Features of Autosomal Recessive Alport Syndrome: A Systematic Review

 , ,
, ,
Abstract
1. Introduction
2. Methods
2.1. Search Strategy and Data Extraction
2.2. Selection of Studies
2.3. Eligibility and Exclusion Criteria
2.4. Statistical Analysis
3. Results
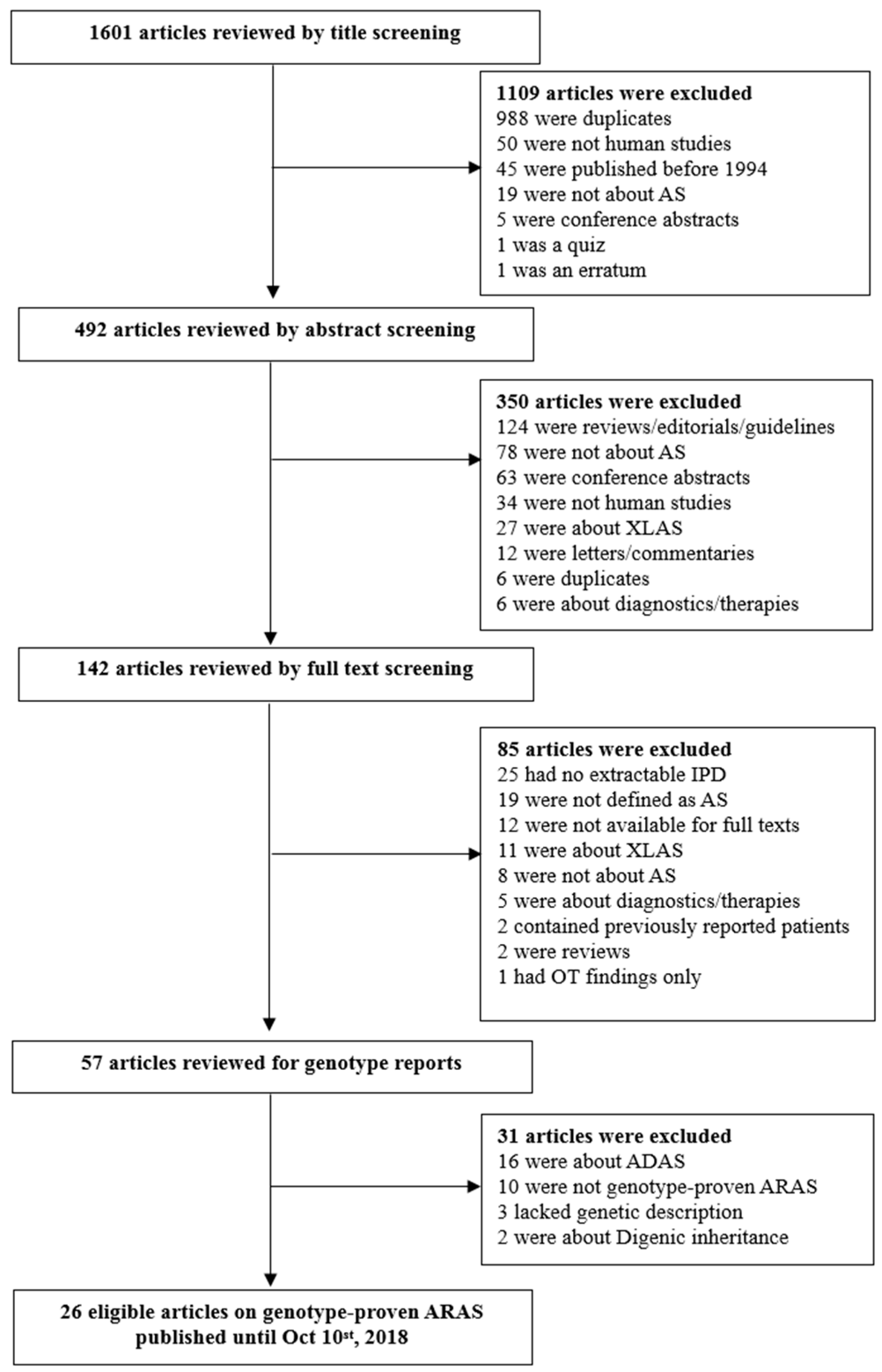
3.1. Study Selection and Characteristics
3.2. Clinical Features
3.3. Genotypephenotype Correlations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nozu, K.; Nakanishi, K.; Abe, Y.; Udagawa, T.; Okada, S.; Okamoto, T.; Kaito, H.; Kanemoto, K.; Kobayashi, A.; Tanaka, E.; et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clin. Exp. Nephrol. 2018. [Google Scholar] [CrossRef]
- Imafuku, A.; Nozu, K.; Sawa, N.; Hasegawa, E.; Hiramatsu, R.; Kawada, M.; Hoshino, J.; Tanaka, K.; Ishii, Y.; Takaichi, K.; et al. Autosomal dominant form of type IV collagen nephropathy exists among patients with hereditary nephritis difficult to diagnose clinicopathologically. Nephrology 2017, 23, 940–947. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. The Prisma Group Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. J. Clin. Epidemiol. 2009, 62, 1006–1012. [Google Scholar] [CrossRef]
- Savige, J.; Gregory, M.; Gross, O.; Kashtan, C.; Ding, J.; Flinter, F. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J. Am. Soc. Nephrol. 2013, 24, 364–375. [Google Scholar] [CrossRef]
- Lemmink, H.H.; Mochizuki, T.; Van Den Heuvel, L.P.W.J.; Schröder, C.H.; Barrientos, A.; Monnens, L.A.H.; Van Oost, B.A.; Brunner, H.G.; Reeders, S.T.; Smeets, H.J.M. Mutations in the type IV collagen a2 (COL4A3) gene in autosomal recessive Alport syndrome. Hum. Mol. Genet. 1994, 3, 1269–1273. [Google Scholar] [CrossRef]
- Mochizuki, T.; Lemmink, H.H.; Mariyama, M.; Antignac, C.; Gubler, M.C.; Pirson, Y.; Verellen-Dumoulin, C.; Chan, B.; Schröder, C.H.; Smeets, H.J.; et al. Identification of mutations in the α3(IV) and α4(IV) collagen genes in autosomal recessive Alport syndrome. Nat. Genet. 1994, 8, 77–82. [Google Scholar] [CrossRef]
- Oka, M.; Nozu, K.; Kaito, H.; Fu, X.J.; Nakanishi, K.; Hashimura, Y.; Morisada, N.; Yan, K.; Matsuo, M.; Yoshikawa, N.; et al. Natural history of genetically proven autosomal recessive Alport syndrome. Pediatr. Nephrol. 2014, 29, 1535–1544. [Google Scholar] [CrossRef]
- Chen, Y.; Colville, D.; Ierino, F.; Symons, A.; Savige, J. Temporal retinal thinning and the diagnosis of Alport syndrome and Thin basement membrane nephropathy. Ophthalmic Genet. 2018, 39, 208–214. [Google Scholar] [CrossRef]
- Savige, J.; Wang, Y.; Crawford, A.; Smith, J.; Symons, A.; Mack, H.; Nicholls, K.; Wilson, D.; Colville, D. Bull’s eye and pigment maculopathy are further retinal manifestations of an abnormal Bruch’s membrane in Alport syndrome. Ophthalmic Genet. 2017, 38, 238–244. [Google Scholar] [CrossRef]
- Nabais Sa, M.J.; Storey, H.; Flinter, F.; Nagel, M.; Sampaio, S.; Castro, R.; Araujo, J.A.; Gaspar, M.A.; Soares, C.; Oliveira, A.; et al. Collagen type IV-related nephropathies in Portugal: Pathogenic COL4A3 and COL4A4 mutations and clinical characterization of 25 families. Clin. Genet. 2015, 88, 456–461. [Google Scholar] [CrossRef]
- Wang, F.; Ding, J.; Zhang, H.; Zhang, Y.; Xiao, H.; Yao, Y.; Zhong, X.; Yu, L. Comparison of phenotypic features between patients with X-linked and autosomal recessive Alport syndrome. J. Peking Univ. Health Sci. 2014, 46, 311–314. [Google Scholar]
- Wang, Y.; Sivakumar, V.; Mohammad, M.; Colville, D.; Storey, H.; Flinter, F.; Dagher, H.; Savige, J. Clinical and genetic features in autosomal recessive and X-linked Alport syndrome. Pediatr. Nephrol. 2014, 29, 391–396. [Google Scholar] [CrossRef]
