Gene Expression Changes Associated with Nintedanib Treatment in Idiopathic Pulmonary Fibrosis Fibroblasts: A Next-Generation Sequencing and Bioinformatics Study

 ,
,  ,
,
Abstract
1. Introduction
2. Experimental Section
2.1. Study Design
2.2. Cultures of IPF Lung Fibroblasts
2.3. Nintedanib Treatment
2.4. Cell Morphological Observation
2.5. Cell Proliferation Assay
2.6. Cell Apoptosis Assay
2.7. Next-Generation Sequencing (NGS) for miRNA and mRNA Expression Profiling
2.8. miRmap Database Analysis
2.9. TargetScan Database Analysis
2.10. miRDB Database Analysis
2.11. STRING Database Analysis
2.12. DAVID Database Analysis
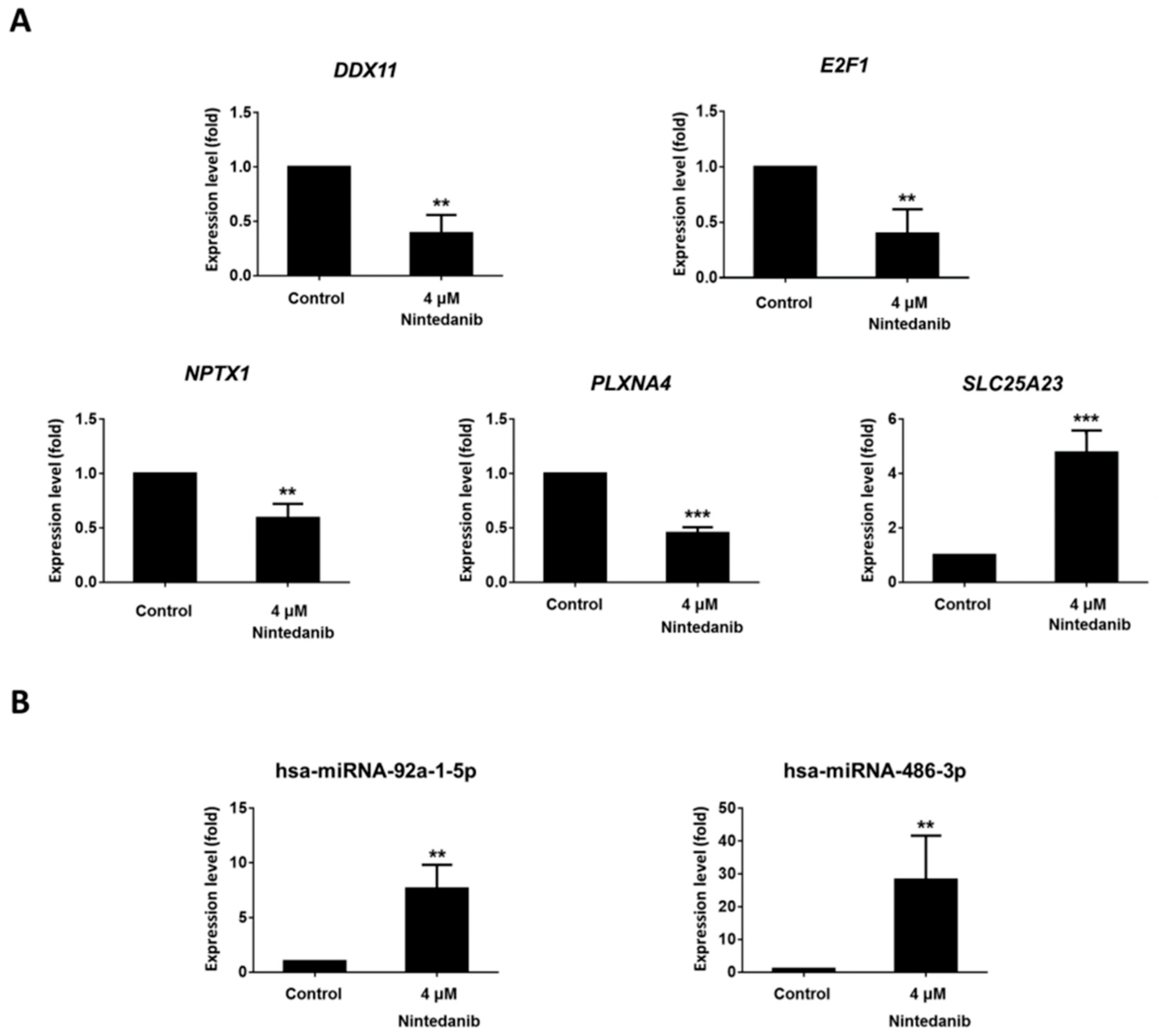
2.13. RT-qPCR
3. Results
3.1. Effects of Nintedanib on Cell Morphology, Proliferation, and Apoptosis in IPF Fibroblasts
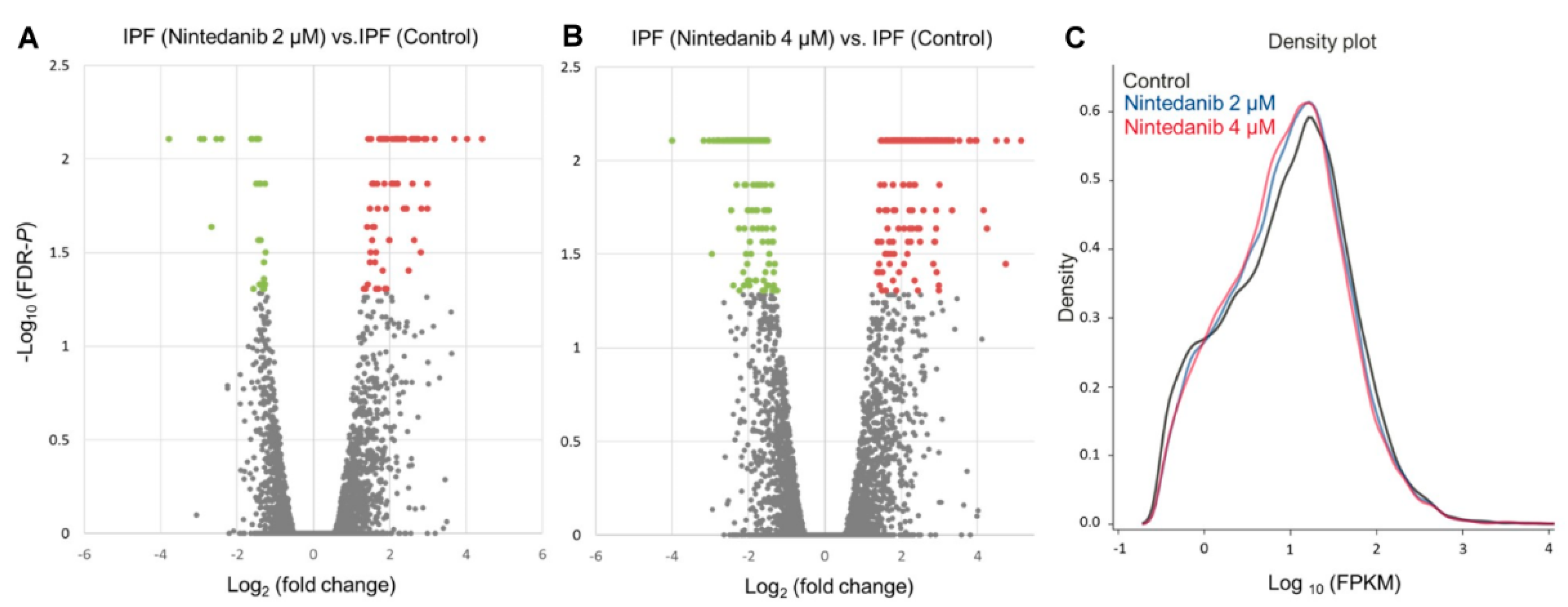
3.2. Overview of Differential Gene Expressions in Nintedanib-Treated IPF Fibroblasts
3.3. Protein–Protein Interaction, Biological Pathway, and Molecular Function Analysis
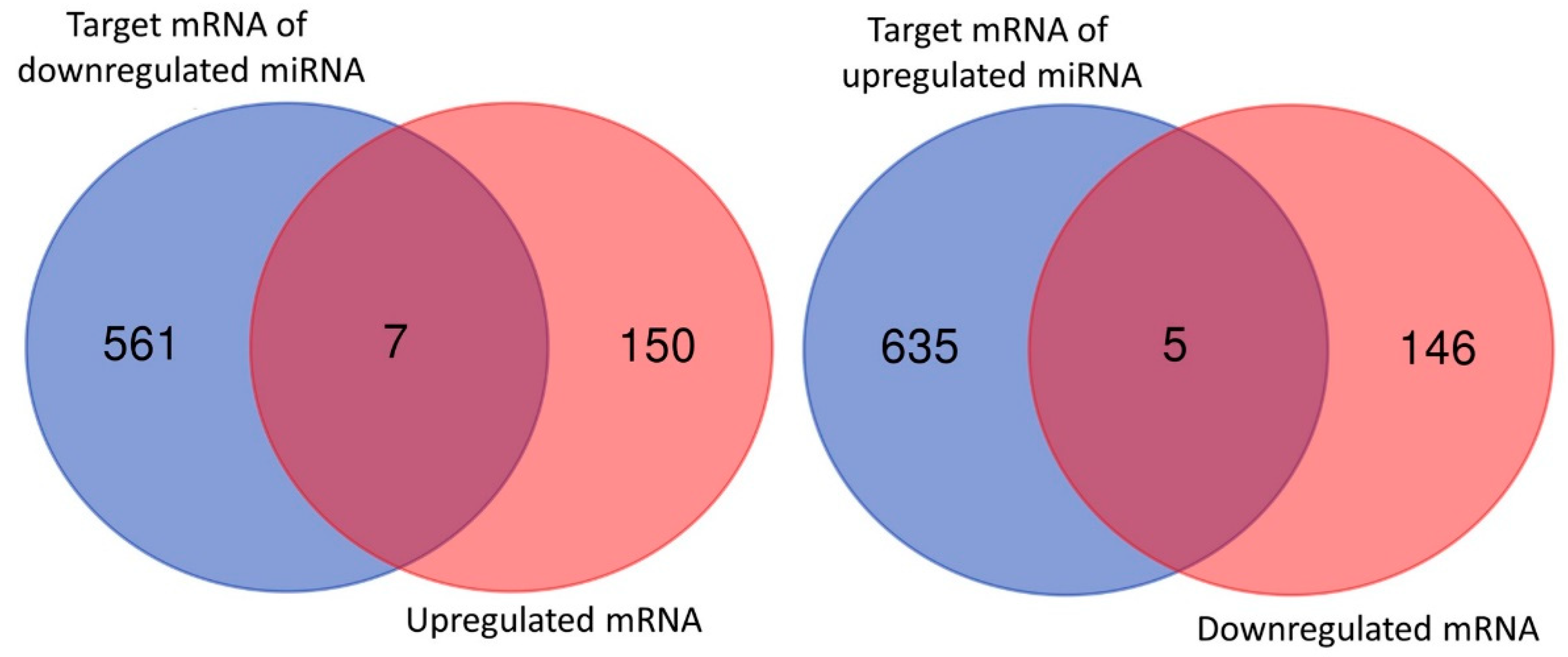
3.4. Dysregulated Genes with Potential miRNA–mRNA Interactions in Nintedanib-Treated IPF Fibroblasts
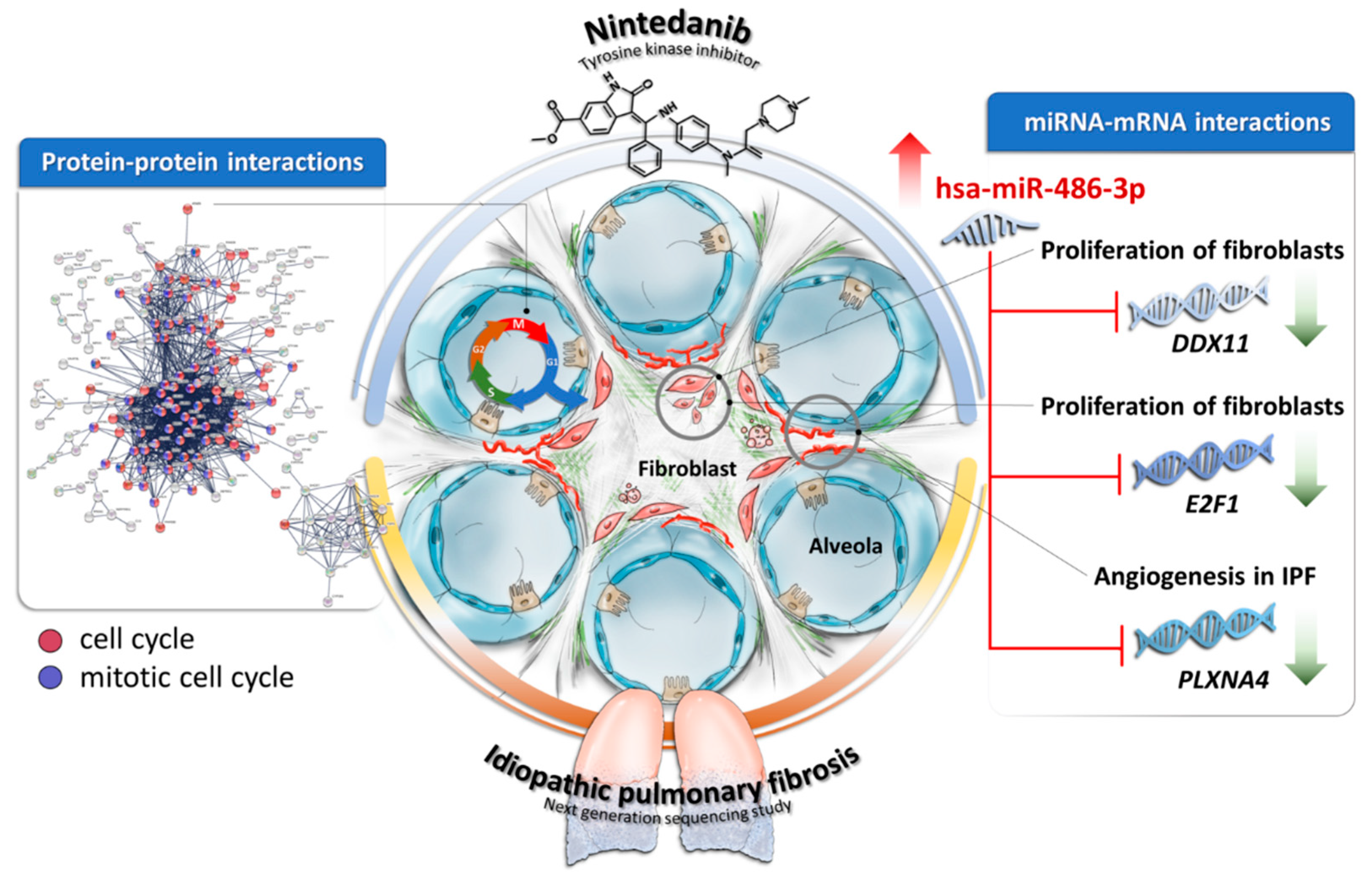
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aburto, M.; Herraez, I.; Iturbe, D.; Jimenez-Romero, A. Diagnosis of idiopathic pulmonary fibrosis: Differential diagnosis. Med. Sci. 2018, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.L.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic pulmonary fibrosis (IPF): An overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef] [PubMed]
