influence the Invasive Properties of Siblings
4.1. Subpopulation Interactions That Promote Single-Cell Invasion
4.2. Subpopulation Interactions That Promote Collective Invasion
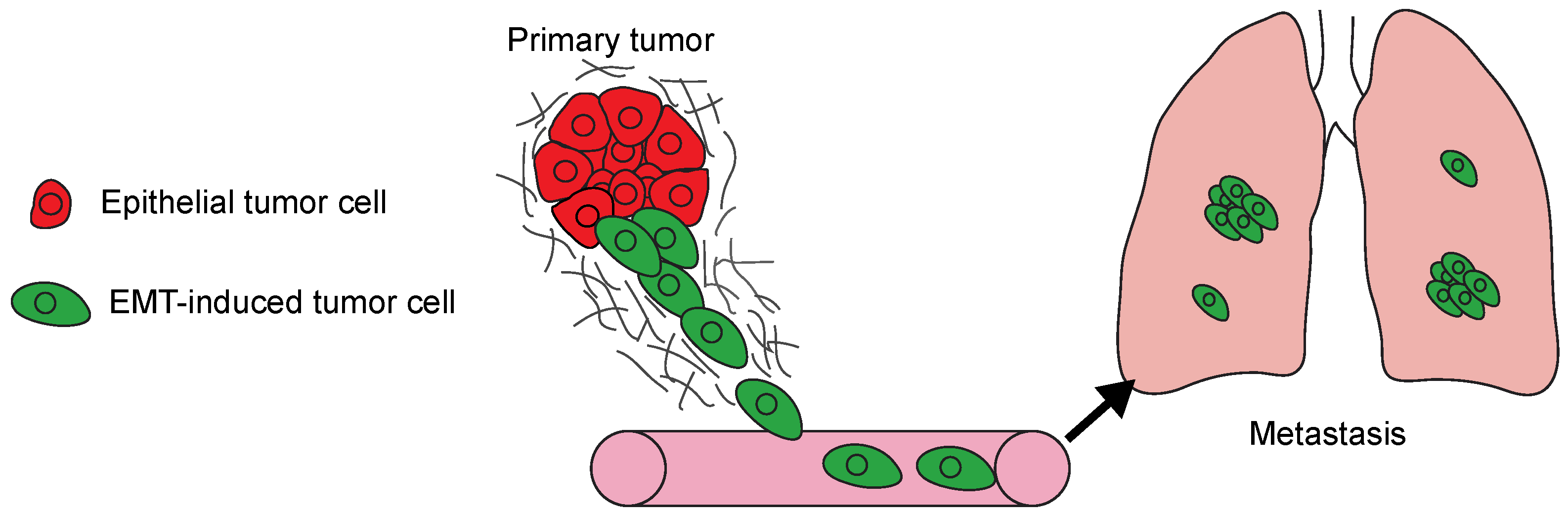
5. EMT Invasion Programs Determine Metastatic Traits
5.1. EMT Activation Promotes Early Dissemination
5.2. There Is EMT Program Heterogeneity in Primary Tumors
5.3. EMT Program Traits Influence Colonization Ability
6. Conclusions
Funding
Conflicts of Interest
References
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging biological principles of metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Res. 1978, 38, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Mc, C.M.; Coman, D.R.; Moore, F.B. Studies on invasiveness of cancer; adhesiveness of malignant cells in various human adenocarcinomas. Cancer 1948, 1, 460–467. [Google Scholar]
- Enterline, H.T.; Coman, D.R. The ameboid motility of human and animal neoplastic cells. Cancer 1950, 3, 1033–1038. [Google Scholar] [CrossRef]
- Zeidman, I.; Mc, C.M.; Coman, D.R. Factors affecting the number of tumor metastases; experiments with a transplantable mouse tumor. Cancer Res. 1950, 10, 357–359. [Google Scholar] [PubMed]
- Nieto, M.A.; Cano, A. The epithelial-mesenchymal transition under control: Global programs to regulate epithelial plasticity. Semin. Cancer Biol. 2012, 22, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Debnath, J.; Brugge, J.S. Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 2005, 5, 675–688. [Google Scholar] [CrossRef]
- Jacks, T.; Weinberg, R.A. Taking the study of cancer cell survival to a new dimension. Cell 2002, 111, 923–925. [Google Scholar] [CrossRef]
- Rorth, P. Collective cell migration. Annu. Rev. Cell Dev. Biol. 2009, 25, 407–429. [Google Scholar] [CrossRef] [PubMed]
- Grunert, S.; Jechlinger, M.; Beug, H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat. Rev. Mol. Cell Biol. 2003, 4, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Weinberg, R.A. Epithelial-mesenchymal plasticity: A central regulator of cancer progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Subramanyam, D.; Blelloch, R.; Derynck, R. Regulation of epithelial-mesenchymal and mesenchymal-epithelial transitions by microRNAs. Curr. Opin. Cell Biol. 2013, 25, 200–207. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a twist1-g3bp2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.M.; Li, A.; Olino, K.; Wolfgang, C.L.; Herman, J.M.; Schulick, R.D.; Iacobuzio-Donahue, C.; Hruban, R.H.; Goggins, M. Loss of e-cadherin expression and outcome among patients with resectable pancreatic adenocarcinomas. Mod. Pathol. 2011, 24, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Horne, H.N.; Sherman, M.E.; Garcia-Closas, M.; Pharoah, P.D.; Blows, F.M.; Yang, X.R.; Hewitt, S.M.; Conway, C.M.; Lissowska, J.; Brinton, L.A.; et al. Breast cancer susceptibility risk associations and heterogeneity by e-cadherin tumor tissue expression. Breast Cancer Res. Treat. 2014, 143, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L.; Chen, M.W.; Xian, L. Prognostic and clinicopathological significance of downregulated e-cadherin expression in patients with non-small cell lung cancer (NSCLC): A meta-analysis. PLoS ONE 2014, 9, e99763. [Google Scholar] [CrossRef] [PubMed]