- Yao, X.D.; Chen, X.; Huang, G.Y.; Yu, Y.T.; Xu, S.T.; Hu, Y.L.; Wang, Q.W.; Chen, H.P.; Zeng, C.H.; Ji, D.X.; et al. Challenge in pathologic diagnosis of Alport syndrome: Evidence from correction of previous misdiagnosis. Orphanet. J. Rare Dis. 2012, 7, 100. [Google Scholar] [CrossRef]
- Temme, J.; Peters, F.; Lange, K.; Pirson, Y.; Heidet, L.; Torra, R.; Grunfeld, J.P.; Weber, M.; Licht, C.; Muller, G.A.; et al. Incidence of renal failure and nephroprotection by RAAS inhibition in heterozygous carriers of X-chromosomal and autosomal recessive Alport mutations. Kidney Int. 2012, 81, 779–783. [Google Scholar] [CrossRef]
- Artuso, R.; Fallerini, C.; Dosa, L.; Scionti, F.; Clementi, M.; Garosi, G.; Massella, L.; Epistolato, M.C.; Mancini, R.; Mari, F.; et al. Advances in Alport syndrome diagnosis using next-generation sequencing. Eur. J. Hum. Genet. 2012, 20, 50–57. [Google Scholar] [CrossRef]
- Pierides, A.; Voskarides, K.; Athanasiou, Y.; Ioannou, K.; Damianou, L.; Arsali, M.; Zavros, M.; Pierides, M.; Vargemezis, V.; Patsias, C.; et al. Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3 COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2009, 24, 2721–2729. [Google Scholar]
- Shaw, E.A.; Colville, D.; Wang, Y.Y.; Zhang, K.W.; Dagher, H.; Fassett, R.; Guymer, R.; Savige, J. Characterization of the peripheral retinopathy in X-linked and autosomal recessive Alport syndrome. Nephrol. Dial. Transplant. 2007, 22, 104–108. [Google Scholar] [CrossRef]
- Wei, G.; Zhihong, L.; Huiping, C.; Caihong, Z.; Zhaohong, C.; Leishi, L. Spectrum of clinical features and type IV collagen α-chain distribution in Chinese patients with Alport syndrome. Nephrol. Dial. Transplant. 2006, 21, 3146–3154. [Google Scholar] [CrossRef]
- Dagher, H.; Yan Wang, Y.; Fassett, R.; Savige, J. Three novel COL4A4 mutations resulting in stop codons and their clinical effects in autosomal recessive Alport syndrome. Hum. Mutat. 2002, 20, 321–322. [Google Scholar] [CrossRef]
- Heidet, L.; Cai, Y.; Guicharnaud, L.; Antignac, C.; Gubler, M.C. Glomerular expression of type IV collagen chains in normal and X-linked Alport syndrome kidneys. Am. J. Pathol. 2000, 156, 1901–1910. [Google Scholar] [CrossRef]
- Torra, R.; Badenas, C.; Cofán, F.; Callis, L.; Pérez-Oller, L.; Darnell, A. Autosomal recessive Alport syndrome: Linkage analysis and clinical features in two families. Nephrol. Dial. Transplant. 1999, 14, 627–630. [Google Scholar] [CrossRef]
- Boye, E.; Mollet, G.; Forestier, L.; Cohen-Solal, L.; Heidet, L.; Cochat, P.; Grünfeld, J.P.; Palcoux, J.B.; Gubler, M.C.; Antignac, C. Determination of the genomic structure of the COL4A4 gene and of novel mutations causing autosomal recessive Alport syndrome. Am. J. Hum. Genet. 1998, 63, 1329–1340. [Google Scholar] [CrossRef]
- Vos, P.; Zietse, R.; van Geel, M.; Brooks, A.S.; Cransberg, K. Diagnosing Alport Syndrome: Lessons from the Pediatric Ward. Nephron 2018, 140, 203–210. [Google Scholar] [CrossRef]
- Braunisch, M.C.; Buttner-Herold, M.; Gunthner, R.; Satanovskij, R.; Riedhammer, K.M.; Herr, P.M.; Klein, H.G.; Wahl, D.; Kuchle, C.; Renders, L.; et al. Heterozygous COL4A3 Variants in Histologically Diagnosed Focal Segmental Glomerulosclerosis. Front. Pediatr. 2018, 6, 171. [Google Scholar] [CrossRef]
- Truong, J.; Deschenes, G.; Callard, P.; Antignac, C.; Niel, O. Macroscopic hematuria with normal renal biopsy-following the chain to the diagnosis: Questions. Pediatr. Nephrol. 2017, 32, 277–278. [Google Scholar] [CrossRef]
- Truong, J.; Deschenes, G.; Callard, P.; Antignac, C.; Niel, O. Macroscopic hematuria with normal renal biopsy-following the chain to the diagnosis: Answers. Pediatr. Nephrol. 2017, 32, 279–281. [Google Scholar] [CrossRef]