- Gomperts, B.N.; Strieter, R.M. Fibrocytes in lung disease. J. Leukoc. Biol. 2007, 82, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug. Discov. 2017, 16, 755–772. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Cabrera, S.; Maldonado, M.; Selman, M. Role of matrix metalloproteinases in the pathogenesis of idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Selman, M. Lung fibroblasts, aging, and idiopathic pulmonary fibrosis. Ann. Am. Thorac. Soc. 2016, 13, S417–S421. [Google Scholar] [CrossRef] [PubMed]
- Hughes, G.; Toellner, H.; Morris, H.; Leonard, C.; Chaudhuri, N. Real world experiences: Pirfenidone and nintedanib are effective and well tolerated treatments for idiopathic pulmonary fibrosis. J. Clin. Med. 2016, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Varone, F.; Sgalla, G.; Iovene, B.; Bruni, T.; Richeldi, L. Nintedanib for the treatment of idiopathic pulmonary fibrosis. Expert Opin. Pharmacother. 2018, 19, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Nintedanib: A review of its use in patients with idiopathic pulmonary fibrosis. Drugs 2015, 75, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. Bibf 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.A. Idiopathic pulmonary fibrosis is a complex genetic disorder. Trans. Am. Clin. Climatol. Assoc. 2016, 127, 34–45. [Google Scholar] [PubMed]
- Helling, B.A.; Gerber, A.N.; Kadiyala, V.; Sasse, S.K.; Pedersen, B.S.; Sparks, L.; Nakano, Y.; Okamoto, T.; Evans, C.M.; Yang, I.V.; et al. Regulation of muc5b expression in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2017, 57, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Nance, T.; Smith, K.S.; Anaya, V.; Richardson, R.; Ho, L.; Pala, M.; Mostafavi, S.; Battle, A.; Feghali-Bostwick, C.; Rosen, G.; et al. Transcriptome analysis reveals differential splicing events in IPF lung tissue. PLoS ONE 2014, 9, e97550. [Google Scholar] [CrossRef] [PubMed]
