Pentoxifylline for Renal Protection in Diabetic Kidney Disease. A Model of Old Drugs for New Horizons


 , , ,
, , ,  ,
,
Abstract
:1. Diabetes Mellitus and Diabetic Kidney Disease
2. Old Drug Repositioning
3. Inflammation in Diabetic Kidney Disease
4. Pentoxifylline: Renoprotection and Targeting Inflammation in Diabetic Kidney Disease
5. Mechanisms Underlying the Renoprotective Effects of PTX
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- International Diabetes Federation. IDF Diabetes Atlas, 8th ed.; International Diabetes Federation: Brussels, Belgium, 2017. [Google Scholar]
- Ritz, E.; Rychlík, I.; Locatelli, F.; Halimi, S. End-stage renal failure in type 2 diabetes: A medical catastrophe of worldwide dimensions. Am. J. Kidney Dis. 1999, 34, 795–808. [Google Scholar] [CrossRef]
- Atkins, R.C.; Zimmet, P. Diabetic kidney disease: act now or pay later. Kidney Int. 2010, 77, 375–377. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.E. Diabetes: treating diabetic nephropathy-still an unresolved issue. Nat. Rev. Endocrinol. 2012, 8, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Gnudi, L.; Gentile, G.; Ruggenenti, P. Oxford Textbook of Clinical Nephrology, 4 ed.; Oxford University Press: Oxford, UK, 2016; Volume 2, pp. 1199–1247. [Google Scholar]
- National Kidney Foundation. KDOQI Clinical Practice Guideline for Diabetes and CKD: 2012 Update. Am. J. Kidney Dis. 2012, 60, 850–886. [Google Scholar] [CrossRef] [PubMed]
- Parving, H.H.; Brenner, B.M.; McMurray, J.J.; de Zeeuw, D.; Haffner, S.M.; Solomon, S.D.; Chaturvedi, N.; Persson, F.; Desai, A.S.; Nicolaides, M.; et al. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N. Engl. J. Med. 2012, 367, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- Fried, L.F.; Emanuele, N.; Zhang, J.H.; Brophy, M.; Conner, T.A.; Duckworth, W.; Leehey, D.J.; McCullough, P.A.; O’Connor, T.; Palevsky, P.M.; et al. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N. Engl. J. Med. 2013, 369, 1892–1903. [Google Scholar] [CrossRef] [PubMed]
- Packham, D.K.; Wolfe, R.; Reutens, A.T.; Berl, T.; Heerspink, H.L.; Rohde, R.; Ivory, S.; Lewis, J.; Raz, I.; Wiegmann, T.B.; et al. Sulodexide fails to demonstrate renoprotection in overt type 2 diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.J.; Greene, T.; Spitalewiz, S.; Blumenthal, S.; Berl, T.; Hunsicker, L.G.; Pohl, M.A.; Rohde, R.D.; Raz, I.; Yerushalmy, Y.; et al. Pyridorin in type 2 diabetic nephropathy. J. Am. Soc. Nephrol. 2012, 23, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.; Green, D.; Jamerson, K.; Ruilope, L.M.; Kuranoff, S.J.; Littke, T.; Viberti, G.; ASCEND Study Group. Avosentan for overt diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 527–535. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Padhy, B.M.; Gupta, Y.K. Drug repositioning: Re-investigating existing drugs for new therapeutic indications. J. Postgrad. Med. 2011, 57, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Persidis, A. The benefits of drug repositioning. Drug Discovery World Spring 2011, 9–12. [Google Scholar]
- US Food & Drug Administration. Drugs@FDA: FDA Approved Drug Products. Available online: www.accessdata.fda.gov (accessed on 15 February 2019).