- Papazachariou, L.; Papagregoriou, G.; Hadjipanagi, D.; Demosthenous, P.; Voskarides, K.; Koutsofti, C.; Stylianou, K.; Ioannou, P.; Xydakis, D.; Tzanakis, I.; et al. Frequent COL4 mutations in familial microhematuria accompanied by later-onset Alport nephropathy due to focal segmental glomerulosclerosis. Clin. Genet. 2017, 92, 517–527. [Google Scholar] [CrossRef]
- Liu, J.H.; Wei, X.X.; Li, A.; Cui, Y.X.; Xia, X.Y.; Qin, W.S.; Zhang, M.C.; Gao, E.Z.; Sun, J.; Gao, C.L.; et al. Novel mutations in COL4A3, COL4A4, and COL4A5 in Chinese patients with Alport Syndrome. PLoS ONE 2017, 12, e0177685. [Google Scholar] [CrossRef]
- Kamijo, M.; Kitamura, M.; Muta, K.; Uramatsu, T.; Obata, Y.; Nozu, K.; Kaito, H.; Iijima, K.; Mukae, H.; Nishino, T. A case of mild phenotype Alport syndrome caused by COL4A3 mutations. CEN Case Rep. 2017, 6, 189–193. [Google Scholar] [CrossRef]
- Ebner, K.; Reintjes, N.; Feldkötter, M.; Körber, F.; Nagel, M.; Dötsch, J.; Hoppe, B.; Weber, L.T.; Beck, B.B.; Liebau, M.C. A case report on the exceptional coincidence of two inherited renal disorders: ADPKD and Alport syndrome. Clin. Nephrol. 2017, 88, 45–51. [Google Scholar] [CrossRef]
- Uchida, N.; Kumagai, N.; Nozu, K.; Fu, X.J.; Iijima, K.; Kondo, Y.; Kure, S. Early RAAS Blockade Exerts Renoprotective Effects in Autosomal Recessive Alport Syndrome. Tohoku J. Exp. Med. 2016, 240, 251–257. [Google Scholar] [CrossRef]
- Nishizawa, Y.; Takei, T.; Miyaoka, T.; Kamei, D.; Mochizuki, T.; Nitta, K. Alport syndrome and pregnancy: Good obstetric and nephrological outcomes in a pregnant woman with homozygous autosomal recessive Alport syndrome. J. Obstet. Gynaecol. Res. 2016, 42, 331–335. [Google Scholar] [CrossRef]
- Gast, C.; Pengelly, R.J.; Lyon, M.; Bunyan, D.J.; Seaby, E.G.; Graham, N.; Venkat-Raman, G.; Ennis, S. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2016, 31, 961–970. [Google Scholar] [CrossRef]
- Sirisena, N.D.; Thalgahagoda, S.; Abeyagunawardena, A.; Neumann, M.; Schmudlach, H.O.; Jayasekara, R.W.; Dissanayake, V.H. Novel COL4A3 gene mutations in a consanguineous family with autosomal recessive Alport syndrome. Nephrology 2015, 20, 580. [Google Scholar] [CrossRef]
- Xie, J.; Wu, X.; Ren, H.; Wang, W.; Wang, Z.; Pan, X.; Hao, X.; Tong, J.; Ma, J.; Ye, Z.; et al. COL4A3 mutations cause focal segmental glomerulosclerosis. J. Mol. Cell Biol. 2014, 6, 498–505. [Google Scholar] [CrossRef]
- Webb, B.D.; Brandt, T.; Liu, L.; Jalas, C.; Liao, J.; Fedick, A.; Linderman, M.D.; Diaz, G.A.; Kornreich, R.; Trachtman, H.; et al. A founder mutation in COL4A3 causes autosomal recessive Alport syndrome in the Ashkenazi Jewish population. Clin. Genet. 2014, 86, 155–160. [Google Scholar] [CrossRef]
- Ramzan, K.; Imtiaz, F.; Taibah, K.; Alnufiee, S.; Akhtar, M.; Al-Hazzaa, S.A.F.; Al-Owain, M. COL4A4-related nephropathy caused by a novel mutation in a large consanguineous Saudi family. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 427–432. [Google Scholar] [CrossRef]
- Fu, X.J.; Morisada, N.; Hashimoto, F.; Taniguchi-Ikeda, M.; Hashimura, Y.; Ohtsubo, H.; Ninchoji, T.; Kaito, H.; Nozu, K.; Takahashi, E.; et al. A patient with autosomal recessive Alport syndrome due to segmental maternal isodisomy. Hum. Genome Var. 2014, 1, 14006. [Google Scholar] [CrossRef]
- Anazi, S.; Al-Sabban, E.; Alkuraya, F.S. Gonadal mosaicism as a rare cause of autosomal recessive inheritance. Clin. Genet. 2014, 85, 278–281. [Google Scholar] [CrossRef]
- Uzak, A.S.; Tokgoz, B.; Dundar, M.; Tekin, M. A novel COL4A3 mutation causes autosomal-recessive Alport syndrome in a large Turkish family. Genet. Test. Mol. Biomarkers 2013, 17, 260–264. [Google Scholar] [CrossRef]