- Pandit, K.V.; Milosevic, J. Microrna regulatory networks in idiopathic pulmonary fibrosis. Biochem. Cell Biol. 2015, 93, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Plantier, L.; Renaud, H.; Respaud, R.; Marchand-Adam, S.; Crestani, B. Transcriptome of cultured lung fibroblasts in idiopathic pulmonary fibrosis: Meta-analysis of publically available microarray datasets reveals repression of inflammation and immunity pathways. Int. J. Mol. Sci. 2016, 17, 2091. [Google Scholar] [CrossRef] [PubMed]
- Kwapiszewska, G.; Gungl, A.; Wilhelm, J.; Marsh, L.M.; Thekkekara Puthenparampil, H.; Sinn, K.; Didiasova, M.; Klepetko, W.; Kosanovic, D.; Schermuly, R.T.; et al. Transcriptome profiling reveals the complexity of pirfenidone effects in idiopathic pulmonary fibrosis. Eur. Respir. J. 2018, 52, 1800564. [Google Scholar] [CrossRef] [PubMed]
- Le Gallo, M.; Lozy, F.; Bell, D.W. Next-generation sequencing. Adv. Exp. Med. Biol. 2017, 943, 119–148. [Google Scholar] [PubMed]
- Wu, P.Y.; Chandramohan, R.; Phan, J.H.; Mahle, W.T.; Gaynor, J.W.; Maher, K.O.; Wang, M.D. Cardiovascular transcriptomics and epigenomics using next-generation sequencing: Challenges, progress, and opportunities. Circ. Cardiovasc. Genet. 2014, 7, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, S.; Kurundkar, A.; Kurundkar, D.; Bernard, K.; Sanders, Y.Y.; Ding, Q.; Antony, V.B.; Zhang, J.; Zmijewski, J.; Thannickal, V.J. Novel mechanisms for the antifibrotic action of nintedanib. Am. J. Respir. Cell Mol. Biol. 2016, 54, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. Mirdeep2 accurately identifies known and hundreds of novel microrna genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Galipon, J.; Ishii, R.; Suzuki, Y.; Tomita, M.; Ui-Tei, K. Differential binding of three major