- Dettelbach, H.R.; Aviado, D.M. Clinical pharmacology of pentoxifylline with special reference to its hemorrheologic effect for the treatment of intermittent claudication. J. Clin. Pharmacol. 1985, 25, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Al-Saad, R.Z.; Hussain, S.A.; Numan, I.T. Dose-response Relationship of the Anti-inflammatory Activity of Pentoxifylline in Experimental Models of Chronic Inflammation. Pharmacologia 2012, 3, 39–45. [Google Scholar] [CrossRef]
- Katsuki, A.; Sumida, Y.; Murashima, S.; Murata, K.; Takarada, Y.; Ito, K.; Fujii, M.; Tsuchihashi, K.; Goto, H.; Nakatani, K.; et al. Serum levels of tumor necrosis factor-alpha are increased in obese patients with noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1998, 83, 859–862. [Google Scholar] [PubMed]
- Pickup, J.C.; Chusney, G.D.; Thomas, S.M.; Burt, D. Plasma interleukin-6, tumour necrosis factor alpha and blood cytokine production in type 2 diabetes. Life Sci. 2000, 67, 291–300. [Google Scholar] [CrossRef]
- Festa, A.; D’Agostino, R.; Howard, G.; Mykkänen, L.; Tracy, R.P.; Haffner, S.M. Inflammation and microalbuminuria in nondiabetic and type 2 diabetic subjects: The Insulin Resistance Atherosclerosis Study. Kidney Int. 2000, 58, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Bruno, G.; Merletti, F.; Biggeri, A.; Bargero, G.; Ferrero, S.; Pagano, G.; Cavallo Perin, P.; Casale Monferrato Study. Progression to overt nephropathy in type 2 diabetes: the Casale Monferrato Study. Diabetes Care 2003, 26, 2150–2155. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.F.; Mora, C.; Maca, M.; García, J. Inflammatory parameters are independently associated with urinary albumin in type 2 diabetes mellitus. Am. J. Kidney Dis. 2003, 42, 53–61. [Google Scholar] [CrossRef]
- Chow, F.; Ozols, E.; Nikolic-Paterson, D.J.; Atkins, R.C.; Tesch, G.H. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int. 2004, 65, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.Y.; Nikolic-Paterson, D.J.; Ozols, E.; Atkins, R.C.; Tesch, G.H. Intercellular adhesion molecule-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. J. Am. Soc. Nephrol. 2005, 16, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Ping, F.; Mu, W.; Hill, P.; Atkins, R.C.; Chadban, S.J. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology (Carlton) 2006, 11, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, G.; Nakano, K.; Sawada, M.; Uno, K.; Shibayama, Y.; Ienaga, K.; Kondo, M. Possible role of tumor necrosis factor and interleukin-1 in the development of diabetic nephropathy. Kidney Int. 1991, 40, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, Y.; Yamamoto, T.; Shibutani, Y.; Aoki, E.; Tsutsumi, Z.; Takahashi, S.; Okamura, H.; Koga, M.; Fukuchi, M.; Hada, T. Elevated levels of interleukin-18 and tumor necrosis factor-alpha in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism 2003, 52, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Ho, A.W.; Tong, P.C.; Yeung, C.Y.; Kong, A.P.; Lun, S.W.; Chan, J.C.; Lam, C.W. Aberrant activation profile of cytokines and mitogen-activated protein kinases in type 2 diabetic patients with nephropathy. Clin. Exp. Immunol. 2007, 149, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Fantuzzi, G.; Reed, D.A.; Dinarello, C.A. IL-12-induced IFN-gamma is dependent on caspase-1 processing of the IL-18 precursor. J. Clin. Invest. 1999, 104, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, K.; Takiyama, Y.; Honjyo, J.; Tateno, M.; Haneda, M. Upregulated IL-18 expression in type 2 diabetic subjects with nephropathy: TGF-beta1 enhanced IL-18 expression in human renal proximal tubular epithelial cells. Diabetes Res. Clin. Pract. 2009, 83, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Noronha, I.L.; Niemir, Z.; Stein, H.; Waldherr, R. Cytokines and growth factors in renal disease. Nephrol Dial Transplant 1995, 10, 775–786. [Google Scholar] [PubMed]