- Storey, H.; Savige, J.; Sivakumar, V.; Abbs, S.; Flinter, F.A. COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J. Am. Soc. Nephrol. 2013, 24, 1945–1954. [Google Scholar] [CrossRef]
- Kaimori, J.Y.; Ichimaru, N.; Isaka, Y.; Hashimoto, F.; Fu, X.; Hashimura, Y.; Kaito, H.; Iijima, K.; Kyo, M.; Namba, T.; et al. Renal transplantations from parents to siblings with autosomal recessive Alport syndrome caused by a rearrangement in an intronic antisense Alu element in the COL4A3 gene led to different outcomes. CEN Case Rep. 2013, 2, 98–101. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, F.; Ding, J.; Zhang, H.; Zhao, D.; Yu, L.; Xiao, H.; Yao, Y.; Zhong, X.; Wang, S. Genotype-phenotype correlations in 17 Chinese patients with autosomal recessive Alport syndrome. Am. J. Med. Genet. A 2012, 158, 2188–2193. [Google Scholar] [CrossRef]
- Cook, C.; Friedrich, C.A.; Baliga, R. Novel COL4A3 mutations in African American siblings with autosomal recessive Alport syndrome. Am. J. Kidney Dis. 2008, 51, e25-8. [Google Scholar] [CrossRef]
- Rana, K.; Tonna, S.; Wang, Y.Y.; Sin, L.; Lin, T.; Shaw, E.; Mookerjee, I.; Savige, J. Nine novel COL4A3 and COL4A4 mutations and polymorphisms identified in inherited membrane diseases. Pediatr. Nephrol. 2007, 22, 652–657. [Google Scholar] [CrossRef]
- Hou, P.; Chen, Y.; Ding, J.; Li, G.; Zhang, H. A novel mutation of COL4A3 presents a different contribution to Alport syndrome and thin basement membrane nephropathy. Am. J. Nephrol. 2007, 27, 538–544. [Google Scholar] [CrossRef]
- Longo, I.; Scala, E.; Mari, F.; Caselli, R.; Pescucci, C.; Mencarelli, M.A.; Speciale, C.; Giani, M.; Bresin, E.; Caringella, D.A.; et al. Autosomal recessive Alport syndrome: An in-depth clinical and molecular analysis of five families. Nephrol. Dial. Transplant. 2006, 21, 665–671. [Google Scholar] [CrossRef]
- Vega, B.T.; Badenas, C.; Ars, E.; Lens, X.; Milà, M.; Darnell, A.; Torra, R. Autosomal Recessive Alport’s Syndrome and Benign Familial Hematuria Are Collagen Type IV Diseases. Am. J. Kidney Dis. 2003, 42, 952–959. [Google Scholar] [CrossRef]
- Jais, J.P.; Knebelmann, B.; Giatras, I.; De Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, K.O.; Flinter, F.; et al. X-linked Alport syndrome: Natural history in 195 families and genotype- phenotype correlations in males. J. Am. Soc. Nephrol. 2000, 11, 649–657. [Google Scholar]
- Jais, J.P.; Knebelmann, B.; Giatras, I.; De Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, K.O.; Flinter, F.; et al. X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport Syndrome Concerted Action” study. J. Am. Soc. Nephrol. 2003, 14, 2603–2610. [Google Scholar] [CrossRef]
- Yamamura, T.; Nozu, K.; Fu, X.J.; Nozu, Y.; Ye, M.J.; Shono, A.; Yamanouchi, S.; Minamikawa, S.; Morisada, N.; Nakanishi, K.; et al. Natural History and Genotype-Phenotype Correlation in Female X-Linked Alport Syndrome. Kidney Int. Rep. 2017, 2, 850–855. [Google Scholar] [CrossRef]
- Gross, O.; Netzer, K.O.; Lambrecht, R.; Seibold, S.; Weber, M. Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: Impact on clinical counselling. Nephrol. Dial. Transplant. 2002, 17, 1218–1227. [Google Scholar] [CrossRef]
- Bekheirnia, M.R.; Reed, B.; Gregory, M.C.; McFann, K.; Shamshirsaz, A.A.; Masoumi, A.; Schrier, R.W. Genotype-phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 2010, 21, 876–883. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author, Year, Reference | N° Patients † | Sex (M/F) | Ethnicity | Renal Manifestation Frequency (Age in Median Years) | Extrarenal Manifestation Frequency (Age in Median Years) | Age at (Median Years) | Genotypes | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HU | PU | ESRD | TPL | Initial Pathology | SNHL | Ocular Lesions | Dx of ARAS | Last F-U | Causative Gene | N° Missense Mutations (N° Patients) | ||||