human adar isoforms to coding and long non-coding transcripts. Genes 2017, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with Tophat and cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Vejnar, C.E.; Zdobnov, E.M. Mirmap: Comprehensive prediction of microrna target repression strength. Nucleic Acids Res. 2012, 40, 11673–11683. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mrnas. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Improving microrna target prediction by modeling with unambiguously identified microrna-target pairs from clip-ligation studies. Bioinformatics 2016, 32, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Wang, X. Mirdb: An online resource for microrna target prediction and functional annotations. Nucleic Acids Res. 2015, 43, D146–D152. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The string database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The david gene functional classification tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Xie, Q.; Hu, C.L.; Jiang, Q.; Shen, H.F.; Schachner, M.; Zhao, W.J. Chl1 is expressed and functions as a malignancy promoter in glioma cells. Front. Mol. Neurosci. 2017, 10, 324. [Google Scholar] [CrossRef] [PubMed]
- Kigel, B.; Rabinowicz, N.; Varshavsky, A.; Kessler, O.; Neufeld, G. Plexin-a4 promotes tumor progression and tumor angiogenesis by enhancement of vegf and bfgf signaling. Blood 2011, 118, 4285–4296. [Google Scholar] [CrossRef] [PubMed]
- Ertosun, M.G.; Hapil, F.Z.; Osman Nidai, O. E2f1 transcription factor and its impact on growth factor and cytokine signaling. Cytokine Growth Factor Rev. 2016, 31, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of e2fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Benaud, C.M.; Dickson, R.B. Adhesion-regulated g1 cell cycle arrest in epithelial cells requires the downregulation of c-myc. Oncogene 2001, 20, 4554–4567. