XPO1 Expression Is a Poor-Prognosis Marker in Pancreatic Adenocarcinoma


Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Expression Data Sets
2.2. Gene Expression Data Analysis
2.3. Statistical Analysis
3. Results
3.1. Patients’ Population
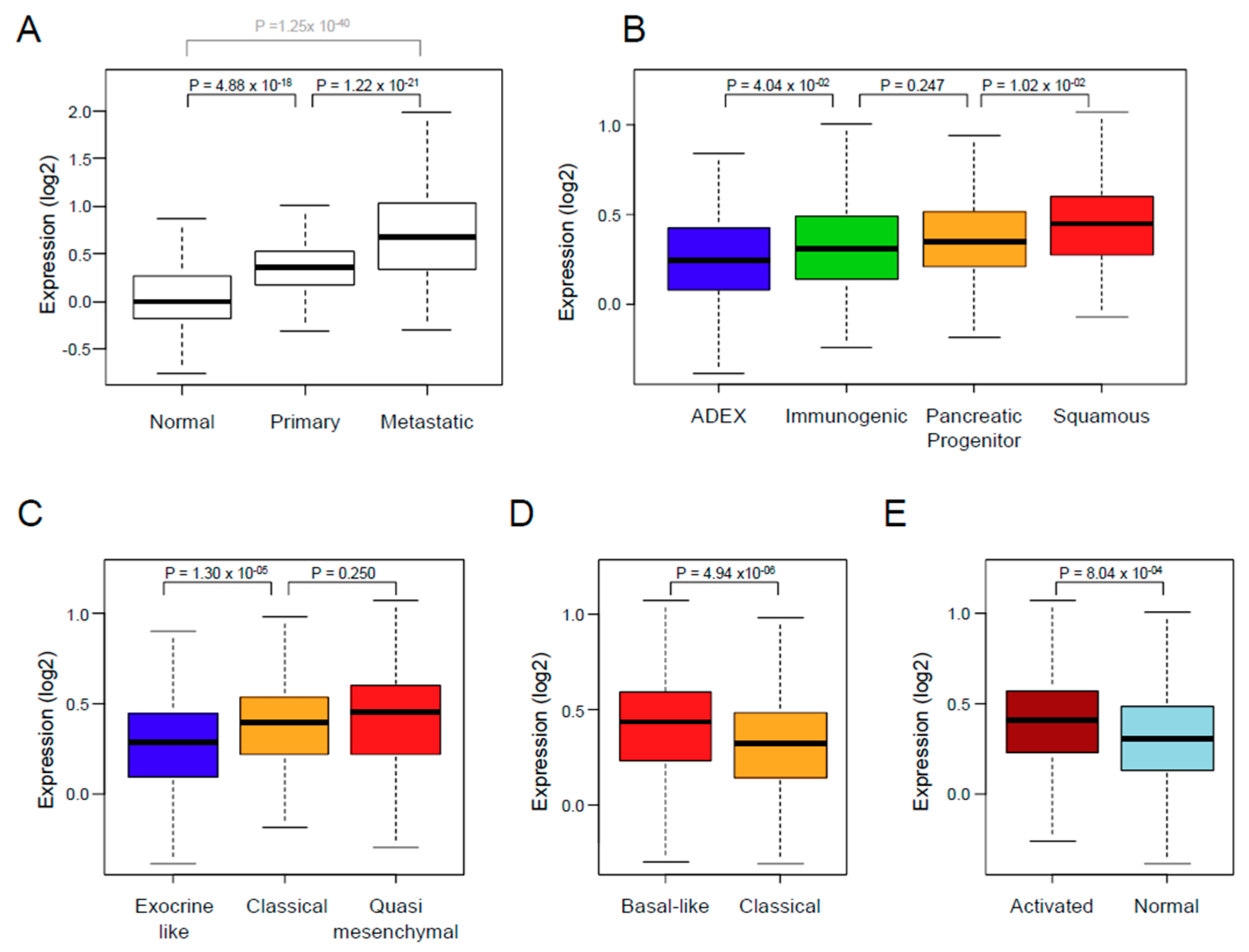
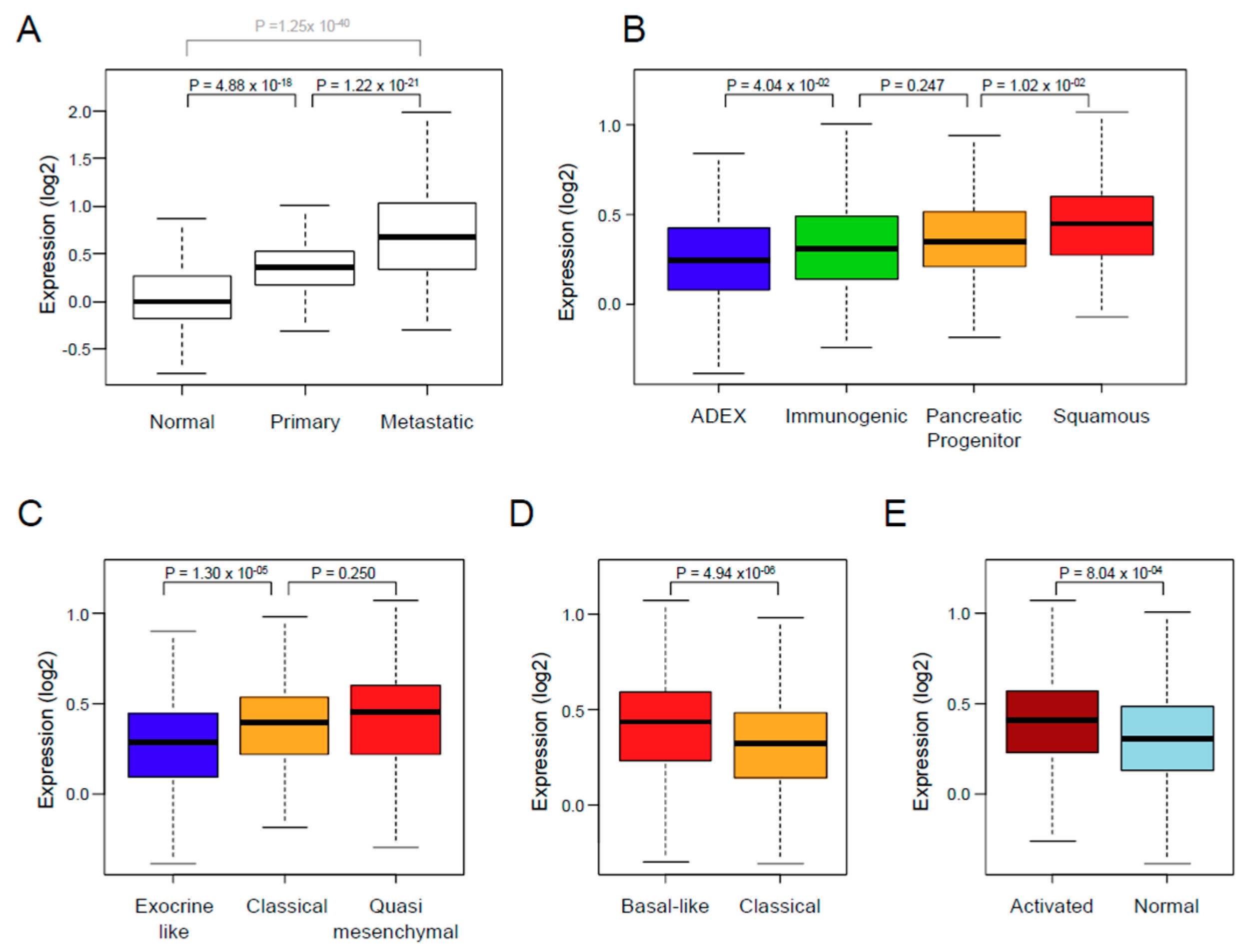
3.2. XPO1 Expression and Clinicopathological Features
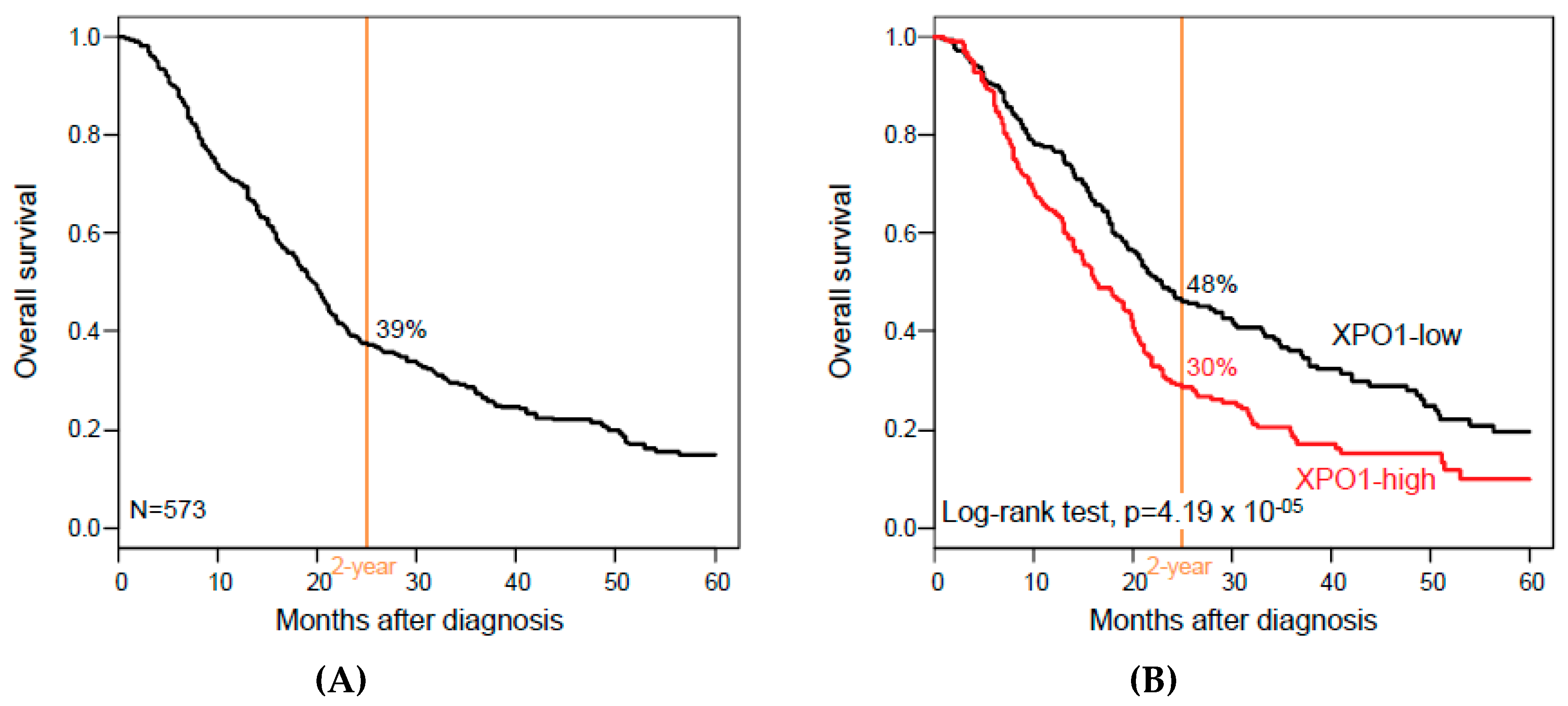
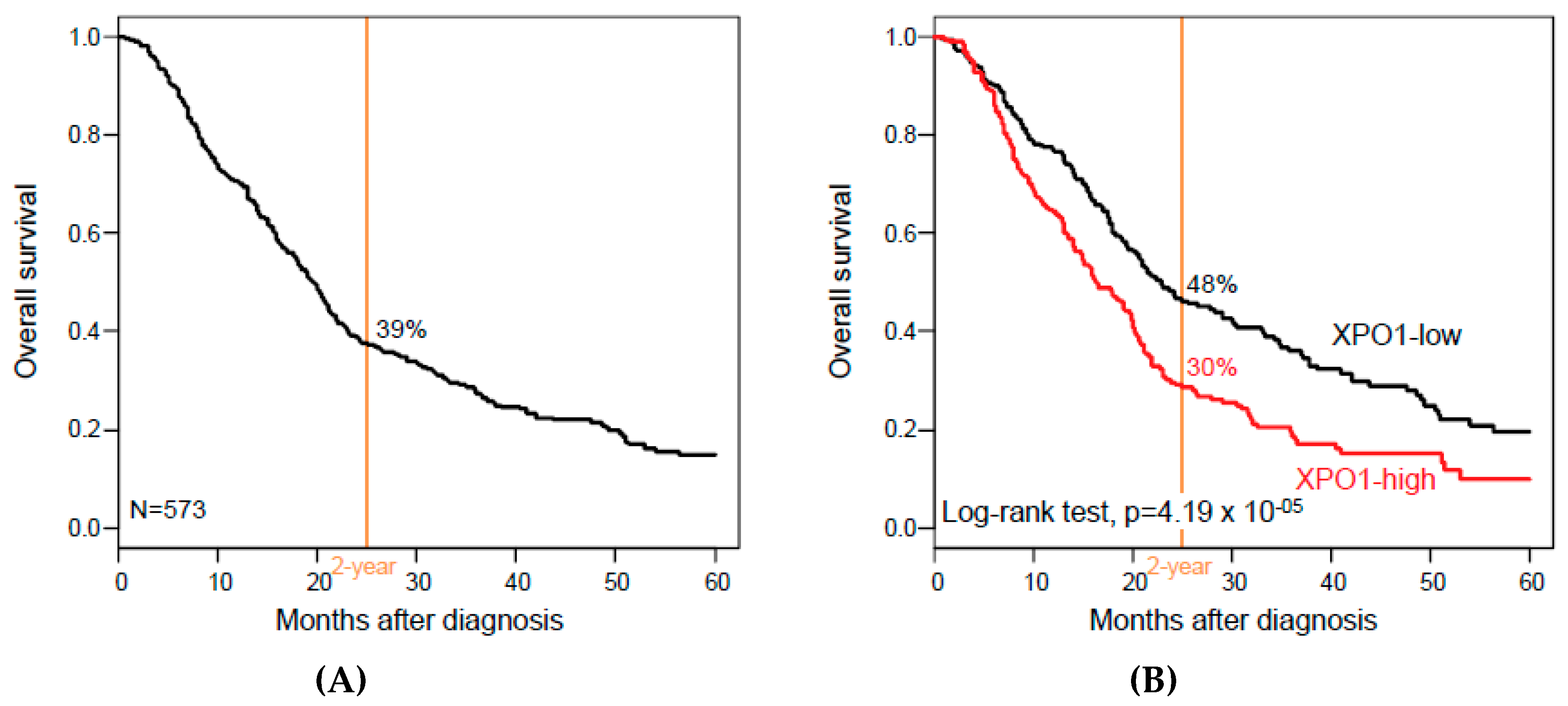
3.3. XPO1 Expression and Overall Survival
3.4. XPO1 Expression and Associated Biological Processes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Witkowski, E.R.; Smith, J.K.; Tseng, J.F. Outcomes following resection of pancreatic cancer. J. Surg. Oncol. 2013, 107, 97–103. [Google Scholar] [CrossRef]
- Hoem, D.; Viste, A. Improving survival following surgery for pancreatic ductal adenocarcinoma—A ten-year experience. Eur. J. Surg. Oncol. 2012, 38, 245–251. [Google Scholar] [CrossRef]
- Birnbaum, D.J.; Adelaide, J.; Mamessier, E.; Finetti, P.; Lagarde, A.; Monges, G.; Viret, F.; Goncalves, A.; Turrini, O.; Delpero, J.R.; et al. Genome profiling of pancreatic adenocarcinoma. Genes Chromosomes Cancer 2011, 50, 456–465. [Google Scholar] [CrossRef]
- Birnbaum, D.J.; Birnbaum, D.; Bertucci, F. Endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2011, 364, 483–484. [Google Scholar] [CrossRef]
- Harada, T.; Okita, K.; Shiraishi, K.; Kusano, N.; Kondoh, S.; Sasaki, K. Interglandular cytogenetic heterogeneity detected by comparative genomic hybridization in pancreatic cancer. Cancer Res. 2002, 62, 835–839. [Google Scholar]
- Harada, T.; Shiraishi, K.; Kusano, N.; Umayahara, K.; Kondoh, S.; Okita, K.; Sasaki, K. Evaluation of the reliability of chromosomal imbalances detected by combined use of universal DNA amplification and comparative genomic hybridization. Jpn. J. Cancer Res. 2000, 91, 1119–1125. [Google Scholar] [CrossRef]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53, 549–554. [Google Scholar] [CrossRef]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Kwei, K.A.; Bashyam, M.D.; Kao, J.; Ratheesh, R.; Reddy, E.C.; Kim, Y.H.; Montgomery, K.; Giacomini, C.P.; Choi, Y.L.; Chatterjee, S.; et al. Genomic profiling identifies GATA6 as a candidate oncogene amplified in pancreatobiliary cancer. PLoS Genet. 2008, 4, e1000081. [Google Scholar] [CrossRef]
- Fukuda, M.; Asano, S.; Nakamura, T.; Adachi, M.; Yoshida, M.; Yanagida, M.; Nishida, E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 1997, 390, 308–311. [Google Scholar] [CrossRef]
- Conforti, F.; Wang, Y.; Rodriguez, J.A.; Alberobello, A.T.; Zhang, Y.W.; Giaccone, G. Molecular Pathways: Anticancer Activity by Inhibition of Nucleocytoplasmic Shuttling. Clin. Cancer Res. 2015, 21, 4508–4513. [Google Scholar] [CrossRef]