- Mariano, F.; Bussolati, B.; Piccoli, G.; Camussi, G. Renal vascular effects of cytokines. Blood Purif 1997, 15, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Qiao, Y.C.; Xu, Y.; Ling, W.; Pan, Y.H.; Huang, Y.C.; Geng, L.J.; Zhao, H.L.; Zhang, X.X. Serum TNF-α concentrations in type 2 diabetes mellitus patients and diabetic nephropathy patients: A systematic review and meta-analysis. Immunol. Lett. 2017, 186, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Khoshakhlagh, P.; Bahrololoumi-Shapourabadi, M.; Mohammadirad, A.; Ashtaral-Nakhai, L.; Minaie, B.; Abdollahi, M. Beneficial effect of phosphodiesterase-5 inhibitor in experimental inflammatory bowel disease; molecular evidence for involvement of oxidative stress. Toxicol. Mech. Methods 2007, 17, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Bustos, C.; Alonso, J.; Alcázar, R.; López-Armada, M.J.; Plaza, J.J.; González, E.; Egido, J. Involvement of tumor necrosis factor-alpha in the pathogenesis of experimental and human glomerulonephritis. Adv. Nephrol. Necker. Hosp. 1995, 24, 53–77. [Google Scholar] [PubMed]
- McCarthy, E.T.; Sharma, R.; Sharma, M.; Li, J.Z.; Ge, X.L.; Dileepan, K.N.; Savin, V.J. TNF-alpha increases albumin permeability of isolated rat glomeruli through the generation of superoxide. J. Am. Soc. Nephrol. 1998, 9, 433–438. [Google Scholar] [PubMed]
- DiPetrillo, K.; Coutermarsh, B.; Gesek, F.A. Urinary tumor necrosis factor contributes to sodium retention and renal hypertrophy during diabetes. Am. J. Physiol. Renal. Physiol. 2003, 284, F113–F121. [Google Scholar] [CrossRef] [PubMed]
- Kalantarinia, K.; Awas, A.S.; Siragy, H.M. Urinary and renal interstitial concentrations of TNF-alpha increase prior to the rise in albuminuria in diabetic rats. Kidney Int. 2003, 64, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, D.; Miyazaki, M.; Naka, R.; Koji, T.; Yagame, M.; Jinde, K.; Endoh, M.; Nomoto, Y.; Sakai, H. In situ hybridization of interleukin 6 in diabetic nephropathy. Diabetes 1995, 44, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Coleman, D.L.; Ruef, C. Interleukin-6: an autocrine regulator of mesangial cell growth. Kidney Int. 1992, 41, 604–606. [Google Scholar] [CrossRef] [PubMed]
- Nosadini, R.; Velussi, M.; Brocco, E.; Bruseghin, M.; Abaterusso, C.; Saller, A.; Dalla Vestra, M.; Carraro, A.; Bortoloso, E.; Sambataro, M.; et al. Course of renal function in type 2 diabetic patients with abnormalities of albumin excretion rate. Diabetes 2000, 49, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Dalla Vestra, M.; Mussap, M.; Gallina, P.; Bruseghin, M.; Cernigoi, A.M.; Saller, A.; Plebani, M.; Fioretto, P. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J. Am. Soc. Nephrol. 2005, 16, S78–S82. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E.; Cooper, M.E. The tubulointerstitium in progressive diabetic kidney disease: more than an aftermath of glomerular injury? Kidney Int. 1999, 56, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Abbate, M.; Remuzzi, G. Progression of renal injury toward interstitial inflammation and glomerular sclerosis is dependent on abnormal protein filtration. Nephrol. Dial. Transplant. 2015, 30, 706–712. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, M.T.; Cesarone, M.R.; Belcaro, G.; Nicolaides, A.N.; Griffin, M.; Incandela, L.; Bucci, M.; Geroulakos, G.; Ramaswami, G.; Vasdekis, S.; et al. Treatment of intermittent claudication with pentoxifylline: a 12-month, randomized trial-walking distance and microcirculation. Angiology 2002, 53, S7–S12. [Google Scholar] [PubMed]
- Aviado, D.M.; Dettelbach, H.R. Pharmacology of pentoxifylline, a hemorheologic agent for the treatment of intermittent claudication. Angiology 1984, 35, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Aviado, D.M.; Porter, J.M. Pentoxifylline: a new drug for the treatment of intermittent claudication. Mechanism of action, pharmacokinetics, clinical efficacy and adverse effects. Pharmacotherapy 1984, 4, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonnaia, I.A.V.; Mamedov, R.; Kozlov, V.V.; Emanuél’, V.L.; Kudriashova, M.I. Effect of trental on indices kidney function in diabetes mellitus. Probl. Endokrinol. 1982, 28, 3–8. [Google Scholar]
- Sinzinger, H. Pentoxifylline enhances formation of prostacyclin from rat vascular and renal tissue. Prostaglandins Leukot. Med. 1983, 12, 217–226. [Google Scholar] [CrossRef]
