Chimeric Antigen Receptor T-Cells: The Future Is Now


Abstract
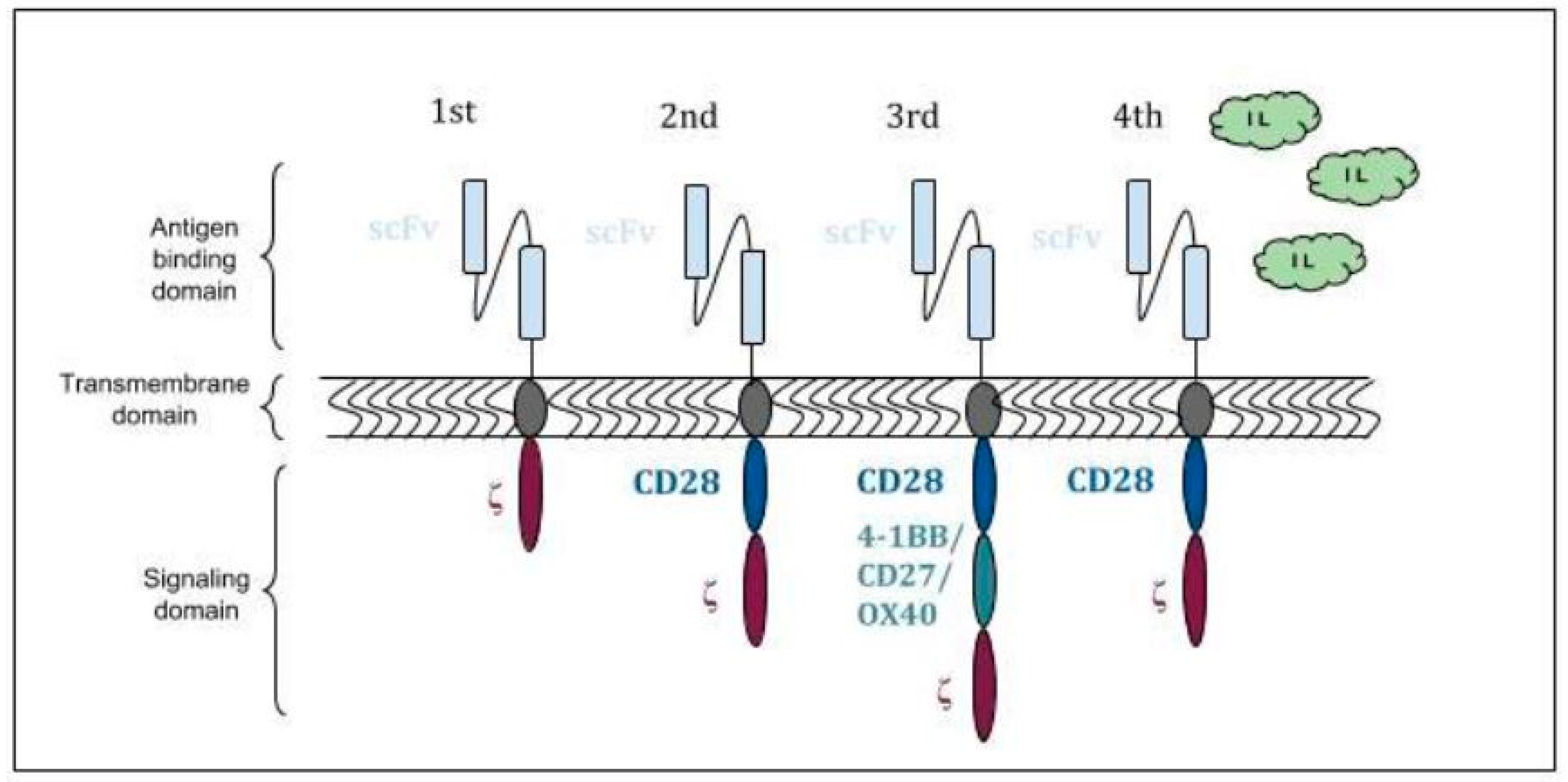
:1. Introduction
2. Structure and Mechanism of Action
3. FDA Approved Indications
4. Acute Lymphoblastic Leukemia (ALL)
5. Non-Hodgkin′s Lymphoma (NHL)
6. Toxicity
7. Mechanisms of Resistance
8. Future indications and Solid Tumors
Author Contributions
Conflicts of Interest
References
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Hoos, A. Development of immuno-oncology drugs - from CTLA4 to PD1 to the next generations. Nat. Rev. Drug Discov. 2016, 15, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Invest. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jaeger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Primary Analysis of Juliet: A Global, Pivotal, Phase 2 Trial of CTL019 in Adult Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Blood 2017, 130, 577. [Google Scholar]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Salter, A.I.; Pont, M.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells: CD19 and the road beyond. Blood 2018, 131, 2621–2629. [Google Scholar] [CrossRef] [PubMed]
- Brocker, T. Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood 2000, 96, 1999–2001. [Google Scholar]
- Jensen, M.C.; Popplewell, L.; Cooper, L.J.; DiGiusto, D.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol. Blood Marrow Transplant. 2010, 16, 1245–1256. [Google Scholar] [CrossRef]
- Krause, A.; Guo, H.F.; Latouche, J.B.; Tan, C.; Cheung, N.K.; Sadelain, M. Antigen-dependent CD28 signaling selectively enhances survival and proliferation in genetically modified activated human primary T lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef]
- Pegram, H.J.; Park, J.H.; Brentjens, R.J. CD28z CARs and armored CARs. Cancer J. 2014, 20, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Yeku, O.O.; Brentjens, R.J. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 2016, 44, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef] [PubMed]
- Roberts, Z.J.; Better, M.; Bot, A.; Roberts, M.R.; Ribas, A. Axicabtagene ciloleucel, a first-in-class CAR T cell therapy for aggressive NHL. Leuk. Lymphoma 2018, 59, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark Res. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Bot, A.; Rossi, J.M.; Jiang, Y.; Navale, L.; Shen, Y.-W.; Sherman, M.; Mardiros, A.; Yoder, S.C.; Go, W.Y.; Rosenberg, S.A.; et al. Cyclophosphamide and Fludarabine Conditioning Chemotherapy Induces a Key Homeostatic Cytokine Profile in Patients Prior to CAR T Cell Therapy. Blood 2015, 126, 4426. [Google Scholar]
- Turtle, C.J.; Berger, C.; Sommermeyer, D.; Hanafi, L.-A.; Pender, B.; Robinson, E.M.; Melville, K.; Budiarto, T.M.; Steevens, N.N.; Chaney, C.; et al. Anti-CD19 Chimeric Antigen Receptor-Modified T Cell Therapy for B Cell Non-Hodgkin Lymphoma and Chronic Lymphocytic Leukemia: Fludarabine and Cyclophosphamide Lymphodepletion Improves In Vivo Expansion and Persistence of CAR-T Cells and Clinical Outcomes. Blood 2015, 126, 184. [Google Scholar]
- US Food and Drug Administration. Approved Products-KYMRIAH (Tisagenlecleucel) [Internet]. 2018. Available online: https://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/ucm573706.htm (accessed on 29 January 2019).
- US Food and Drug Administration. Approved Products-YESCARTA (Axicabtagene Ciloleucel) [Internet]. 2018. Available online: https://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/ucm581222.htm (accessed on 29 January 2019).