- Tan, D.S.; Bedard, P.L.; Kuruvilla, J.; Siu, L.L.; Razak, A.R. Promising SINEs for embargoing nuclear-cytoplasmic export as an anticancer strategy. Cancer Discov. 2014, 4, 527–537. [Google Scholar] [CrossRef]
- Okamura, M.; Inose, H.; Masuda, S. RNA Export through the NPC in Eukaryotes. Genes 2015, 6, 124–149. [Google Scholar] [CrossRef]
- Liu, X.; Niu, M.; Xu, X.; Cai, W.; Zeng, L.; Zhou, X.; Yu, R.; Xu, K. CRM1 is a direct cellular target of the natural anti-cancer agent plumbagin. J. Pharmacol. Sci. 2014, 124, 486–493. [Google Scholar] [CrossRef]
- Yao, Y.; Dong, Y.; Lin, F.; Zhao, H.; Shen, Z.; Chen, P.; Sun, Y.J.; Tang, L.N.; Zheng, S.E. The expression of CRM1 is associated with prognosis in human osteosarcoma. Oncol. Rep. 2009, 21, 229–235. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Sanita, P.; Vitale, F.; Marampon, F.; Ventura, L.; Landesman, Y.; McCauley, D.; Kauffman, M.; Shacham, S.; et al. KPT-330, a potent and selective exportin-1 (XPO-1) inhibitor, shows antitumor effects modulating the expression of cyclin D1 and survivin [corrected] in prostate cancer models. BMC Cancer 2015, 15, 941. [Google Scholar] [CrossRef]
- Van der Watt, P.J.; Maske, C.P.; Hendricks, D.T.; Parker, M.I.; Denny, L.; Govender, D.; Birrer, M.J.; Leaner, V.D. The Karyopherin proteins, Crm1 and Karyopherin beta1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int. J. Cancer 2009, 124, 1829–1840. [Google Scholar] [CrossRef]
- Noske, A.; Weichert, W.; Niesporek, S.; Roske, A.; Buckendahl, A.C.; Koch, I.; Sehouli, J.; Dietel, M.; Denkert, C. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer 2008, 112, 1733–1743. [Google Scholar] [CrossRef]
- Shen, A.; Wang, Y.; Zhao, Y.; Zou, L.; Sun, L.; Cheng, C. Expression of CRM1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery 2009, 65, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Y.; Yue, L.; Qiu, W.S.; Wang, L.W.; Zhou, X.H.; Sun, Y.J. Prognostic value of CRM1 in pancreas cancer. Clin. Investig. Med. 2009, 32, E315. [Google Scholar] [CrossRef]
- Zhou, F.; Qiu, W.; Yao, R.; Xiang, J.; Sun, X.; Liu, S.; Lv, J.; Yue, L. CRM1 is a novel independent prognostic factor for the poor prognosis of gastric carcinomas. Med. Oncol. 2013, 30, 726. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Landesman, Y.; Acharya, C.; Calle, Y.; Zhong, M.Y.; Cea, M.; Tannenbaum, D.; Cagnetta, A.; Reagan, M.; Munshi, A.A.; et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: Molecular mechanisms and therapeutic implications. Leukemia 2014, 28, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Kornblau, S.M.; Ruvolo, V.; Dilip, A.; Duvvuri, S.; Davis, R.E.; Zhang, M.; Wang, Z.; Coombes, K.R.; Zhang, N.; et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood 2013, 121, 4166–4174. [Google Scholar] [CrossRef]