- Doherty, G.M.; Jensen, J.C.; Alexander, H.R.; Buresh, C.M.; Norton, J.A. Pentoxifylline suppression of tumor necrosis factor gene transcription. Surgery 1991, 110, 192–198. [Google Scholar] [PubMed]
- Voisin, L.; Breuillé, D.; Ruot, B.; Rallière, C.; Rambourdin, F.; Dalle, M.; Obled, C. Cytokine modulation by PX differently affects specific acute phase proteins during sepsis in rats. Am. J. Physiol. 1998, 275, R1412–R1419. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Heeg, M.; Kochsiek, T.; Siemers, G.; Zeisberg, M.; Müller, G.A. Effects of pentoxifylline, pentifylline and gamma-interferon on proliferation, differentiation, and matrix synthesis of human renal fibroblasts. Nephrol. Dial. Transplant. 2000, 15, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, O.M.; Baiuomy, A.R.; El-Shenawy, S.M.; Arbid, M.S. The anti-inflammatory effects of the phosphodiesterase inhibitor pentoxifylline in the rat. Pharmacol. Res. 2003, 4, 331–340. [Google Scholar] [CrossRef]
- Dávila-Esqueda, M.E.; Martínez-Morales, F. Pentoxifylline diminishes the oxidative damage to renal tissue induced by streptozotocin in the rat. Exp. Diabesity Res. 2004, 5, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Navarro-González, J.F.; Mora-Fernández, C. The role of inflammatory cytokines in diabetic nephropathy. J. Am. Soc. Nephrol. 2008, 19, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Donate-Correa, J.; Martín-Núñez, E.; Muros de Fuentes, M.; Mora-Fernández, C.; Navarro-González, J.F. Inflammatory cytokines in diabetic nephropathy. J. Diabetes Res. 2015, 2015, 948417. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Thompson, P.; Beutler, B. Dexamethasone and pentoxifylline inhibit endotoxin-induced cachectin/tumor necrosis factor synthesis at separate points in the signaling pathway. J. Exp. Med. 1990, 172, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.F.; Mora, C.; Muros, M.; García, J. Urinary tumour necrosis factor-alpha excretion independently correlates with clinical markers of glomerular and tubulointerstitial injury in type 2 diabetic patients. Nephrol. Dial. Transplant. 2006, 21, 3428–3434. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.F.; Milena, F.J.; Mora, C.; León, C.; García, J. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am. J. Nephrol. 2006, 26, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Mikhail, A.; Lethbridge, M.W.; Kemeny, D.M.; Macdougall, I.C. Pentoxifylline improves hemoglobin levels in patients with erythropoietin-resistant anemia in renal failure. J. Am. Soc. Nephrol. 2004, 15, 1877–1882. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.L.; Dias de Oliveira, R.T.; Mamonib, R.L.; Rizzi-Coelho, O.; Nicolau, J.C.; Blotta, M.H.; Serrano, C.V., Jr. Pentoxifylline reduces pro-inflammatory and increases anti-inflammatory activity in patients with coronary artery disease-A randomized placebo-controlled study. Atherosclerosis 2008, 196, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Mohammadpour, A.H.; Falsoleiman, H.; Shamsara, J.; Abadi, G.A.; Rasooli, R.; Ramezani, M. Pentoxifylline decreases serum level of adhesion molecules in atherosclerosis patients. Iran Biomed. J. 2014, 17, 23–27. [Google Scholar]
- Navarro, J.F.; Mora, C.; Rivero, A.; Gallego, E.; Chahin, J.; Macía, M.; Méndez, M.L.; García, J. Urinary protein excretion and serum tumor necrosis factor in diabetic patients with advanced renal failure: effects of pentoxifylline administration. Am. J. Kidney Dis. 1999, 33, 458–463. [Google Scholar] [CrossRef]
- Aminorroaya, A.; Janghorbani, M.; Rezvanian, H.; Aminian, T.; Gharavi, M.; Amini, M. Comparison of the effect of pentoxifylline and captopril on proteinuria in patients with type 2 diabetes mellitus. Nephron. Clin. Pract. 2005, 99, c73–c77. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Morán, M.; Guerrero-Romero, F. Pentoxifylline is as effective as captopril in the reduction of microalbuminuria in non-hypertensive type 2 diabetic patients–A randomized, equivalent trial. Clin. Nephrol. 2005, 64, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.F.; Mora, C.; Muros, M.; García, J. Additive antiproteinuric effect of pentoxifylline in patients with type 2 diabetes under angiotensin II receptor blockade: a short-term, randomized, controlled trial. J. Am. Soc. Nephrol. 