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Sheng, G.; Warner, B.W. Epidermal growth factor-induced rapid retinoblastoma phosphorylation at ser780 and ser795 is mediated by erk1/2 in small intestine epithelial cells. J. Biol. Chem. 2005, 280, 35992–35998. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.; Ordonez-Ercan, D.; Dasgupta, P.; Chellappan, S. Induction of human metallothionein 1g promoter by vegf and heavy metals: Differential involvement of e2f and metal transcription factors. Oncogene 2005, 24, 2204–2217. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallstrom, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F. The mitochondrial transporter family slc25: Identification, properties and physiopathology. Mol. Asp. Med. 2013, 34, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.E.; Chandramoorthy, H.C.; Shanmughapriya, S.; Zhang, X.Q.; Vallem, S.; Doonan, P.J.; Malliankaraman, K.; Guo, S.; Rajan, S.; Elrod, J.W.; et al. Slc25a23 augments mitochondrial Ca2+ uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol. Biol. Cell 2014, 25, 936–947. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Fold Change | Gene Symbol | Fold Change | miRmap Score | TargetScan Total Context++ Score | miRDB |
|---|---|---|---|---|---|---|
| hsa-miR-486-3p | 2.31 | DDX11 | −4.05 | 99.389 | −0.50 | 59 |
| hsa-miR-486-3p | 2.31 | E2F1 | −5.78 | 99.597 | −0.85 | 65 |
| hsa-miR-486-3p | 2.31 | NPTX1 | −2.89 | 99.908 | −0.62 | 95 |
| hsa-miR-486-3p | 2.31 | PLXNA4 | −6.94 | 99.700 | −0.18 | 70 |
| hsa-miR-92a-1-5p | −2.71 | SLC25A23 | 2.68 | 99.527 | −1.27 | 65 |
| hsa-miR-1275 | −4.74 | PRELP | 3.87 | 99.724 | 65 | |