- Kuruvilla, J.; Savona, M.; Baz, R.; Mau-Sorensen, P.M.; Gabrail, N.; Garzon, R.; Stone, R.; Wang, M.; Savoie, L.; Martin, P.; et al. Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin lymphoma. Blood 2017, 129, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Wolff, B.; Sekimoto, T.; Schreiner, E.P.; Yoneda, Y.; Yanagida, M.; Horinouchi, S.; Yoshida, M. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 1998, 242, 540–547. [Google Scholar] [CrossRef]
- Newlands, E.S.; Rustin, G.J.; Brampton, M.H. Phase I trial of elactocin. Br. J. Cancer 1996, 74, 648–649. [Google Scholar] [CrossRef]
- Niu, M.; Wu, S.; Mao, L.; Yang, Y. CRM1 is a cellular target of curcumin: New insights for the myriad of biological effects of an ancient spice. Traffic 2013, 14, 1042–1052. [Google Scholar] [CrossRef]
- Sakakibara, K.; Saito, N.; Sato, T.; Suzuki, A.; Hasegawa, Y.; Friedman, J.M.; Kufe, D.W.; Vonhoff, D.D.; Iwami, T.; Kawabe, T. CBS9106 is a novel reversible oral CRM1 inhibitor with CRM1 degrading activity. Blood 2011, 118, 3922–3931. [Google Scholar] [CrossRef]
- Niu, M.; Chong, Y.; Han, Y.; Liu, X. Novel reversible selective inhibitor of nuclear export shows that CRM1 is a target in colorectal cancer cells. Cancer Biol. Ther. 2015, 16, 1110–1118. [Google Scholar] [CrossRef]
- Kanbur, T.; Kara, M.; Kutluer, M.; Sen, A.; Delman, M.; Alkan, A.; Otas, H.O.; Akcok, I.; Cagir, A. CRM1 inhibitory and antiproliferative activities of novel 4’-alkyl substituted klavuzon derivatives. Bioorg. Med. Chem. 2017, 25, 4444–4451. [Google Scholar] [CrossRef] [PubMed]
- Etchin, J.; Sun, Q.; Kentsis, A.; Farmer, A.; Zhang, Z.C.; Sanda, T.; Mansour, M.R.; Barcelo, C.; McCauley, D.; Kauffman, M.; et al. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia 2013, 27, 66–74. [Google Scholar] [CrossRef]
- Lapalombella, R.; Sun, Q.; Williams, K.; Tangeman, L.; Jha, S.; Zhong, Y.; Goettl, V.; Mahoney, E.; Berglund, C.; Gupta, S.; et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood 2012, 120, 4621–4634. [Google Scholar] [CrossRef]
- Van Neck, T.; Pannecouque, C.; Vanstreels, E.; Stevens, M.; Dehaen, W.; Daelemans, D. Inhibition of the CRM1-mediated nucleocytoplasmic transport by N-azolylacrylates: Structure-activity relationship and mechanism of action. Bioorg. Med. Chem. 2008, 16, 9487–9497. [Google Scholar] [CrossRef]
- Gerecitano, J. SINE (selective inhibitor of nuclear export)—Translational science in a new class of anti-cancer agents. J. Hematol. Oncol. 2014, 7, 67. [Google Scholar] [CrossRef]