- Pui, C.-H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J. Clin. Oncol. 2015, 33, 2938–2948. [Google Scholar] [CrossRef]
- Fielding, A.K.; Richards, S.M.; Chopra, R.; Lazarus, H.M.; Litzow, M.R.; Buck, G.; Durrant, I.J.; Luger, S.M.; Marks, D.I.; Franklin, I.M.; et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood 2007, 109, 944–950. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Spratt, K.; et al. Intent to treat leukemia remission by CD19CAR T cells of defined formulation and dose in children and young adults. Blood 2017. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res. 2013, 1, 26–31. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Carpenter, R.O.; Kassim, S.H.; Rose, J.J.; Telford, W.G.; Hakim, F.T.; Halverson, D.C.; Fowler, D.H.; Hardy, N.M.; et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013, 122, 4129–4139. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy-assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar]
- Hay, K.A.; Hanafi, L.-A.; Li, D.; Gust, J.; Liles, W.C.; Wurfel, M.M.; López, J.A.; Chen, J.; Chung, D.; Harju-Baker, S.; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017, 130, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.A.; Berger, C. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor–modified T cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Conrad Liles, W.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tasian,, S.K.; Gardner, R.A. CD19-redirected chimeric antigen receptor-modified T cells: A promising immunotherapy for children and adults with B-cell acute lymphoblastic leukemia (ALL). Ther. Adv. Hematol. 2015, 6, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2014, 257, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Brentjens, R.J. Tumors evading CARs-the chase is on. Nat. Med. 2018, 24, 1492–1493. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Sharifzadeh, Z.; Rahbarizadeh, F.; Shokrgozar, M.A.; Ahmadvand, D.; Mahboudi, F.; Jamnani, F.R.; Moghimi, S.M. Genetically engineered T cells bearing chimeric nanoconstructed receptors harboring TAG-72-specific camelid single domain antibodies as targeting agents. Cancer Lett. 2013, 334, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Ratnayake, M.; Savoldo, B.; Perlaky, L.; Dotti, G.; Wels, W.S.; Bhattacharjee, M.B.; Gilbertson, R.J.; Shine, H.D.; Weiss, H.L.; et al. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res. 2007, 67, 5957–5964. [Google Scholar] [CrossRef]
- Chow, K.K.H.; Naik, S.; Kakarla, S.; Brawley, V.S.; Shaffer, D.R.; Yi, Z.; Rainusso, N.; Wu, M.-F.; Liu, H.; Kew, Y.; et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol. Ther. 2013, 21, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Poussin, M.; Hagemann, I.S.; Coukos, G.; Sandaltzopoulos, R.; Scholler, N.; Powell, D.J., Jr. Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol. Ther. 2012, 20, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Heitzeneder, S.; Mackall, C.L. Harnessing the Immunotherapy Revolution for the Treatment of Childhood Cancers. Cancer Cell 2017, 31, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Sommermeyer, D.; Hill, T.; Shamah, S.M.; Salter, A.I.; Chen, Y.; Mohler, K.M.; Riddell, S.R. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia 2017, 31, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Bagashev, A.; Sotillo, E.; Tang, C.-H.A.; Black, K.L.; Perazzelli, J.; Seeholzer, S.H.; Argon, Y.; Barrett, D.M.; Grupp, S.A.; Hu, C.-C.A.; et al. CD19 alterations emerging after CD19-directed immunotherapy cause retention of the misfolded protein in the endoplasmic reticulum. Mol. Cell. Biol. 2018, 38, e00383-18. [Google Scholar] [CrossRef] [PubMed]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef]