2005, 16, 2119–2126. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Morán, M.; González-González, G.; Bermúdez-Barba, M.V.; Medina de la Garza, C.E.; Tamez-Pérez, H.E.; Martínez-Martínez, F.J.; Guerrero-Romero, F. Effects of pentoxifylline on the urinary protein excretion profile of type 2 diabetic patients with microproteinuria: A double-blind, placebo-controlled randomized trial. Clin. Nephrol. 2006, 66, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.M.; Lin, S.L.; Chiang, W.C.; Wu, K.D.; Tsai, T.J. Pentoxifylline ameliorates proteinuria through suppression of renal monocyte chemoattractant protein-1 in patients with proteinuric primary glomerular diseases. Kidney Int. 2006, 69, 1410–1415. [Google Scholar] [CrossRef] [PubMed]
- Shu, K.H.; Wu, M.J.; Chen, C.H.; Cheng, C.H.; Lian, J.D.; Lu, Y.S. Effect of pentoxifylline on graft function of renal transplant recipients complicated with chronic allograft nephropathy. Clin. Nephrol. 2007, 67, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Diskin, C.J.; Stokes, T.J.; Dansby, L.M.; Radcliff, L.; Carter, T.B. Will the addition of pentoxifylline reduce proteinuria in patients with diabetic glomerulosclerosis refractory to maximal doses of both an angiotensin converting enzyme inhibitor and an angiotensin receptor blocker? J. Nephrol. 2007, 20, 410–416. [Google Scholar] [PubMed]
- Badri, S.; Dashti-Khavidaki, S.; Ahmadi, F.; Mahdavi-Mazdeh, M.; Abbasi, M.R.; Khalili, H. Effect of add-on pentoxifylline on proteinuria in membranous glomerulonephritis: A 6-month placebo-controlled trial. Clin. Drug Investig. 2013, 33, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Perkins, R.M.; Aboudara, M.C.; Uy, A.L.; Olson, S.W.; Cushner, H.M.; Yuan, C.M. Effect of pentoxifylline on GFR decline in CKD: A pilot, double-blind, randomized, placebo-controlled trial. Am. J. Kidney Dis. 2009, 53, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Goicoechea, M.; García de Vinuesa, S.; Quiroga, B.; Verdalles, U.; Barraca, D.; Yuste, C.; Panizo, N.; Verde, E.; Muñoz, M.A.; Luño, J. Effects of pentoxifylline on inflammatory parameters in chronic kidney disease patients: a randomized trial. J. Nephrol. 2012, 25, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Chen, Y.M.; Chiang, W.C.; Wu, K.D.; Tsai, T.J. Effect of pentoxifylline in addition to losartan on proteinuria and GFR in CKD: a 12-month randomized trial. Am. J. Kidney Dis. 2008, 52, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Navarro-González, J.F.; Mora-Fernández, C.; Muros de Fuentes, M.; Chahin, J.; Méndez, M.L.; Gallego, E.; Macía, M.; del Castillo, N.; Rivero, A.; Getino, M.A.; García, P.; Jarque, A.; García, J. Effect of pentoxifylline on renal function and urinary albumin excretion in patients with diabetic kidney disease: the PREDIAN trial. J. Am. Soc. Nephrol. 2015, 26, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Lai, T.; Chen, P.; Lai, C.; Wu, V.; Chiang, W.; Chen, Y.M.; Wu, K.D.; Tsai, T.J. Renoprotective effect of combining pentoxifylline with angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker in advanced chronic kidney disease. J. Formos Med. Assoc. 2015, 113, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Navarro-González, J.F.; Sánchez-Niño, M.D.; Donate-Correa, J.; Martín-Núñez, E.; Ferri, C.; Pérez-Delgado, N.; Górriz, J.L.; Martínez-Castelao, A.; Ortiz, A.; Mora-Fernández, C. Effects of pentoxifylline on soluble Klotho concentrations and renal tubular cell expression in diabetic kidney disease. Diabetes Care 2018, 41, 1817–1820. [Google Scholar] [CrossRef] [PubMed]
- Gentile, G.; Remuzzi, G.; Ruggenenti, P. Dual renin-angiotensin system blockade for nephroprotection: Still under scrutiny. Nephron. 2015, 129, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.C.; Wu, C.J.; Lin, C.J.; Pan, C.F.; Chen, C.Y.; Huang, T.M.; Wu, C.H.; Lin, S.L.; Chen, Y.M.; Chen, L.; et al. Pentoxifylline decreases dialysis risk in patients with advanced chronic kidney disease. Clin. Pharmacol. Ther. 2015, 98, 442–449. [Google Scholar] [PubMed]