- Jardin, F.; Pujals, A.; Pelletier, L.; Bohers, E.; Camus, V.; Mareschal, S.; Dubois, S.; Sola, B.; Ochmann, M.; Lemonnier, F.; et al. Recurrent mutations of the exportin 1 gene (XPO1) and their impact on selective inhibitor of nuclear export compounds sensitivity in primary mediastinal B-cell lymphoma. Am. J. Hematol. 2016, 91, 923–930. [Google Scholar] [CrossRef]
- Kashyap, T.; Argueta, C.; Aboukameel, A.; Unger, T.J.; Klebanov, B.; Mohammad, R.M.; Muqbil, I.; Azmi, A.S.; Drolen, C.; Senapedis, W.; et al. Selinexor, a Selective Inhibitor of Nuclear Export (SINE) compound, acts through NF-kappaB deactivation and combines with proteasome inhibitors to synergistically induce tumor cell death. Oncotarget 2016, 7, 78883–78895. [Google Scholar] [CrossRef] [PubMed]
- Muqbil, I.; Aboukameel, A.; Elloul, S.; Carlson, R.; Senapedis, W.; Baloglu, E.; Kauffman, M.; Shacham, S.; Bhutani, D.; Zonder, J.; et al. Anti-tumor activity of selective inhibitor of nuclear export (SINE) compounds, is enhanced in non-Hodgkin lymphoma through combination with mTOR inhibitor and dexamethasone. Cancer Lett. 2016, 383, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Abdul Razak, A.R.; Mau-Soerensen, M.; Gabrail, N.Y.; Gerecitano, J.F.; Shields, A.F.; Unger, T.J.; Saint-Martin, J.R.; Carlson, R.; Landesman, Y.; McCauley, D.; et al. First-in-Class, First-in-Human Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4142–4150. [Google Scholar] [CrossRef] [PubMed]
- Conforti, F.; Zhang, X.; Rao, G.; De Pas, T.; Yonemori, Y.; Rodriguez, J.A.; McCutcheon, J.N.; Rahhal, R.; Alberobello, A.T.; Wang, Y.; et al. Therapeutic Effects of XPO1 Inhibition in Thymic Epithelial Tumors. Cancer Res. 2017, 77, 5614–5627. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Chen, W.; Zhong, Y.; Hou, X.; Fang, S.; Liu, C.Y.; Wang, G.; Yu, T.; Huang, Y.Y.; Ouyang, X.; et al. Nuclear Export of Ubiquitinated Proteins Determines the Sensitivity of Colorectal Cancer to Proteasome Inhibitor. Mol. Cancer Ther. 2017, 16, 717–728. [Google Scholar] [CrossRef]
- Chen, Y.; Camacho, S.C.; Silvers, T.R.; Razak, A.R.; Gabrail, N.Y.; Gerecitano, J.F.; Kalir, E.; Pereira, E.; Evans, B.R.; Ramus, S.J.; et al. Inhibition of the Nuclear Export Receptor XPO1 as a Therapeutic Target for Platinum-Resistant Ovarian Cancer. Clin. Cancer Res. 2017, 23, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, R.; Zhang, Y.X.; Czaplinski, J.T.; Anatone, A.J.; Sicinska, E.T.; Fletcher, J.A.; Demetri, G.D.; Wagner, A.J. Preclinical activity of selinexor, an inhibitor of XPO1, in sarcoma. Oncotarget 2016, 7, 16581–16592. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Hattori, N.; Chien, W.; Sun, Q.; Sudo, M.; GL, E.L.; Ding, L.; Lim, S.L.; Shacham, S.; Kauffman, M.; et al. KPT-330 has antitumour activity against non-small cell lung cancer. Br. J. Cancer 2014, 111, 281–291. [Google Scholar] [CrossRef]