- Christou, N.; Perraud, A.; Blondy, S.; Jauberteau, M.O.; Battu, S.; Mathonnet, M. E-cadherin: A potential biomarker of colorectal cancer prognosis. Oncol. Lett. 2017, 13, 4571–4576. [Google Scholar] [CrossRef] [PubMed]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, zeb and bhlh factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of e-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.A.; Ilina, O.; Gritsenko, P.G.; Bult, P.; Span, P.N.; Friedl, P. Collective invasion in ductal and lobular breast cancer associates with distant metastasis. Clin. Exp. Metastasis 2017, 34, 421–429. [Google Scholar] [CrossRef] [PubMed]
- de Beco, S.; Gueudry, C.; Amblard, F.; Coscoy, S. Endocytosis is required for e-cadherin redistribution at mature adherens junctions. Proc. Natl. Acad. Sci. USA 2009, 106, 7010–7015. [Google Scholar] [CrossRef]
- Hong, S.; Troyanovsky, R.B.; Troyanovsky, S.M. Spontaneous assembly and active disassembly balance adherens junction homeostasis. Proc. Natl. Acad. Sci. USA 2010, 107, 3528–3533. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. Emt subtype influences epithelial plasticity and mode of cell migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Guilford, P.; Thiery, J.P. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci. 2012, 125, 4417–4422. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in emt and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. Emt, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Philippar, U.; Roussos, E.T.; Oser, M.; Yamaguchi, H.; Kim, H.D.; Giampieri, S.; Wang, Y.; Goswami, S.; Wyckoff, J.B.; Lauffenburger, D.A.; et al. A mena invasion isoform potentiates EGF-induced carcinoma cell invasion and metastasis. Dev. Cell 2008, 15, 813–828. [Google Scholar] [CrossRef]
- Shin, S.; Buel, G.R.; Nagiec, M.J.; Han, M.J.; Roux, P.P.; Blenis, J.; Yoon, S.O. Erk2 regulates epithelial-to-mesenchymal plasticity through dock10-dependent rac1/foxo1 activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2967–2976. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during emt. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Nieberler, M.; Reuning, U.; Reichart, F.; Notni, J.; Wester, H.J.; Schwaiger, M.; Weinmuller, M.; Rader, A.; Steiger, K.; Kessler, H. Exploring the role of RGD-recognizing integrins in cancer. Cancers 2017, 9, 116. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Wyckoff, J.; Condeelis, J. Cell migration in tumors. Curr. Opin. Cell Biol. 2005, 17, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Nistico, P.; Bissell, M.J.; Radisky, D.C. Epithelial-mesenchymal transition: General principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Akatsuka, T.; Imanaka-Yoshida, K. Tenascin-c and integrins in cancer. Cell Adh. Migr. 2015, 9, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Topalovski, M.; Brekken, R.A. Matrix control of pancreatic cancer: New insights into fibronectin signaling. Cancer Lett. 2016, 381, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Moreno, V.; Marshall, C.J. The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr. Opin. Cell Biol. 2010, 22, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Cukierman, E. Modeling tissue morphogenesis and cancer in 3d. Cell 2007, 130, 601–610. [Google Scholar] [CrossRef]