| Vos, 2018 [23] | 2 | 2/0 | n/a | 2/2 (2) | n/a | n/a | n/a | 1 AS 1 normal | n/a | n/a | n/a | n/a | 1 COL4A3, 1 COL4A4 | 0 missense (1) 1 missense (1) |
| Braunish, 2018 [24] | 1 | 0/1 | Caucasian | 1/1 (12) | 1/1 (n/a) | 0/1 | 0/1 | FSGS | 1/1 (34) | 0/1 | 34 | 34 | 1 COL4A3 | 2 missense (1) |
| Truong, 2017 [25,26] | 1 | 1/0 | Caucasian | 1/1 (n/a) | 1/1 (4.5) | 0/1 | 0/1 | Normal | n/a | n/a | n/a | 10 | 1 COL4A3 | 0 missense (1) |
| Papazachariou, 2017 [27] | 7 | 4/3 | Caucasian | 7/7 (19) | 7/7 (n/a) | 2/7 (21) | 1/7 (22) | 3 FSGS 1TBMD | 2/7 (n/a) | n/a | n/a | n/a | 7 COL4A4 | 0 missense (2) 2 missense (5) |
| Liu,2017 [28] | 3 | n/a | Asian | n/a | n/a | n/a | n/a | n/a | 3/3 (n/a) | n/a | n/a | n/a | 2 COL4A3, 1 COL4A4 | 2 missense (3) |
| Kamijo, 2017 [29] | 1 | 1/0 | Asian | 1/1 (n/a) | 1/1 (n/a) | 0/1 | n/a | AS | 1/1 (37) | 0/1 | 41 | 41 | 1 COL4A3 | 0 missense (1) |
| Ebner, 2017 [30] | 2 | 2/0 | Caucasian | 2/2 (0.7) | 2/2 (n/a) | 0/2 | n/a | AS | 2/2 (6) | 1/2 (n/a) | n/a | 19 | 2 COL4A3 | 0 missense (2) |
| Uchida, 2016 [31] | 4 | 3/1 | Asian | 4/4 (n/a) | 4/4 (n/a) | 2/4 (30) | n/a | AS | 1/4 (n/a) | 1/4 (n/a) | 6.5 | 24.5 | 4 COL4A3 | 2 missense (4) |
| Nishizawa, 2016 [32] | 1 | 0/1 | Asian | 1/1 (6) | 1/1 (23) | n/a | n/a | AS | n/a | n/a | 27 | n/a | 1 COL4A4 | 2 missense (1) |
| Gast, 2016 [33] | 2 | 1/1 | n/a | 2/2 (n/a) | n/a | 0/2 | n/a | 2 FSGS | n/a | n/a | 33 | n/a | 2 COL4A3 | 2 missense (2) |
| Sirisena, 2015 [34] | 4 | 1/3 | Caucasian | 4/4 (n/a) | 4/4 (n/a) | 1/4 (15) | n/a | 2 MPGN 1FSGS | 3/4 (n/a) | 0/4 | 42 | n/a | 4 COL4A3 | 0 missense (4) |
| Xie, 2014 [35] | 2 | 2/0 | Asian | 2/2 (1) | 2/2 (19) | n/a | n/a | n/a | 2/2 (n/a) | n/a | n/a | n/a | 1 COL4A3, 1 COL4A4 | 0 missense (1) 2 missense (1) |
| Webb, 2014 [36] | 3 | 0/3 | Caucasian | 3/3 (2) | 3/3 (2) | n/a | n/a | AS | 3/3 (n/a) | 0/3 | n/a | n/a | 3 COL4A3 | 0 missense (3) |
| Ramzan, 2014 [37] | 3 | 1/2 | Caucasian | 3/3 (n/a) | 3/3 (n/a) | 3/3 (20) | 1/3 (25) | 1 AS | 3/3 (7) | 2/3 (31) | n/a | 31 | 3 COL4A4 | 0 missense (3) |
| Oka, 2014 [7] | 30 | 14/16 | Asian | 30/30 (n/a) | n/a | 13/30 | n/a | 2 TBMD 24 AS | 12/30 (14.5) | 3/30 (n/a) | 18 | n/a | 23 COL4A3, 7 COL4A3 | 0 missense (6) 1 missense (13) 2 missense (11) |
| Fu, 2014 [38] | 1 | 1/0 | Asian | 1/1 (2) | 1/1 (2) | 1/1 (n/a) | 1/1 (22) | TBMD | 1/1 (17) | 0/1 | 7 | n/a | 1 COL4A3 | 2 missense (1) |
| Anazi, 2014 [39] | 3 | 1/2 | Caucasian | 3/3 (n/a) | 3/3 (5.5) | 1/3 (12) | 2/3 (12) | AS | 1/3 (n/a) | 0/3 | n/a | n/a | 3 COL4A4 | 0 missense (3) |
| Uzak, 2013 [40] | 4 | 3/1 | Caucasian | 4/4 (n/a) | 4.4 (n/a) | 4/4 (15) | 3/4 (25) | n/a | 4/4 (n/a) | 4/4 (n/a) | 29 | n/a | 4 COL4A3 | 0 missense (4) |
| Storey, 2013 [41] | 40 | 19/21 | Caucasian ‡ | 40/40 (n/a) | n/a | 20/40 (22.5) | n/a | 1 TBMD 39 AS | 23/40 (n/a) | 10/40 (n/a) | 31 | n/a | 20 COL4A3, 20 COL4A4 | 0 missense (20) 1 missense (12) 2 missense (8) |
| Kaimori, 2013 [42] | 2 | 1/1 | Asian | 2/2 (n/a) | 2/2 (n/a) | 2/2 (20.5) | 2/2 (19.5) | AS | 1/2 (5) | 1/2 (n/a) | 10 | n/a | 2 COL4A3 | 0 missense (2) |
| Zhang, 2012 [43] | 15 | 8/7 | Asian | 15/15 (3.8) | n/a | n/a | n/a | n/a | 7/15 (n/a) | 1/15 (n/a) | 7.5 | n/a | 13 COL4A3, 2 COL4A4 | 0 missense (8) 1 missense (5) 2 missense (2) |
| Cook, 2008 [44] | 2 | 0/2 | African | 2/2 (n/a) | 2/2 (n/a) | 1/2 (14) | 1/1 (15) | n/a | 2/2 (12) | 0/2 | 12.5 | 15.5 | 2 COL4A3 | 0 missense (2) |