- McCormick, B.B.; Sydor, A.; Akbari, A.; Fergusson, D.; Doucette, S.; Knoll, G. The effect of pentoxifylline on proteinuria in diabetic kidney disease: a meta-analysis. Am. J. Kidney Dis. 2008, 52, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.L.; Shen, Y.; Sun, Z.L.; Zha, Y. Efficacy and safety of combining pentoxifylline with angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker in diabetic nephropathy: a meta-analysis. Int. Urol. Nephrol. 2015, 47, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Grande, J.P. Cyclic nucleotide phosphodiesterase (PDE) inhibitors: novel therapeutic agents for progressive renal disease. Exp. Biol. Med. (Maywood) 2007, 232, 38–51. [Google Scholar] [PubMed]
- Ward, A.; Clissold, S. Pentoxifylline: a review of its pharmacodynamics and pharmacokinetic properties and its therapeutic efficacy. Drugs 1987, 34, 50–97. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Chen, R.H.; Chen, Y.M.; Chiang, W.C.; Tsai, T.J.; Hsieh, B.S. Pentoxifylline inhibits platelet-derived growth factor-stimulated cyclin D1 expression in mesangial cells by blocking Akt membrane translocation. Mol. Pharmacol. 2003, 64, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.M.; Chiang, W.C.; Lin, S.L.; Wu, K.D.; Tsai, T.J.; Hsieh, B.S. Dual regulation of tumor necrosis factor-alpha-induced CCL2/monocyte chemoattractant protein-1 expression in vascular smooth muscle cells by nuclear factor-kappaB and activator protein-1: Modulation by type III phosphodiesterase inhibition. J. Pharmacol. Exp. Ther. 2004, 309, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.M.; Wu, K.D.; Tsai, T.J.; Hsieh, B.S. Pentoxifylline inhibits PDGF-induced proliferation of and TGF-beta-stimulated collagen synthesis by vascular smooth muscle cells. J. Mol. Cell. Cardiol. 1999, 31, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Tenor, H.; Schudt, C. Analysis of PDE isoenzyme profiles in cells and tissues by pharmacological methods. In Phoshodiesterase inhibitors; Schudt, C., Dent, G., Rabe, K.F., Eds.; Academic Press Inc.: San Diego, CA, USA, 1996; pp. 21–40. [Google Scholar]
- Chen, Y.M.; Ng, Y.Y.; Lin, S.L.; Chiang, W.C.; Lan, H.Y.; Tsai, T.J. Pentoxifylline suppresses renal tumor necrosis factor-alpha and ameliorates experimental crescentic glomerulonephritis in rats. Nephrol. Dial. Transplant. 2004, 19, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Garcia, F.A.; Rebouças, J.F.; Balbino, T.Q.; da Silva, T.G.; de Carvalho-Júnior, C.H.; Cerqueira, G.S.; Brito, G.A.; Viana, G.S. Pentoxifylline reduces the inflammatory process in diabetic rats: relationship with decreases of pro-inflammatory cytokines and inducible nitric oxide synthase. J. Inflamm. 2015, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Aizawa, H.; Shiraki-Iida, T.; Nagai, R.; Kuro-o, M.; Nabeshima, Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem. Biophys. Res. Commun. 1998, 242, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Liu, S.; Morgenthaler, N.G.; Wong, M.D.; Tavintharan, S.; Sum, C.F.; Lim, S.C. Association of plasma soluble α-klotho with pro-endothelin-1 in patients with type 2 diabetes. Atherosclerosis 2014, 233, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wang, Q.; Lv, C.; Qin, N.; Lei, S.; Yuan, Q.; Wang, G. The changes of serum sKlotho and NGAL levels and their correlation in type 2 diabetes mellitus patients with different stages of urinary albumin. Diabetes Res. Clin. Pract. 2014, 106, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Asai, O.; Nakatani, K.; Tanaka, T.; Sakan, H.; Imura, A.; Yoshimoto, S.; Samejima, K.; Yamaguchi, Y.; Matsui, M.; Akai, Y.; et al. Decreased renal α-Klotho expression in early diabetic nephropathy in humans and mice and its possible role in urinary calcium excretion. Kidney Int. 2012, 81, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Song, S.H.; Kim, I.J.; Lee, E.Y.; Lee, S.M.; Chung, C.H.; Kwak, I.S.; Lee, E.K.; Kim, Y.K. Decreased plasma α-Klotho predict progression of nephropathy with type 2 diabetic patients. J. Diabetes Complications 2016, 30, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Suárez-Alvarez, B.; Lopez-Larrea, C.; Jakubowski, A.; Blanco, J.; Ramírez, R.; Selgas, R.; Ruiz-Ortega, M.; et al. The inflammatory cytokines TWEAK and TNFα reduce renal klotho expression through NF-κB. J. Am. Soc. Nephrol. 