- Azmi, A.S.; Aboukameel, A.; Bao, B.; Sarkar, F.H.; Philip, P.A.; Kauffman, M.; Shacham, S.; Mohammad, R.M. Selective inhibitors of nuclear export block pancreatic cancer cell proliferation and reduce tumor growth in mice. Gastroenterology 2013, 144, 447–456. [Google Scholar] [CrossRef]
- Gao, J.; Azmi, A.S.; Aboukameel, A.; Kauffman, M.; Shacham, S.; Abou-Samra, A.B.; Mohammad, R.M. Nuclear retention of Fbw7 by specific inhibitors of nuclear export leads to Notch1 degradation in pancreatic cancer. Oncotarget 2014, 5, 3444–3454. [Google Scholar] [CrossRef]
- Kazim, S.; Malafa, M.P.; Coppola, D.; Husain, K.; Zibadi, S.; Kashyap, T.; Crochiere, M.; Landesman, Y.; Rashal, T.; Sullivan, D.M.; et al. Selective Nuclear Export Inhibitor KPT-330 Enhances the Antitumor Activity of Gemcitabine in Human Pancreatic Cancer. Mol. Cancer Ther. 2015, 14, 1570–1581. [Google Scholar] [CrossRef]
- Azmi, A.S.; Li, Y.; Muqbil, I.; Aboukameel, A.; Senapedis, W.; Baloglu, E.; Landesman, Y.; Shacham, S.; Kauffman, M.G.; Philip, P.A.; et al. Exportin 1 (XPO1) inhibition leads to restoration of tumor suppressor miR-145 and consequent suppression of pancreatic cancer cell proliferation and migration. Oncotarget 2017, 8, 82144–82155. [Google Scholar] [CrossRef]
- Kashyap, T.; Argueta, C.; Unger, T.; Klebanov, B.; Debler, S.; Senapedis, W.; Crochiere, M.L.; Lee, M.S.; Kauffman, M.; Shacham, S.; et al. Selinexor reduces the expression of DNA damage repair proteins and sensitizes cancer cells to DNA damaging agents. Oncotarget 2018, 9, 30773–30786. [Google Scholar] [CrossRef]
- Saulino, D.M.; Younes, P.S.; Bailey, J.M.; Younes, M. CRM1/XPO1 expression in pancreatic adenocarcinoma correlates with survivin expression and the proliferative activity. Oncotarget 2018, 9, 21289–21295. [Google Scholar] [CrossRef]
- Mahipal, A.; Coppola, D.; Gupta, S.; Centeno, B.; Malafa, M.P. CRM1 expression in pancreatic carcinoma. J. Clin. Oncol. 2013, 31, e15115. [Google Scholar] [CrossRef]
- Van den Broeck, A.; Vankelecom, H.; Van Eijsden, R.; Govaere, O.; Topal, B. Molecular markers associated with outcome and metastasis in human pancreatic cancer. J. Exp. Clin. Cancer Res. 2012, 31, 68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; He, P.; Tan, H.; Budhu, A.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Yfantis, H.G.; Lee, D.H.; Maitra, A.; et al. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin. Cancer Res. 2013, 19, 4983–4993. [Google Scholar] [CrossRef]