| Rana, 2007 [45] | 1 | 1/0 | n/a | 1/1 (n/a) | 1/1 (n/a) | 1/1 (n/a) | n/a | n/a | 1/1 (n/a) | 1/1 (n/a) | 55 | 55 | 1 COL4A3 | 0 missense (1) |
| Hou, 2007 [46] | 1 | 1/0 | Asian | 1/1 (n/a) | 1/1 (n/a) | 1/1 (30) | n/a | AS | 1/1 (n/a) | 1/1 (n/a) | 28 | n/a | 1 COL4A3 | 2 missense (1) |
| Longo, 2006 [47] | 6 | 2/4 | Caucasian | 6/6 (7) | 6/6 (21) | 3/6 (20.5) | 3/6 (22) | 1 TBMD 2AS | 2/6 (27.5) | 1/6 (32) | 27.5 | 31 | 1 COL4A3, 5 COL4A4 | 0 missense (2) 2 missense (4) |
| Vega, 2003 [48] | 7 | 2/5 | n/a | 7/7 (n/a) | 7/7 (n/a) | 3/7 (26.5) | n/a | n/a | 6/7 (n/a) | 2/7 (n/a) | n/a | n/a | 6 COL4A3, 1 COL4A4 | 0 missense (3) 2 missense (4) |
| Total (n = 148) | Without Missense (n = 69) | With Missense (n = 79) | p | |
|---|---|---|---|---|
| N˚ (%) | N˚ (%) | N˚ (%) | Without vs. With | |
| Causative gene | ||||
| COL4A3 | 96 (65%) | 46 | 50 | 0.668 |
| COL4A4 | 52 (35%) | 23 | 29 | |
| Ethnicity | ||||
| Caucasian | 53 | 32/52 (62%) | 21/63 (33%) | 0.001 |
| Asian | 60 | 18/52 (35%) | 42/63 (67%) | |
| African | 2 | 2/52 (4%) | 0/63 (0%) | |
| n/a | 33 | 17 | 16 | |
| Family | ||||
| Consanguineous | 31/102 (30%) | 27/57 (47%) | 6/39 (15%) | <0.001 |
| Positive family Hx | 44/57 (77%) | 27/35 (77%) | 14/22 (63%) | 0.991 |
| Sex | ||||
| Male | 71/145 (49%) | 36/69 (52%) | 35/76 (44%) | 0.461 |
| Female | 74/145 (51%) | 33/69 (48%) | 41/76 (52%) | |
| n/a | 3 (2%) | 0 | 3 (4%) | |
| Age (median, years) | ||||
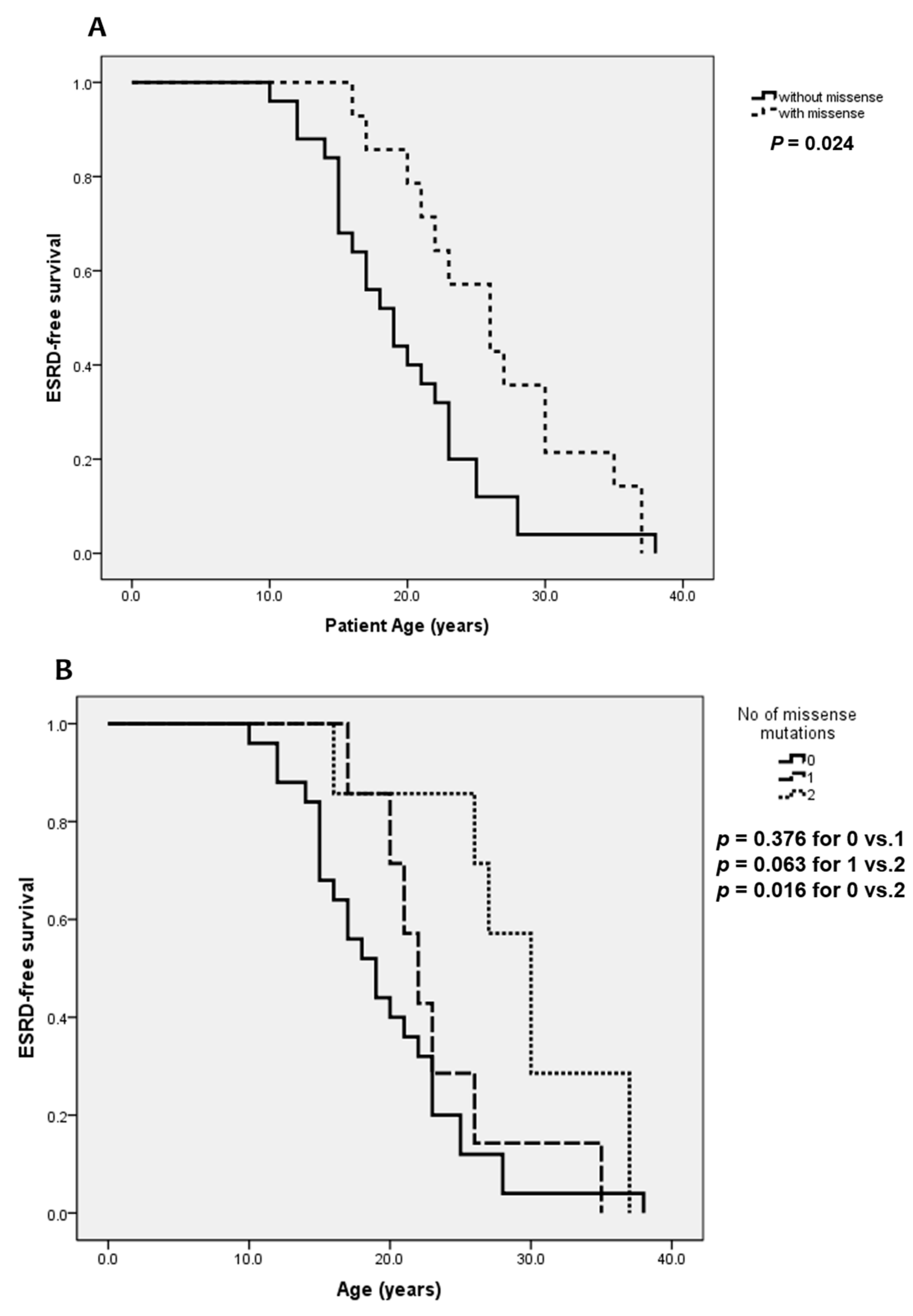
| HU | 2.5 | 2.0 | 5.6 | 0.004 |
| PU | 6.5 | 3.8 | 20 | 0.044 |
| Diagnosis | 20 | 20 | 19.5 | 0.231 |
| ESRD | 21 | 19 | 26 | 0.006 |
| TPL | 20 | 19 | 25.5 | 0.088 |
| SNHL | 13 | 6.5 | 18 | 0.019 |
| OT | 32 (2 cases) | 32 (2 cases) | n/a | - |
| Last F-U | 27 | 19 | 27 | 0.516 |
| Renal | ||||
| HU | 93/93 | 54/54 | 39/39 | - |
| PU | 89/89 | 53/53 | 36/36 | - |
| ESRD | 59/95 (62%) | 34/48 (71%) | 25/47 (54%) | 0.076 |
| TPL | 14/21 (67%) | 12/17 (71%) | 2/4 (50%) | 0.587 |
| Extrarenal | ||||
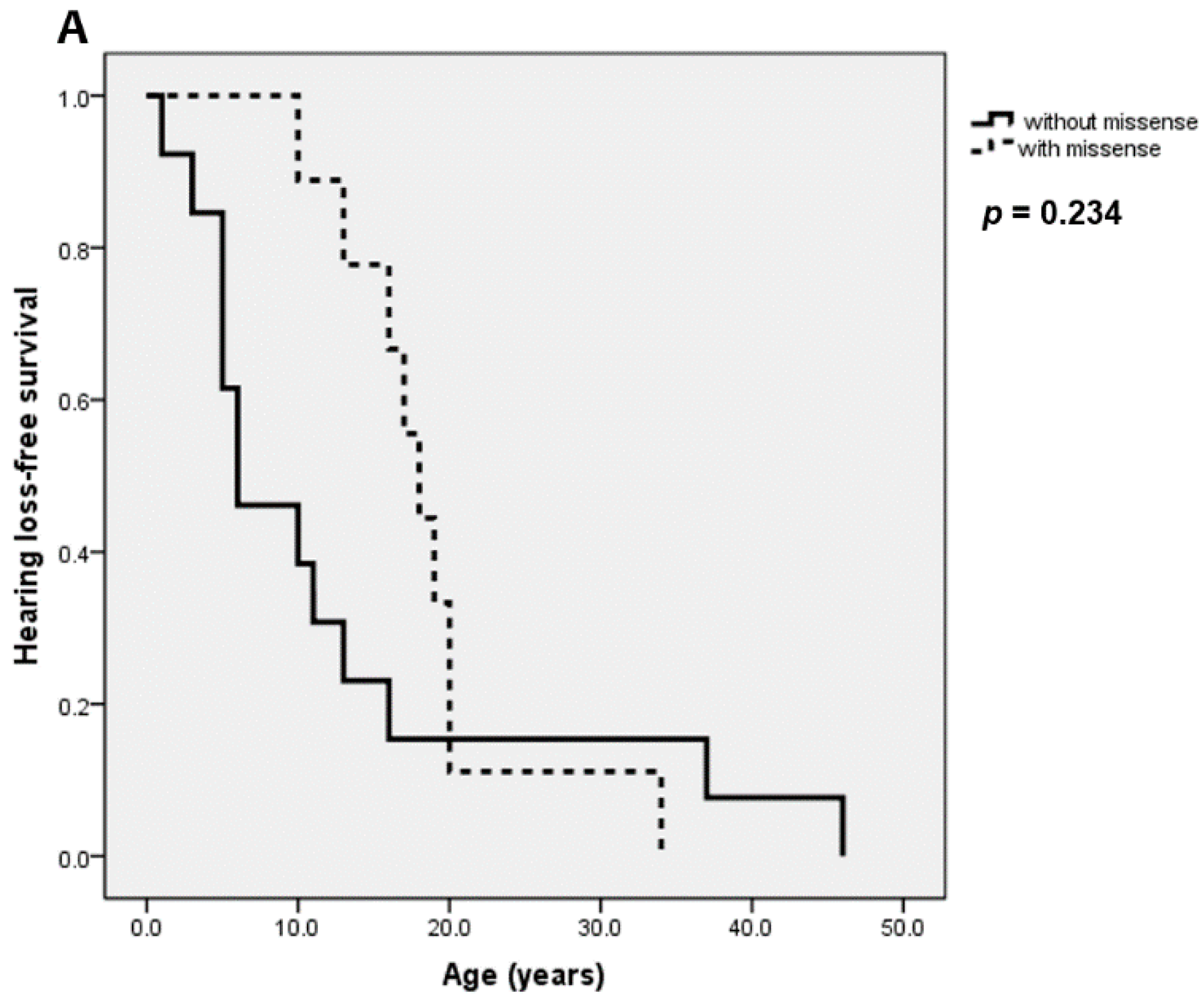
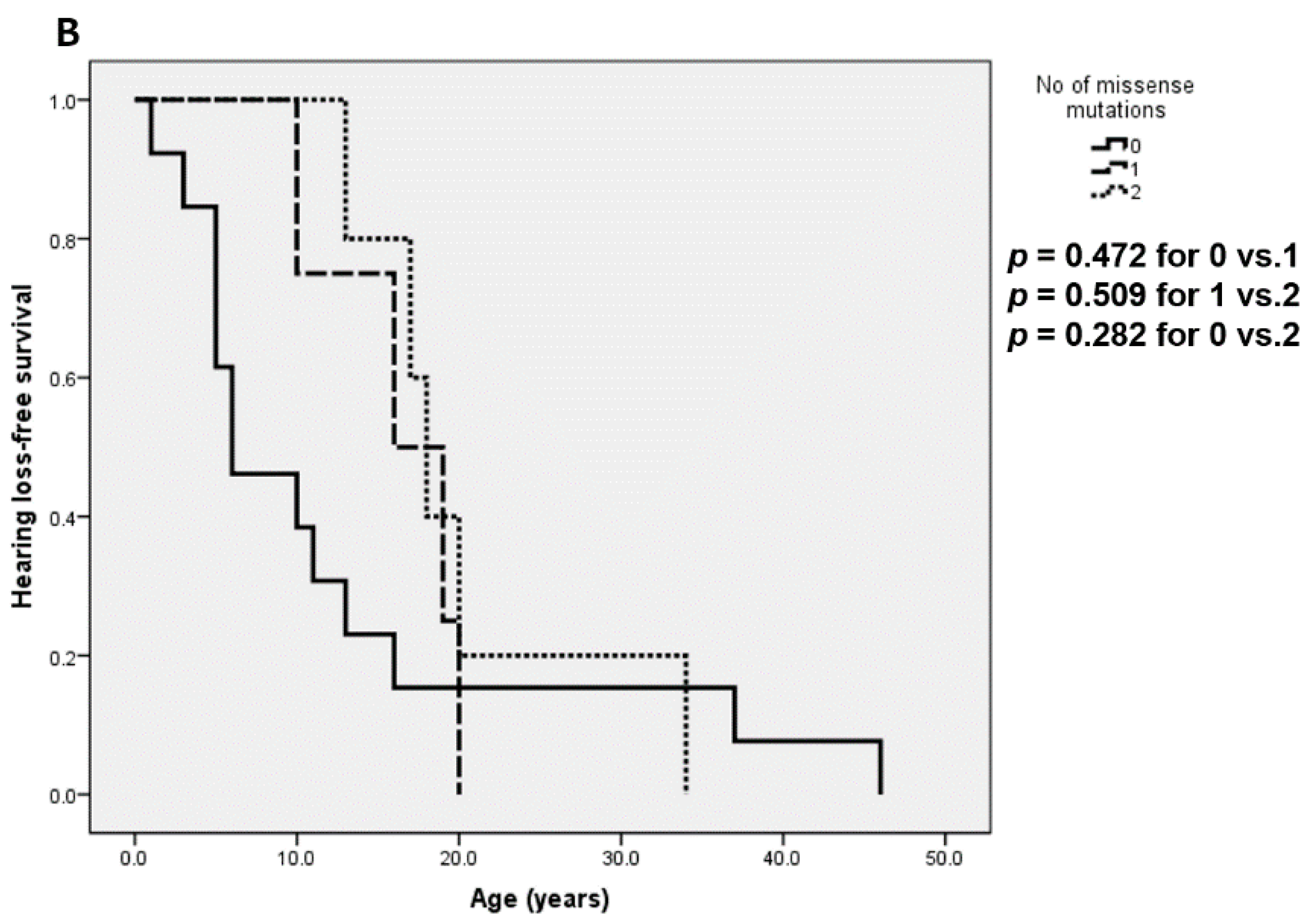
| SNHL | 82/129 (64%) | 54/62 (87%) | 28/67 (42%) | <0.001 |
| OT | 15/88 (17%) | 12/42 (29%) | 3/46 (6%) | 0.006 |
| Outcome | ||||
| Alive | 147 | 69 | 78 | |
| Death | 1 | 0 | 1 | |
| n/a | 0 | 0 | 0 |
| Study | N˚ patients † | Age at Biopsy (year) | LM | EM | Collagen IV Stain in GBM | Initial–Pathology Diagnosis | Mutation | Zygosity | N˚ Missense |
|---|---|---|---|---|---|---|---|---|---|
| Vos, 2018 [23] | 2/2 | n/a | Normal | Splitting | α4:(−) | AS | Truncating | H | 0 |
| Vos, 2018 [23] | n/a | Normal | Normal | Normal | Normal | Splicing/missense | ch | 1 | |
| Braunisch, 2018 [24] | 1/1 | 21 (1st) ‡ 32 (2nd) ‡ | Normal (1st) 4/11 GS (2nd) | Normal (1st) lamellation (2nd) | n/a | Nonspecific (1st) → AS (2nd) | Missense | ch | 2 |
| Truong, 2017 [25,26] | 1/1 | 4.6 | Normal | Normal | Normal | Normal | Duplication | H | 0 |
| Papazachariou, 2017 [27] | 4/7 | n/a | FSGS | n/a | n/a | FSGS | Truncating | H | 0 |
| n/a | FSGS | n/a | n/a | FSGS | Truncating | H | 0 | ||
| n/a | Normal | Thinning | n/a | TBMD | Missense | H | 2 | ||
| n/a | FSGS | n/a | n/a | FSGS | Missense | H | 2 | ||
| Liu, 2017 [28] | 3/3 | 15 | FSGS | Lamellation | α3:mosaic, α5:mosaic | AS/FSGS | Missense | ch | 2 |
| 13 | FSGS | Lamellation | α3:mosaic, α5:(−) | AS/FSGS | Missense | H | 2 | ||
| 18 | Lamellation | α3:mosaic, α5:mosaic | AS | Missense | H | 2 | |||
| Kamijo, 2017 [29] | 1/1 | 39 | GS + SS | Lamellation splitting | α5:(+) | Atypical AS | Splicing | ch | 0 |
| Ebner, 2017 [30] | 1/2 | 4 | n/a | Irregularity | n/a | AS | Truncating | H | 0 |