2011, 22, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Banerjee, S.; Dey, N.; LeJeune, W.S.; Sarkar, P.S.; Brobey, R.; Rosenblatt, K.P.; Tilton, R.G.; Choudhary, S. Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via RelA (serine)536 phosphorylation. Diabetes 2011, 60, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, Y.; Ishikawa, K.; Yasuda, O.; Oguro, R.; Hanasaki, H.; Kida, I.; Takemura, Y.; Ohishi, M.; Katsuya, T.; Rakugi, H. Klotho suppresses TNF-alpha-induced expression of adhesion molecules in the endothelium and attenuates NF-kappaB activation. Endocrine 2009, 35, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Jheng, H.F.; Tsai, P.J.; Chuang, Y.L.; Shen, Y.T.; Tai, T.A.; Chen, W.C.; Chou, C.K.; Ho, L.C.; Tang, M.J.; Lai, K.T.; et al. Albumin stimulates renal tubular inflammation through an HSP70-TLR4 axis in mice with early diabetic nephropathy. Dis Model Mech 2015, 8, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fernández, B.; Izquierdo, M.C.; Valiño-Rivas, L.; Nastou, D.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Albumin downregulates Klotho in tubular cells. Nephrol Dial Transplant 2018, 33, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
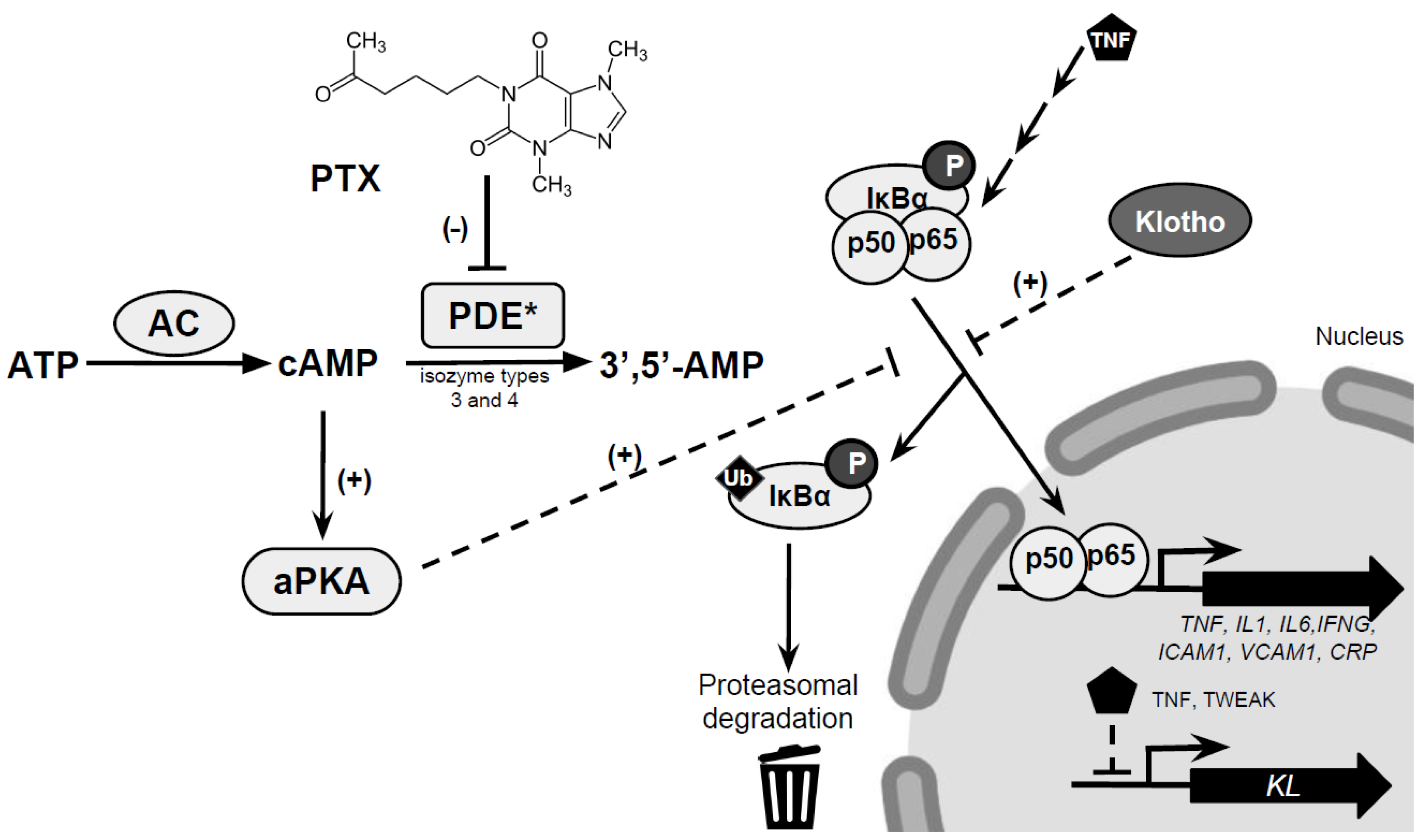

{kind=link}
| Drug | Original indication | Reposition |
|---|---|---|
| Amantadine | Influenza | Parkinson’s disease |
| Amphotericin | Antifungal | Leishmaniasis |
| Aspirin | Inflammation, pain | Antiplatelet |
| Bromocriptine | Parkinson’s disease | Diabetes mellitus |
| Bupropion | Depression | Smoking cessation |
| Colchicine | Gout | Recurrent pericarditis |
| Finasteride | Benign prostatic hyperplasia | Male pattern baldness |
| Gabapentin | Epilepsy | Neuropathic pain |
| Methotrexate | Cancer | Psoriasis, rheumatoid arthritis |
| Miltefosine | Cancer | Visceral leishmaniasis |
| Minoxidil | Hypertension | Male pattern baldness |
| Propranolol | Hypertension | Migraine prophylaxis |
| Sildenafil | Angina | Erectile dysfunction, pulmonary hypertension |
| Thalidomide | Morning sickness | Erythema nodosum leprosum |
| Zidovudine | Cancer | HIV/AIDS |
| Ref. | Type of Study | Type of Intervention | Population | PTX Dose, Duration | Background RAAS Blockade | Main Findings | Anti-Inflammatory Effect |
|---|---|---|---|---|---|---|---|
| [65] | Randomized, controlled, open-label trial. | PTX vs. untreated | DM patients, n = 24 Albuminuria > 300 mg/24 h; creatinine clearance < 35 mL/min | 400 mg/day, 6 months. | No. | 59.3% proteinuria reduction in PTX-group (p < 0.001) | 42.2% TNFα reduction in PTX-group (p < 0.001) |
| [66] | Randomized, controlled, open-label trial. | PTX vs. Captopril | DM patients, n = 39 Albuminuria > 300 mg/24 h; eGFR > 60 mL/min | 1200 mg/day, 8 weeks | No. | PTX and Captopril reduced proteinuria; 40% in PTX-group (p < 0.05) and 38.5% in Captopril-group (p < 0.01) | Not reported |
| [67] | Randomized, controlled, open-label trial. | PTX vs. Captopril | DM patients, n = 130 UAE 20–200 μg/min. | 1200 mg/day, 6 months. | No. | PTX and Captopril reduced proteinuria; 77.2% in PTX-group and 76.6 % in Captopril-group (p < 0.01 for both) | Not reported |
| [68] | Randomized, controlled, open-label trial. | PTX vs. untreated | DM patients, n = 61 Albuminuria > 300 mg/24 h; eGFR > 90 mL/min | 1200 mg/day, 4 months. | ARB. | 12.1% proteinuria reduction in PTX-group (p < 0.001) | 28.1% and 28.8% reductions in serum and urinary TNFα, respectively (p < 0.01). TNFα changes were related to UAE |
| [69] | Randomized, double-blind controlled trial. | PTX vs. placebo | DM patients, n = 40 UAE 20–200 μg/min. | 1200 mg/day, 4 months. | No. | 73.8% and 84.6% reductions in urinary levels of both high and low molecular weight proteins (p < 0.05) | Not reported |
| [70] | Prospective trial | All in PTX | Patients with GN; non-diabetic, n = 17 Spot proteinuria > 1.5 g/g Cr; eGFR 24–115 mL/min/1.73 m2 | 800 mg/day, 6 months. | No. | 36.5% and 33.9% reductions in spot and 24 h proteinuria (g/g Cr) (p < 0.01) | 46% MCP-1 decrease (p < 0.01) |
| [71] | Prospective trial | All in PTX | CAN patients, n = 17 UAE 20–200 μg/min., mean eGFR 38 ± 8 mL/min/1.73 m2 | 1200 mg/day, 6 months. | No. | 19.6% reduction of proteinuria at 3rd month (p < 0.05) and improved graft survival | 5.3% and 43.75% reductions in CD4+ cells bearing TNFα and IL10, respectively (p < 0.05) |