- Zhang, G.; Schetter, A.; He, P.; Funamizu, N.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Hassan, R.; Yfantis, H.G.; Lee, D.H.; et al. DPEP1 inhibits tumor cell invasiveness, enhances chemosensitivity and predicts clinical outcome in pancreatic ductal adenocarcinoma. PLoS ONE 2012, 7, e31507. [Google Scholar] [CrossRef] [PubMed]
- Stratford, J.K.; Bentrem, D.J.; Anderson, J.M.; Fan, C.; Volmar, K.A.; Marron, J.S.; Routh, E.D.; Caskey, L.S.; Samuel, J.C.; Der, C.J.; et al. A six-gene signature predicts survival of patients with localized pancreatic ductal adenocarcinoma. PLoS Med. 2010, 7, e1000307. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.T.; Davis-Yadley, A.H.; Huang, P.Y.; Husain, K.; Centeno, B.A.; Permuth-Wey, J.; Pimiento, J.M.; Malafa, M. Prognostic Fifteen-Gene Signature for Early Stage Pancreatic Ductal Adenocarcinoma. PLoS ONE 2015, 10, e0133562. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.K.; Ramaker, R.C.; Gertz, J.; Davis, N.S.; Johnston, B.E.; Oliver, P.G.; Sexton, K.C.; Greeno, E.W.; Christein, J.D.; Heslin, M.J.; et al. RNA sequencing of pancreatic adenocarcinoma tumors yields novel expression patterns associated with long-term survival and reveals a role for ANGPTL4. Mol. Oncol. 2016, 10, 1169–1182. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef]
- Birnbaum, D.J.; Finetti, P.; Lopresti, A.; Gilabert, M.; Poizat, F.; Raoul, J.L.; Delpero, J.R.; Moutardier, V.; Birnbaum, D.; Mamessier, E.; et al. A 25-gene classifier predicts overall survival in resectable pancreatic cancer. BMC Med. 2017, 15, 170. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3. [Google Scholar] [CrossRef]
- Hochberg, Y.; Benjamini, Y. More powerful procedures for multiple significance testing. Stat. Med. 1990, 9, 811–818. [Google Scholar] [CrossRef]
- McShane, L.M.; Altman, D.G.; Sauerbrei, W.; Taube, S.E.; Gion, M.; Clark, G.M. REporting recommendations for tumour MARKer prognostic studies (REMARK). Br. J. Cancer 2005, 93, 387–391. [Google Scholar] [CrossRef]
- Jain, P.; Kanagal-Shamanna, R.; Wierda, W.; Keating, M.; Sarwari, N.; Rozovski, U.; Thompson, P.; Burger, J.; Kantarjian, H.; Patel, K.P.; et al. Clinical and molecular characteristics of XPO1 mutations in patients with chronic lymphocytic leukemia. Am. J. Hematol. 2016, 91, E478–E479. [Google Scholar] [CrossRef]
- Van der Watt, P.J.; Zemanay, W.; Govender, D.; Hendricks, D.T.; Parker, M.I.; Leaner, V.D. Elevated expression of the nuclear export protein, Crm1 (exportin 1), associates with human oesophageal squamous cell carcinoma. Oncol. Rep. 2014, 32, 730–738. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Charateristics | N | Global (N = 573) | XPO1 ‘Low’ (N = 275) | XPO1 ‘High’ (N = 298) | p-Value |