| Uchida, 2016 [31] | 2/4 | 7 | Mesangial proliferation | Lamellation | α5:(−) | AS | Missense | ch | 2 |
| 6 | Mesangial proliferation | Lamellation | α5:(−) | AS | Missense | ch | 2 | ||
| Nishizawa, 2016 [32] | 1/1 | 27 | Normal | Lamellation | α5:reduced | AS | Missense | H | 2 |
| Gast, 2016 [33] | 2/2 | n/a | FSGS | Splitting | n/a | FSGS | Missense | ch | 2 |
| n/a | Normal | n/a | n/a | Normal | Missense | ch | 2 | ||
| Sirisena, 2015 [34] | 1/4 | 14 | FSGS | n/a | n/a | FSGS | Truncating | H | 0 |
| Xie, 2014 [35] | 2/2 | n/a | n/a | n/a | α3:(−), α5:(−) | AS | Truncating | H | 0 |
| n/a | n/a | n/a | α3:(−), α5:(−) | AS | Missense | H | 2 | ||
| Webb, 2014 [36] | 1/3 | 3 | n/a | Lamellation | n/a | AS | Deletion | H | 0 |
| Ramzan, 2014 [37] | 1/3 | 15 | n/a | Lamellation | n/a | AS | Truncating | H | 0 |
| Oka, 2014 [7] | 30/30 | n/a | n/a | BWC | α3:(+), α4:(+), α5:(+) | AS | Missense | ch | 2 |
| n/a | n/a | BWC | α5:(+) | AS | Splicing/missense | ch | 1 | ||
| n/a | n/a | BWC | α5:(−) | AS | Splicing/truncating | ch | 0 | ||
| n/a | n/a | BWC | α5:(−) | AS | Missense | H | 2 | ||
| n/a | n/a | BWC | α5:(−) | AS | Truncating | ch | 0 | ||
| n/a | n/a | BWC | α5:(−) | AS | Missense | ch | 2 | ||
| n/a | n/a | BWC | n/a | AS | Missense | ch | 2 | ||
| n/a | n/a | n/a | n/a | AS | Missense | ch | 2 | ||
| n/a | n/a | BWC | n/a | AS | Missense | ch | 2 | ||
| n/a | n/a | n/a | n/a | AS | Missense | ch | 2 | ||
| n/a | n/a | BWC | n/a | AS | Deletion/missense | ch | 1 | ||
| n/a | n/a | BWC | n/a | AS | Deletion/missense | ch | 1 | ||
| n/a | n/a | TBMD | α5:(−) | n/a | Missense/truncating | ch | 1 | ||
| n/a | n/a | BWC | α5:(+) | AS | Missense | H | 2 | ||
| n/a | n/a | BWC | n/a | AS | Splicing | H | 2 | ||
| n/a | n/a | n/a | n/a | AS | Splicing | H | 2 | ||
| n/a | n/a | BWC | α5:(−) | AS | Missense/insertion | ch | 1 | ||
| n/a | n/a | TBMD | α5:(−) | n/a | Missense/splicing | ch | 1 | ||
| n/a | n/a | BWC | α5:(−) | AS | Truncating/missense | ch | 1 | ||
| n/a | n/a | BWC | α5:(−) | AS | Missense/deletion | ch | 1 | ||
| n/a | n/a | BWC | α5:(−) | AS | Missense | H | 2 | ||
| n/a | n/a | BWC | α3:(−), α4:(−), α5:(−) | AS | Missense/deletion | ch | 1 | ||
| n/a | n/a | BWC | α3:(+), α4:(+), α5:(+) | AS | Missense/truncating | ch | 1 | ||
| n/a | n/a | BWC | n/a | AS | Deletion | H | 2 | ||
| n/a | n/a | n/a | n/a | AS | deletion | H | 2 | ||
| n/a | n/a | BWC | α3:(−), α4:(−), α5:(−) | AS | Truncating/missense | ch | 1 | ||
| n/a | n/a | BWC | α5:(−) | AS | Missense | ch | 2 | ||
| n/a | n/a | BWC | α5:(−) | AS | Missense/truncating | ch | 1 | ||
| n/a | n/a | BWC | α5:(−) | AS | Missense | ch | 2 | ||
| n/a | n/a | BWC | α5:(−) | AS | Missense/deletion | ch | 1 | ||
| Fu, 2014 [38] | 1/1 | 7 | n/a | Thinning | α5:(−) | AS | Missense | H | 2 |
| Anazi, 2014 [39] | 2/3 | 8.5 | n/a | Lamellation | n/a | AS | Truncating | ch | 0 |
| n/a | n/a | Lamellation | n/a | AS | Truncating | ch | 0 | ||
| Hou, 2007 [46] | 1/1 | 26 | n/a | Lamellation | α3:(−), α4:(−), α5:(−) | AS | Missense | H | 2 |
| Longo, 2006 [47] | 3/6 | 5 | n/a | Thinning | n/a | TBMD | Missense | ch | 2 |
| 24 | n/a | Lamellation | n/a | AS | Missense | H | 2 | ||
| 22 | n/a | Splitting | n/a | AS | Missense | H | 2 | ||
| Pooled data | 60 (49 EM) | 14 median years | 7/49 FSGS 7 normal | 42/49 AS 5 TBMD 2 normal | 23/34 absent α5 3/34 abnormal α5 8/34 normal α5 | 47/58 AS 5 FSGS 4 normal 2 TBMD | |||
| N° Missense Mutations | 0 Missense (n = 69) | 1 Missense (n = 31) | 2 Missense (n = 48) | p |
|---|---|---|---|---|
| N˚ (%) | N˚ (%) | N˚ (%) | 0 vs. 2 | |