| [72] | Open-label, controlled trial | PTX vs. untreated | Diabetic glomerulosclerosis patients, n = 14 Proteinuria > 1.5 g/24 h; Cr clearance > 15 mL/min | 400–800 mg/day, 1 year | ACEIs/ARBs. | PTX not reduced proteinuria or improved renal function | Not reported |
| [73] | Randomized, double-blind, controlled trial | PTX vs. placebo | Patients with GN, n = 18 proteinuria > 500 mg/24 h, mean eGFR 71.2 ± 30.6 mL/min/1.73 m2 | 800–1200 mg/day, 6 months. | ACEIs/ARBs. | 56% reduction of proteinuria without affecting GFR | Not reported |
| [74] | Randomized, double-blind, controlled trial | PTX vs. placebo | CKD patients, n = 40 mean eGFR 29.5 ± 10.1 mL/min/1.73 m2, proteinuria greater than 1 g/24 h | 800 mg/day, 1 year | ACEIs/ARBs. | PTX stabilized GFR. No reduction of proteinuria | Not reported |
| [75] | Randomized, controlled trial | PTX vs. untreated | CKD patients, n = 91 albuminuria > 300 mg/24 h, eGFR <60 mL/min/1.73 m2 | 800 mg/day, 1 year | ACEIs/ARBs. | PTX stabilized GFR. No reduction of proteinuria. | 45.5 %, 11.1 %, and 57.4 % reductions in TNFα, fibrinogen and hsCRP, respectively (p < 0.05) |
| [76] | Randomized, controlled trial. | PTX vs. untreated | CKD patients, n = 56 Proteinuria > 500 mg/g of Cr; eGFR 10–60 mL/min/1.73 m2 | 400–800 mg/day, 1 year | ARB. | 8.7% reduction of proteinuria compared to the control group (p < 0.001) stabilized GFR | Decrease in proteinuria was in conjunction with the decrease in TNFα and MCP1 (R = 0.64 and R = 0.55, respectively; p < 0.001 for both) |
| [77] | Randomized, controlled trial. | PTX vs. untreated | DM patients, n = 166 Albuminuria > 30 mg/24 h, eGFR 60–15 mL/min/1.73 m2 | 1200 mg/day, 2 years. | ARB. | Compared to the control group, 67.9% and 14.9% reduction in GFR decrease (p < 0.001) and proteinuria (p = 0.001) in the PTX-group, respectively. | 10.6% reduction in urinary TNFα. |
| [78] | Single-center retrospective study | PTX vs. untreated | CKD patients, n = 661 Mean proteinuria 1102 mg/g of Cr, eGFR < 45 mL/min/1.73 m2 | 400–800 mg/day, 1 year. | ACEIs/ARBs. | PTX group showed a better renal outcome in patients with higher proteinuria (p = 0.005). | Not reported |
| [79] | Randomized, controlled trial. Post-hoc analysis. | PTX vs. untreated | DM patients, n = 166 Albuminuria > 30 mg/24 h, eGFR 60–15 mL/min/1.73 m2 | 1200 mg/day, 2 years. | ARB. | Compared to the control group, 5.9% and 9.3% increase in serum (p < 0.05) and urine Klotho (p < 0.001) in the PTX-group, respectively. | Changes in TNFα associated with changes of urinary Klotho (R2 = 0.60; p < 0.0001). |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donate-Correa, J.; Tagua, V.G.; Ferri, C.; Martín-Núñez, E.; Hernández-Carballo, C.; Ureña-Torres, P.; Ruiz-Ortega, M.; Ortiz, A.; Mora-Fernández, C.; Navarro-González, J.F. Pentoxifylline for Renal Protection in Diabetic Kidney Disease. A Model of Old Drugs for New Horizons. J. Clin. Med. 2019, 8, 287. https://doi.org/10.3390/jcm8030287
Donate-Correa J, Tagua VG, Ferri C, Martín-Núñez E, Hernández-Carballo C, Ureña-Torres P, Ruiz-Ortega M, Ortiz A, Mora-Fernández C, Navarro-González JF. Pentoxifylline for Renal Protection in Diabetic Kidney Disease. A Model of Old Drugs for New Horizons. Journal of Clinical Medicine. 2019; 8(3):287. https://doi.org/10.3390/jcm8030287
Chicago/Turabian StyleDonate-Correa, Javier, Víctor G. Tagua, Carla Ferri, Ernesto Martín-Núñez, Carolina Hernández-Carballo, Pablo Ureña-Torres, Marta Ruiz-Ortega, Alberto Ortiz, Carmen Mora-Fernández, and Juan F. Navarro-González. 2019. "Pentoxifylline for Renal Protection in Diabetic Kidney Disease. A Model of Old Drugs for New Horizons" Journal of Clinical Medicine 8, no. 3: 287. https://doi.org/10.3390/jcm8030287
APA StyleDonate-Correa, J., Tagua, V. G., Ferri, C., Martín-Núñez, E., Hernández-Carballo, C., Ureña-Torres, P., Ruiz-Ortega, M., Ortiz, A., Mora-Fernández, C., & Navarro-González, J. F. (2019). Pentoxifylline for Renal Protection in Diabetic Kidney Disease. A Model of Old Drugs for New Horizons. Journal of Clinical Medicine, 8(3), 287. https://doi.org/10.3390/jcm8030287