- Sahai, E.; Marshall, C.J. Differing modes of tumour cell invasion have distinct requirements for rho/rock signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef]
- Wolf, K.; Mazo, I.; Leung, H.; Engelke, K.; von Andrian, U.H.; Deryugina, E.I.; Strongin, A.Y.; Brocker, E.B.; Friedl, P. Compensation mechanism in tumor cell migration: Mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 2003, 160, 267–277. [Google Scholar] [CrossRef]
- Beerling, E.; Seinstra, D.; de Wit, E.; Kester, L.; van der Velden, D.; Maynard, C.; Schafer, R.; van Diest, P.; Voest, E.; van Oudenaarden, A.; et al. Plasticity between epithelial and mesenchymal states unlinks emt from metastasis-enhancing stem cell capacity. Cell Rep. 2016, 14, 2281–2288. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhu, X.; Cui, K.; Mancuso, J.; Federley, R.; Fischer, K.; Teng, G.J.; Mittal, V.; Gao, D.; Zhao, H.; et al. In vivo visualization and characterization of epithelial-mesenchymal transition in breast tumors. Cancer Res. 2016, 76, 2094–2104. [Google Scholar] [CrossRef]
- Shamir, E.R.; Pappalardo, E.; Jorgens, D.M.; Coutinho, K.; Tsai, W.T.; Aziz, K.; Auer, M.; Tran, P.T.; Bader, J.S.; Ewald, A.J. Twist1-induced dissemination preserves epithelial identity and requires e-cadherin. J. Cell Biol. 2014, 204, 839–856. [Google Scholar] [CrossRef]
- Shamir, E.R.; Coutinho, K.; Georgess, D.; Auer, M.; Ewald, A.J. Twist1-positive epithelial cells retain adhesive and proliferative capacity throughout dissemination. Biol. Open 2016, 5, 1216–1228. [Google Scholar] [CrossRef]
- Ilina, O.; Campanello, L.; Gritsenko, P.G.; Vullings, M.; Wang, C.; Bult, P.; Losert, W.; Friedl, P. Intravital microscopy of collective invasion plasticity in breast cancer. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible tgfbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296. [Google Scholar] [CrossRef]
- Lehmann, S.; Te Boekhorst, V.; Odenthal, J.; Bianchi, R.; van Helvert, S.; Ikenberg, K.; Ilina, O.; Stoma, S.; Xandry, J.; Jiang, L.; et al. Hypoxia induces a hif-1-dependent transition from collective-to-amoeboid dissemination in epithelial cancer cells. Curr. Biol. 2017, 27, 392–400. [Google Scholar] [CrossRef]
- Condeelis, J.; Segall, J.E. Intravital imaging of cell movement in tumours. Nat. Rev. Cancer 2003, 3, 921–930. [Google Scholar] [CrossRef]
- Cheung, K.J.; Ewald, A.J. Illuminating breast cancer invasion: Diverse roles for cell–cell interactions. Curr. Opin. Cell Biol. 2014, 30, 99–111. [Google Scholar] [CrossRef]
- Friedl, P.; Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef]
- Yu, X.; Zech, T.; McDonald, L.; Gonzalez, E.G.; Li, A.; Macpherson, I.; Schwarz, J.P.; Spence, H.; Futo, K.; Timpson, P.; et al. N-wasp coordinates the delivery and f-actin-mediated capture of mt1-mmp at invasive pseudopods. J. Cell Biol. 2012, 199, 527–544. [Google Scholar] [CrossRef]
- Stuelten, C.H.; Parent, C.A.; Montell, D.J. Cell motility in cancer invasion and metastasis: Insights from simple model organisms. Nat. Rev. Cancer 2018, 18, 296–312. [Google Scholar] [CrossRef]
- Wolf, K.; Wu, Y.I.; Liu, Y.; Geiger, J.; Tam, E.; Overall, C.; Stack, M.S.; Friedl, P. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 2007, 9, 893–904. [Google Scholar] [CrossRef]
