Annexin A1 as Neuroprotective Determinant for Blood-Brain Barrier Integrity in Neonatal Hypoxic-Ischemic Encephalopathy

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Section
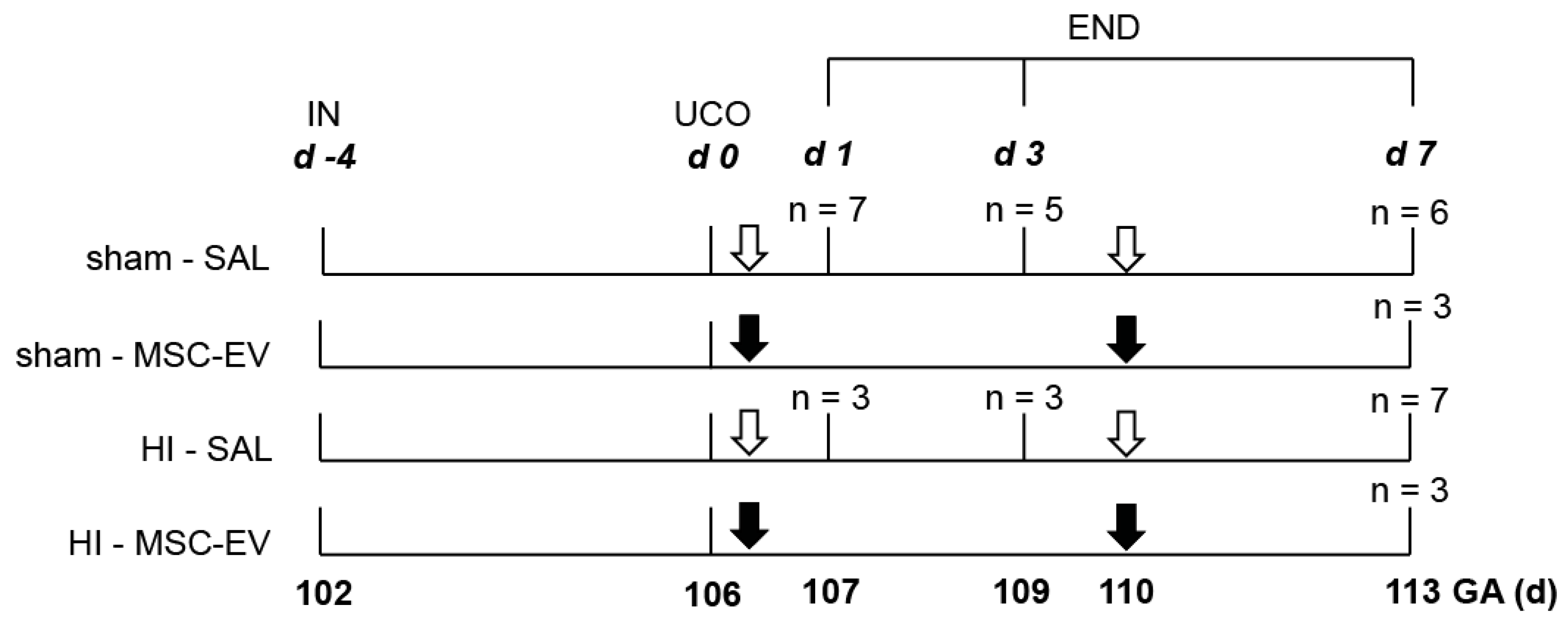
2.1. Study Approval and Experimental Design
2.2. Mesenchymal Stem/Stromal Cell-Extracellular Vesicles