|---|---|---|---|---|---|
| Age at diagnosis (years) | 0.348 | ||||
| ≤60 | 106 | 106 (32%) | 47 (29%) | 59 (34%) | |
| >60 | 230 | 230 (68%) | 116 (71%) | 114 (66%) | |
| Sex | 0.159 | ||||
| female | 159 | 159 (47%) | 83 (51%) | 76 (43%) | |
| male | 180 | 180 (53%) | 80 (49%) | 100 (57%) | |
| AJCC Stage | 0.507 | ||||
| 1 | 53 | 53 (11%) | 30 (13%) | 23 (9%) | |
| 2 | 413 | 413 (85%) | 191 (83%) | 222 (86%) | |
| 3 | 10 | 10 (2%) | 4 (2%) | 6 (2%) | |
| 4 | 12 | 12 (2%) | 6 (3%) | 6 (2%) | |
| Pathological type | 0.853 | ||||
| ductal | 401 | 401 (93%) | 196 (92%) | 205 (93%) | |
| other | 31 | 31 (7%) | 16 (8%) | 15 (7%) | |
| Pathological grade | 0.179 | ||||
| 1 | 23 | 23 (9%) | 13 (10%) | 10 (8%) | |
| 2 | 144 | 144 (57%) | 78 (61%) | 66 (52%) | |
| 3 | 85 | 85 (33%) | 37 (29%) | 48 (38%) | |
| 4 | 2 | 2 (1%) | 0 (0%) | 2 (2%) | |
| Pathological tumor size (pT) | 0.538 | ||||
| pT1 | 16 | 16 (4%) | 8 (5%) | 8 (4%) | |
| pT2 | 54 | 54 (15%) | 30 (17%) | 24 (12%) | |
| pT3 | 285 | 285 (78%) | 132 (76%) | 153 (80%) | |
| pT4 | 11 | 11 (3%) | 4 (2%) | 7 (4%) | |
| Pathological lymph node status (pN) | 1.97 × 10−2 | ||||
| negative | 128 | 128 (30%) | 73 (36%) | 55 (25%) | |
| positive | 296 | 296 (70%) | 131 (64%) | 165 (75%) | |
| Collisson subtypes | 9.07 × 10−4 | ||||
| classical | 257 | 257 (45%) | 114 (41%) | 143 (48%) | |
| exocrine-like | 199 (35%) | 116 (42%) | 83 (28%) | ||
| quasi-mesenchymal | 117 | 117 (20%) | 45 (16%) | 72 (24%) | |
| Moffit subtypes, ‘tumor’ | 4.55 × 10−4 | ||||
| basal-like | 229 | 229 (40%) | 89 (32%) | 140 (47%) | |
| classical | 344 | 344 (60%) | 186 (68%) | 158 (53%) | |
| Moffit subtypes, ‘stroma’ | 4.69 × 10−4 | ||||
| Activated | 324 | 324 (59%) | 132 (51%) | 192 (66%) | |
| Normal | 222 | 222 (41%) | 125 (49%) | 97 (34%) | |
| Bailey subtypes | 3.20 × 10−5 | ||||
| ADEX | 124 | 124 (22%) | 77 (28%) | 47 (16%) | |
| immunogenic | 99 | 99 (17%) | 53 (19%) | 46 (15%) | |
| pancreatic progenitor | 141 | 141 (25%) | 70 (25%) | 71 (24%) | |
| squamous | 209 | 209 (36%) | 75 (27%) | 134 (45%) | |
| Follow-up, months (range) | 573 | 16 (1-156) | 17 (1-156) | 13 (1-126) | 0.08 |
| 2-year OS (95% CI) | 573 | 39% (35–44) | 48% (42–56) | 30% (24–36) | 4.19 × 10−5 |
| Median OS, months (range) | 573 | 19 (1–156) | 23 (1–156) | 16 (1–126) | 1.67 × 10−3 |
| Characteristics | Univariate | Multivariate | Multivariate | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | HR (95% CI) | p-Value | N | HR (95% CI) | p-Value | N | HR (95% CI) | p-Value | ||
| Age at diagnosis (years) | >60 vs. ≤60 | 336 | 1.14 (0.83–1.56) | 0.410 | ||||||
| Sex | male vs. female | 339 | 1.12 (0.85–1.49) | 0.421 | ||||||