| hsa-miR-1275 | −4.74 | HRK | 5.36 | 99.881 | 87 | |
| hsa-miR-100-3p | −2.52 | GJA3 | 3.04 | 99.326 | −0.39 | |
| hsa-miR-1275 | −4.74 | ZBED3 | 3.46 | 99.669 | −0.94 | |
| hsa-miR-1275 | −4.74 | ANTXR1 | 2.69 | 99.535 | −0.66 | |
| hsa-miR-486-3p | 2.31 | SUV39H1 | −4.35 | 99.421 | −0.47 | |
| hsa-miR-1275 | −4.74 | MBP | 8.97 | 99.886 | ||
| hsa-miR-326 | 4.64 | PLXNA4 | −6.94 | 99.932 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheu, C.-C.; Chang, W.-A.; Tsai, M.-J.; Liao, S.-H.; Chong, I.-W.; Kuo, P.-L. Gene Expression Changes Associated with Nintedanib Treatment in Idiopathic Pulmonary Fibrosis Fibroblasts: A Next-Generation Sequencing and Bioinformatics Study. J. Clin. Med. 2019, 8, 308. https://doi.org/10.3390/jcm8030308
Sheu C-C, Chang W-A, Tsai M-J, Liao S-H, Chong I-W, Kuo P-L. Gene Expression Changes Associated with Nintedanib Treatment in Idiopathic Pulmonary Fibrosis Fibroblasts: A Next-Generation Sequencing and Bioinformatics Study. Journal of Clinical Medicine. 2019; 8(3):308. https://doi.org/10.3390/jcm8030308
Chicago/Turabian StyleSheu, Chau-Chyun, Wei-An Chang, Ming-Ju Tsai, Ssu-Hui Liao, Inn-Wen Chong, and Po-Lin Kuo. 2019. "Gene Expression Changes Associated with Nintedanib Treatment in Idiopathic Pulmonary Fibrosis Fibroblasts: A Next-Generation Sequencing and Bioinformatics Study" Journal of Clinical Medicine 8, no. 3: 308. https://doi.org/10.3390/jcm8030308
APA StyleSheu, C.-C., Chang, W.-A., Tsai, M.-J., Liao, S.-H., Chong, I.-W., & Kuo, P.-L. (2019). Gene Expression Changes Associated with Nintedanib Treatment in Idiopathic Pulmonary Fibrosis Fibroblasts: A Next-Generation Sequencing and Bioinformatics Study. Journal of Clinical Medicine, 8(3), 308. https://doi.org/10.3390/jcm8030308