- Bronsert, P.; Enderle-Ammour, K.; Bader, M.; Timme, S.; Kuehs, M.; Csanadi, A.; Kayser, G.; Kohler, I.; Bausch, D.; Hoeppner, J.; et al. Cancer cell invasion and EMT marker expression: A three-dimensional study of the human cancer-host interface. J. Pathol. 2014, 234, 410–422. [Google Scholar] [CrossRef]
- Hegerfeldt, Y.; Tusch, M.; Brocker, E.B.; Friedl, P. Collective cell movement in primary melanoma explants: Plasticity of cell–cell interaction, beta1-integrin function, and migration strategies. Cancer Res. 2002, 62, 2125–2130. [Google Scholar]
- Nguyen-Ngoc, K.V.; Cheung, K.J.; Brenot, A.; Shamir, E.R.; Gray, R.S.; Hines, W.C.; Yaswen, P.; Werb, Z.; Ewald, A.J. Ecm microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, 2595–2604. [Google Scholar] [CrossRef]
- Alexander, S.; Koehl, G.E.; Hirschberg, M.; Geissler, E.K.; Friedl, P. Dynamic imaging of cancer growth and invasion: A modified skin-fold chamber model. Histochem. Cell Biol. 2008, 130, 1147–1154. [Google Scholar] [CrossRef]
- Friedl, P.; Locker, J.; Sahai, E.; Segall, J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012, 14, 777–783. [Google Scholar] [CrossRef]
- Westcott, J.M.; Prechtl, A.M.; Maine, E.A.; Dang, T.T.; Esparza, M.A.; Sun, H.; Zhou, Y.; Xie, Y.; Pearson, G.W. An epigenetically distinct breast cancer cell subpopulation promotes collective invasion. J. Clin. Investig. 2015, 125, 1927–1943. [Google Scholar] [CrossRef]
- Konen, J.; Summerbell, E.; Dwivedi, B.; Galior, K.; Hou, Y.; Rusnak, L.; Chen, A.; Saltz, J.; Zhou, W.; Boise, L.H.; et al. Image-guided genomics of phenotypically heterogeneous populations reveals vascular signalling during symbiotic collective cancer invasion. Nat. Commun. 2017, 8, 15078. [Google Scholar] [CrossRef]
- Dang, T.T.; Prechtl, A.M.; Pearson, G.W. Breast cancer subtype-specific interactions with the microenvironment dictate mechanisms of invasion. Cancer Res. 2011, 71, 6857–6866. [Google Scholar] [CrossRef]
- Labernadie, A.; Kato, T.; Brugues, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; Gonzalez-Tarrago, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic e-cadherin/n-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef]
- Celia-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef]
- Gadea, G.; Sanz-Moreno, V.; Self, A.; Godi, A.; Marshall, C.J. Dock10-mediated cdc42 activation is necessary for amoeboid invasion of melanoma cells. Curr. Biol. 2008, 18, 1456–1465. [Google Scholar] [CrossRef]
- Zeltz, C.; Gullberg, D. The integrin-collagen connection—A glue for tissue repair? J. Cell Sci. 2016, 129, 653–664. [Google Scholar] [CrossRef]
- Teckchandani, A.; Mulkearns, E.E.; Randolph, T.W.; Toida, N.; Cooper, J.A. The clathrin adaptor dab2 recruits eh domain scaffold proteins to regulate integrin beta1 endocytosis. Mol. Biol. Cell 2012, 23, 2905–2916. [Google Scholar] [CrossRef]
- Eckert, M.A.; Lwin, T.M.; Chang, A.T.; Kim, J.; Danis, E.; Ohno-Machado, L.; Yang, J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011, 19, 372–386. [Google Scholar] [CrossRef]
- Maeda, M.; Johnson, K.R.; Wheelock, M.J. Cadherin switching: Essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J. Cell Sci. 2005, 118, 873–887. [Google Scholar] [CrossRef]