| AJCC Stage | 2 vs. 1 | 488 | 2.00 (1.32–3.02) | 3.98 × 10−3 | 417 | 1.46 (0.83–2.55) | 0.188 | |||
| 3 vs. 1 | 2.95 (1.27–6.83) | 417 | 1.46 (0.54–3.94) | 0.454 | ||||||
| 4 vs. 1 | 2.96 (1.27–6.90) | 417 | 0.98 (0.13–7.37) | 0.983 | ||||||
| Pathological type | other vs. ductal | 432 | 0.94 (0.54–1.64) | 0.822 | ||||||
| Pathological grade | 2 vs. 1 | 254 | 1.69 (0.67–4.23) | 0.056 | ||||||
| 3 vs. 1 | 2.58 (1.02–6.51) | |||||||||
| 4 vs. 1 | 2.92 (0.56–15.2) | |||||||||
| Pathological tumor size (pT) | 2 vs. 1 | 366 | 1.92 (0.80–4.61) | 0.097 | ||||||
| 3 vs. 1 | 2.35 (1.04–5.32) | |||||||||
| 4 vs. 1 | 3.40 (1.18–9.82) | |||||||||
| Pathological lymph node status (pN) | 1 vs. 0 | 424 | 1.85 (1.38–2.48) | 3.80 × 10−5 | 417 | 1.51 (1.07–2.13) | 1.87 × 10–2 | |||
| Collisson subtypes | exocrine-like vs. classical | 573 | 0.99 (0.77–1.26) | 7.62 × 10−3 | 546 | 1.00 (0.71–1.41) | 0.997 | |||
| quasi-mesenchymal vs. classical | 1.47 (1.12–1.91) | 546 | 0.93 (0.67–1.30) | 0.674 | ||||||
| Moffit subtypes, ‘tumor’ | classical vs. basal-like | 573 | 0.63 (0.51–0.77) | 1.69 × 10−5 | 546 | 1.01 (0.74–1.38) | 0.961 | |||
| Moffit subtypes, ‘stroma’ | normal vs. activated | 546 | 0.67 (0.53–0.84) | 4.33 × 10−4 | 546 | 0.79 (0.62–1.01) | 0.060 | |||
| Bailey subtypes | immunogenic vs. ADEX | 573 | 0.89 (0.62–1.26) | 1.26 × 10−6 | 546 | 0.87 (0.55–1.38) | 0.566 | |||
| pancreatic progenitor vs. ADEX | 0.98 (0.71–1.35) | 546 | 0.95 (0.62–1.45) | 0.805 | ||||||
| squamous vs. ADEX | 1.74 (1.31–2.32) | 546 | 1.57 (1.02–2.43) | 4.24 × 10−2 | ||||||
| XPO1 | high vs. low | 573 | 1.56 (1.26–1.93) | 4.78 × 10−5 | 417 | 1.6 (1.23–2.09) | 5.07 × 10−4 | 546 | 1.49 (1.18–1.87) | 6.68 × 10−4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birnbaum, D.J.; Finetti, P.; Birnbaum, D.; Mamessier, E.; Bertucci, F. XPO1 Expression Is a Poor-Prognosis Marker in Pancreatic Adenocarcinoma. J. Clin. Med. 2019, 8, 596. https://doi.org/10.3390/jcm8050596
Birnbaum DJ, Finetti P, Birnbaum D, Mamessier E, Bertucci F. XPO1 Expression Is a Poor-Prognosis Marker in Pancreatic Adenocarcinoma. Journal of Clinical Medicine. 2019; 8(5):596. https://doi.org/10.3390/jcm8050596
Chicago/Turabian StyleBirnbaum, David Jérémie, Pascal Finetti, Daniel Birnbaum, Emilie Mamessier, and François Bertucci. 2019. "XPO1 Expression Is a Poor-Prognosis Marker in Pancreatic Adenocarcinoma" Journal of Clinical Medicine 8, no. 5: 596. https://doi.org/10.3390/jcm8050596
APA StyleBirnbaum, D. J., Finetti, P., Birnbaum, D., Mamessier, E., & Bertucci, F. (2019). XPO1 Expression Is a Poor-Prognosis Marker in Pancreatic Adenocarcinoma. Journal of Clinical Medicine, 8(5), 596. https://doi.org/10.3390/jcm8050596