- Alcantara, M.; Tesio, M.; June, C.H.; Houot, R. CAR T-cells for T-cell malignancies: Challenges in distinguishing between therapeutic, normal and neoplastic T-cells. Leukemia 2018, 32, 2307–2315. [Google Scholar] [CrossRef]
- Scarfò, I.; Maus, M.V. Current approaches to increase CAR T cell potency in solid tumors: Targeting the tumor microenvironment. J. Immunother Cancer 2017, 5, 28. [Google Scholar] [CrossRef]
- Thistlethwaite, F.C.; Gilham, D.E.; Guest, R.D.; Rothwell, D.G.; Pillai, M.; Burt, D.J.; Byatte, A.J.; Kirillova, N.; Valle, J.W.; Sharma, S.K.; et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017, 66, 1425–1436. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.J.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.J.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Sleijfer, S.; van Steenbergen, S.; van Elzakker, P.; van Krimpen, B.; Groot, C.; Vulto, A.; den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Manning, L. Faculty of 1000 Evaluation for Human Epidermal Growth Factor Receptor 2 (HER2)-Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma; F1000-Post-Publication Peer Review of the Biomedical Literature: London, UK, 2017. [Google Scholar]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lin, Q.; Song, Y.; Liu, D. Universal CARs, universal T cells and universal CAR T cells. J. Hematol. Oncol. 2018, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.L.; van der Stegen, S.; Lee, T.; Mansilla-Soto, J.; Chang, C.-W.; Sasaki, J.; Husain, M.; Peralta, E.; Hardy, I.; Ortiz, E.; et al. Abstract LB-108: Generation of off-the-shelf TCR-less CAR-targeted cytotoxic T cells from renewable pluripotent cells for cancer immunotherapy. Cancer Res. 2018, 78. [Google Scholar] [CrossRef]

{kind=link}
| CAR T-Cell Product | CAR Construct | FDA Approved Indications |
|---|---|---|
| Tisagenlecleucel (KYMRIAH) | CD19scFv/4-1BB/CD3ζ | • B-Cell acute lymphoblastic leukemia (ALL) that is refractory or in the second relapse in patients up to age 25 years [18] • Adult patients with (r/r) large B-Cell lymphoma after two or more lines of systemic therapy including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high-grade B-cell lymphoma and DLBCL arising from follicular lymphoma [18]. |
| Axicabtagene ciloleucel (YESCARTA) | CD19scFv/CD28/CD3ζ | • Adult patients with (r/r) large B-cell lymphoma after two or more lines of systemic therapy, DLBCL not otherwise specified, primary mediastinal large B-cell lymphoma, high-grade B-cell lymphoma and DLBCL arising from follicular lymphoma [19]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mchayleh, W.; Bedi, P.; Sehgal, R.; Solh, M. Chimeric Antigen Receptor T-Cells: The Future Is Now. J. Clin. Med. 2019, 8, 207. https://doi.org/10.3390/jcm8020207
Mchayleh W, Bedi P, Sehgal R, Solh M. Chimeric Antigen Receptor T-Cells: The Future Is Now. Journal of Clinical Medicine. 2019; 8(2):207. https://doi.org/10.3390/jcm8020207
Chicago/Turabian StyleMchayleh, Wassim, Prabhjot Bedi, Rajesh Sehgal, and Melhem Solh. 2019. "Chimeric Antigen Receptor T-Cells: The Future Is Now" Journal of Clinical Medicine 8, no. 2: 207. https://doi.org/10.3390/jcm8020207
APA StyleMchayleh, W., Bedi, P., Sehgal, R., & Solh, M. (2019). Chimeric Antigen Receptor T-Cells: The Future Is Now. Journal of Clinical Medicine, 8(2), 207. https://doi.org/10.3390/jcm8020207