| Causative gene | ||||
| COL4A3 | 46 | 20 | 30 | 0.642 |
| COL4A4 | 23 | 11 | 18 | |
| Ethnicity | ||||
| Caucasian | 32/52 (62%) | 5/23 (22%) | 16/40 (40%) | 0.002 |
| Asian | 18/52 (35%) | 18/23 (78%) | 24/40 (60%) | |
| African | 2/52 (4%) | 0/23 (0%) | 0/40 (0%) | |
| ND | 17 | 8 | 8 | |
| Family | ||||
| Consanguineous | 27/57 (47%) | 0/18 (0%) | 6/21 (29%) | 0.028 |
| Positive family Hx | 27/35 (77%) | 3/6 (50%) | 14/16 (88%) | 0.387 |
| Sex | ||||
| Male | 36 (52%) | 14/31 (45%) | 21/45 (47%) | 0.565 |
| Female | 33 (48%) | 17/31 (55%) | 24/45 (53%) | |
| n/a | 0 | 0 | 3 | |
| Age (median, years) | ||||
| HU | 2.0 | 3.4 | 10.5 | 0.005 |
| PU | 3.8 | n/a | 20 | 0.044 |
| Diagnosis | 20 | 17 | 20 | 0.612 |
| ESRD | 19 | 22 | 30 | 0.005 |
| TPL | 19 | n/a | 25.5 | 0.088 |
| SNHL | 6.5 | 17.5 | 18 | 0.038 |
| OT | 32 (2 cases) | n/a | n/a | - |
| Last F-U | 19 | n/a | 27 | 0.516 |
| Renal | ||||
| HU | 54/54 | 13/13 | 26/26 | - |
| PU | 53/53 | n/a | 26/26 | - |
| ESRD | 34/48 (71%) | 11/20 (55%) | 14/27 (52%) | 0.100 |
| TPL | 12/17 (71%) | n/a | 2/4 (50%) | 0.587 |
| Extrarenal | ||||
| SNHL | 54/62 (87%) | 10/25 (40%) | 18/42 (43%) | <0.001 |
| OT | 12/42 (29%) | 2/20 (10%) | 1/26 (3%) | 0.012 |
| Outcome | ||||
| Alive | 69 | 31 | 47 | - |
| Death | 0 | 0 | 1 | |
| n/a | 0 | 0 | 0 |
| Author | N˚ ARAS | ESRD | SNHL | OT | |||
|---|---|---|---|---|---|---|---|
| N˚ | N˚ (%) | Median Age | N˚ (%) | Median Age | N˚ (%) | Median Age | |
| Chen, 2018 ‡ [8] | 4 | n/a | 23 | 3/4 (75%) | n/a | 4/4 (100%) | n/a |
| Savige, 2017 ‡ [9] | 13 | 12/13 (92%) | n/a | 12/13 (92%) | n/a | 13/13 (100%) | n/a |
| Nabais, 2015 [10] | 15 | 15/15 (100%) | 23 | 9/10 (90%) | 32 | 3/9 (30%) | 30 |
| Wang, 2014 [11] | 14 | n/a | n/a | 6/9 (67%) | n/a | 0/8 (0%) | n/a |
| Wang, 2014 ‡ [12] | 15 | 14/15 (93%) | 27.2 | 15/15 (100%) | n/a | 13/15 (87%) | n/a |
| Yao, 2012 [13] | 24 | n/a | n/a | 8/24 (30%) | n/a | 7/12 (58%) | n/a |
| Temme, 2012 [14] | 29 | 3/29 | n/a | n/a | n/a | n/a | n/a |
| Artuso, 2012 [15] | 2 | 1/2 (50) | n/a | n/a | n/a | n/a | n/a |
| Pierides, 2009 [16] | 42 | 18/42 (43%) | n/a | n/a | n/a | n/a | n/a |
| Shaw, 2007 [17] | 7 | 6/6 (100%) | 25 | 7/7 (100%) | 32 | 7/7 (100%) | 32 |
| Wei, 2006 [18] | 13 | 3/13 (23%) | 17 | 6/12 (50%) | 22 | 3/12 (25%) | 26 |
| Dagher, 2002 ‡ [19] | 11 | 8/11 (72%) | 24 | 10/11 (91%) | n/a | 10/11 (91%) | n/a |
| Heidet, 2000 [20] | 60 | 44/60 (73%) | 22 | 27/35 (77%) | n/a | 16/26 (62%) | n/a |
| Torra, 1999 [21] | 5 | 2/5 (40%) | 33 | 4/5 (80%) | 13.5 | 0/5 (0%) | n/a |
| Boye, 1998 [22] | 10 | 8/31 (26%) | n/a | n/a | n/a | n/a | n/a |
| Pooled data | 264 | 134/242 (55%) | 23.5 | 107/145 (74%) | 27 | 76/115 (66%) | 30 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.M.; Nozu, K.; Choi, D.E.; Kang, H.G.; Ha, I.-S.; Cheong, H.I. Features of Autosomal Recessive Alport Syndrome: A Systematic Review. J. Clin. Med. 2019, 8, 178. https://doi.org/10.3390/jcm8020178
Lee JM, Nozu K, Choi DE, Kang HG, Ha I-S, Cheong HI. Features of Autosomal Recessive Alport Syndrome: A Systematic Review. Journal of Clinical Medicine. 2019; 8(2):178. https://doi.org/10.3390/jcm8020178
Chicago/Turabian StyleLee, Jiwon M., Kandai Nozu, Dae Eun Choi, Hee Gyung Kang, II-Soo Ha, and Hae II Cheong. 2019. "Features of Autosomal Recessive Alport Syndrome: A Systematic Review" Journal of Clinical Medicine 8, no. 2: 178. https://doi.org/10.3390/jcm8020178
APA StyleLee, J. M., Nozu, K., Choi, D. E., Kang, H. G., Ha, I.-S., & Cheong, H. I. (2019). Features of Autosomal Recessive Alport Syndrome: A Systematic Review. Journal of Clinical Medicine, 8(2), 178. https://doi.org/10.3390/jcm8020178