- Li, C.F.; Chen, J.Y.; Ho, Y.H.; Hsu, W.H.; Wu, L.C.; Lan, H.Y.; Hsu, D.S.; Tai, S.K.; Chang, Y.C.; Yang, M.H. Snail-induced claudin-11 prompts collective migration for tumour progression. Nat. Cell Biol. 2019, 21, 251–262. [Google Scholar] [CrossRef]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2019, 194, 161–184. [Google Scholar] [CrossRef]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for rhogtpases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Pearson, G.W.; Hunter, T. Real-time imaging reveals that noninvasive mammary epithelial acini can contain motile cells. J. Cell Biol. 2007, 179, 1555–1567. [Google Scholar] [CrossRef]
- Schliekelman, M.J.; Taguchi, A.; Zhu, J.; Dai, X.; Rodriguez, J.; Celiktas, M.; Zhang, Q.; Chin, A.; Wong, C.H.; Wang, H.; et al. Molecular portraits of epithelial, mesenchymal and hybrid states in lung adenocarcinoma and their relevance to survival. Cancer Res. 2015. [Google Scholar] [CrossRef]
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155, 1639–1651. [Google Scholar] [CrossRef]
- Dang, T.T.; Westcott, J.M.; Maine, E.A.; Kanchwala, M.; Xing, C.; Pearson, G.W. Deltanp63alpha induces the expression of fat2 and slug to promote tumor invasion. Oncotarget 2016, 7, 28592–28611. [Google Scholar] [CrossRef]
- Dang, T.T.; Esparza, M.A.; Maine, E.A.; Westcott, J.M.; Pearson, G.W. Deltanp63alpha promotes breast cancer cell motility through the selective activation of components of the epithelial-to-mesenchymal transition program. Cancer Res. 2015, 75, 3925–3935. [Google Scholar] [CrossRef]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef]
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. Microrna-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18144–18149. [Google Scholar] [CrossRef]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct emt programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef]
- Rios, A.C.; Capaldo, B.D.; Vaillant, F.; Pal, B.; van Ineveld, R.; Dawson, C.A.; Chen, Y.; Nolan, E.; Fu, N.Y.; Group, D.; et al. Intraclonal plasticity in mammary tumors revealed through large-scale single-cell resolution 3d imaging. Cancer Cell 2019, 35, 618–632. [Google Scholar] [CrossRef]
- Celia-Terrassa, T.; Meca-Cortes, O.; Mateo, F.; Martinez de Paz, A.; Rubio, N.; Arnal-Estape, A.; Ell, B.J.; Bermudo, R.; Diaz, A.; Guerra-Rebollo, M.; et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J. Clin. Investig. 2012, 122, 1849–1868. [Google Scholar] [CrossRef]
- Celia-Terrassa, T.; Bastian, C.; Liu, D.D.; Ell, B.; Aiello, N.M.; Wei, Y.; Zamalloa, J.; Blanco, A.M.; Hang, X.; Kunisky, D.; et al. Hysteresis control of epithelial-mesenchymal transition dynamics conveys a distinct program with enhanced metastatic ability. Nat. Commun. 2018, 9, 5005. [Google Scholar] [CrossRef]
- Calbo, J.; van Montfort, E.; Proost, N.; van Drunen, E.; Beverloo, H.B.; Meuwissen, R.; Berns, A. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell 2011, 19, 244–256. [Google Scholar] [CrossRef]
- Neelakantan, D.; Zhou, H.; Oliphant, M.U.J.; Zhang, X.; Simon, L.M.; Henke, D.M.; Shaw, C.A.; Wu, M.F.; Hilsenbeck, S.G.; White, L.D.; et al. EMT cells increase breast cancer metastasis via paracrine gli activation in neighbouring tumour cells. Nat. Commun. 2017, 8, 15773. [Google Scholar] [CrossRef]
- Husemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmuller, G.; et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. Emt and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in HER2(+) mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef]
- Hosseini, H.; Obradovic, M.M.S.; Hoffmann, M.; Harper, K.L.; Sosa, M.S.; Werner-Klein, M.; Nanduri, L.K.; Werno, C.; Ehrl, C.; Maneck, M.; et al. Early dissemination seeds metastasis in breast cancer. Nature 2016, 540, 552–558. [Google Scholar] [CrossRef]
- Rhim, A.D.; Thege, F.I.; Santana, S.M.; Lannin, T.B.; Saha, T.N.; Tsai, S.; Maggs, L.R.; Kochman, M.L.; Ginsberg, G.G.; Lieb, J.G.; et al. Detection of circulating pancreas epithelial cells in patients with pancreatic cystic lesions. Gastroenterology 2014, 146, 647–651. [Google Scholar] [CrossRef]
- Casasent, A.K.; Schalck, A.; Gao, R.; Sei, E.; Long, A.; Pangburn, W.; Casasent, T.; Meric-Bernstam, F.; Edgerton, M.E.; Navin, N.E. Multiclonal invasion in breast tumors identified by topographic single cell sequencing. Cell 2018, 172, 205–217. [Google Scholar] [CrossRef]
- Reichert, M.; Bakir, B.; Moreira, L.; Pitarresi, J.R.; Feldmann, K.; Simon, L.; Suzuki, K.; Maddipati, R.; Rhim, A.D.; Schlitter, A.M.; et al. Regulation of epithelial plasticity determines metastatic organotropism in pancreatic cancer. Dev. Cell 2018, 45, 696–711. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, 854–863. [Google Scholar] [CrossRef]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef]
- Ocana, O.H.; Corcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer PRRX1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Castano, Z.; San Juan, B.P.; Spiegel, A.; Pant, A.; DeCristo, M.J.; Laszewski, T.; Ubellacker, J.M.; Janssen, S.R.; Dongre, A.; Reinhardt, F.; et al. Il-1beta inflammatory response driven by primary breast cancer prevents metastasis-initiating cell colonization. Nat. Cell Biol. 2018, 20, 1084–1097. [Google Scholar] [CrossRef]
- Jolly, M.K.; Mani, S.A.; Levine, H. Hybrid epithelial/mesenchymal phenotype(s): The ‘fittest’ for metastasis? Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 151–157. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Aiello, N.M.; Brabletz, T.; Kang, Y.; Nieto, M.A.; Weinberg, R.A.; Stanger, B.Z. Upholding a role for EMT in pancreatic cancer metastasis. Nature 2017, 547, 7–8. [Google Scholar] [CrossRef]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a role for emt in breast cancer metastasis. Nature 2017, 547, 1–3. [Google Scholar] [CrossRef]



© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pearson, G.W. Control of Invasion by Epithelial-to-Mesenchymal Transition Programs during Metastasis. J. Clin. Med. 2019, 8, 646. https://doi.org/10.3390/jcm8050646
Pearson GW. Control of Invasion by Epithelial-to-Mesenchymal Transition Programs during Metastasis. Journal of Clinical Medicine. 2019; 8(5):646. https://doi.org/10.3390/jcm8050646
Chicago/Turabian StylePearson, Gray W. 2019. "Control of Invasion by Epithelial-to-Mesenchymal Transition Programs during Metastasis" Journal of Clinical Medicine 8, no. 5: 646. https://doi.org/10.3390/jcm8050646
APA StylePearson, G. W. (2019). Control of Invasion by Epithelial-to-Mesenchymal Transition Programs during Metastasis. Journal of Clinical Medicine, 8(5), 646. https://doi.org/10.3390/jcm8050646