2.3. MSC-EV Analysis Using Tunable Resistive Pulse Sensing (TRPS)
2.4. Western Blot of MSC-EVs
2.5. Immunohistochemistry and Analysis
2.6. Endothelial Cell Isolation and Culture
2.7. Trans-Endothelial Electrical Resistance (TEER)
2.8. Statistical Analysis
3. Results
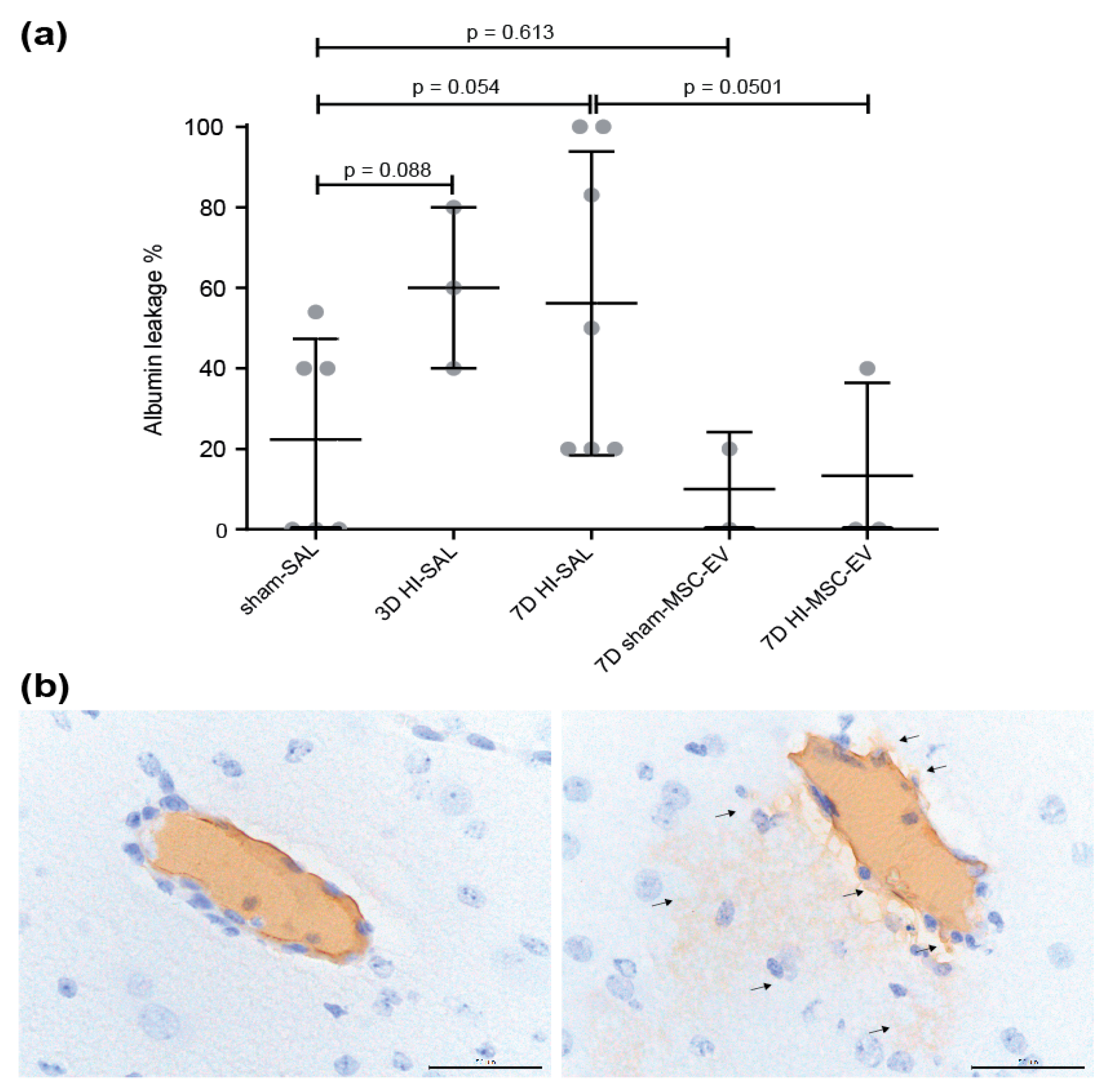
3.1. MSC-EVs Tended to Prevent Albumin Leakage in the Fetal Ovine Brain following Global Hypoxia-Ischemia
3.2. MSC-EVs and ANXA1 Restored Endothelial Resistance/Barrier Integrity following Oxygen-Glucose Deprivation in FPR Dependent Manner
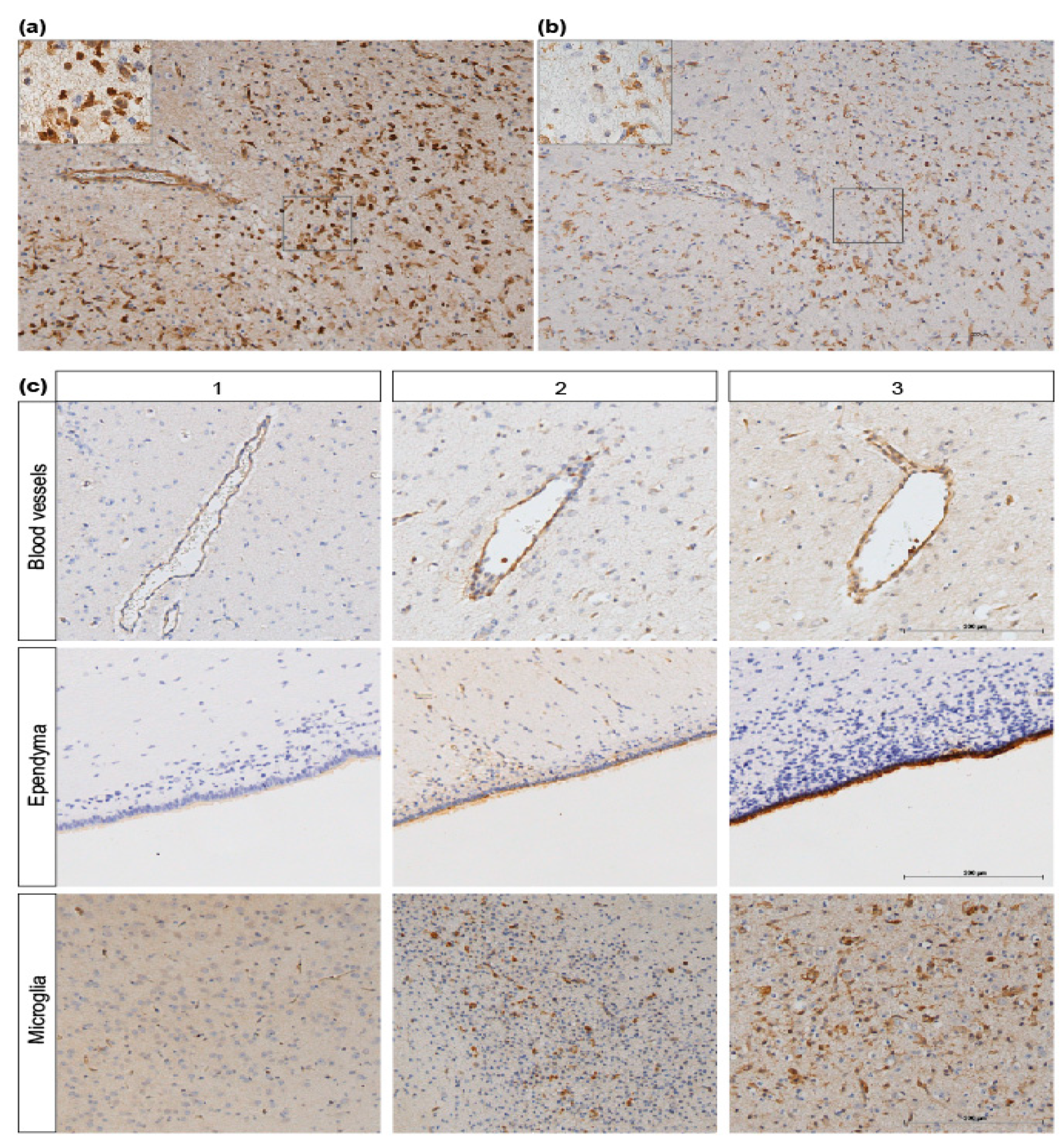
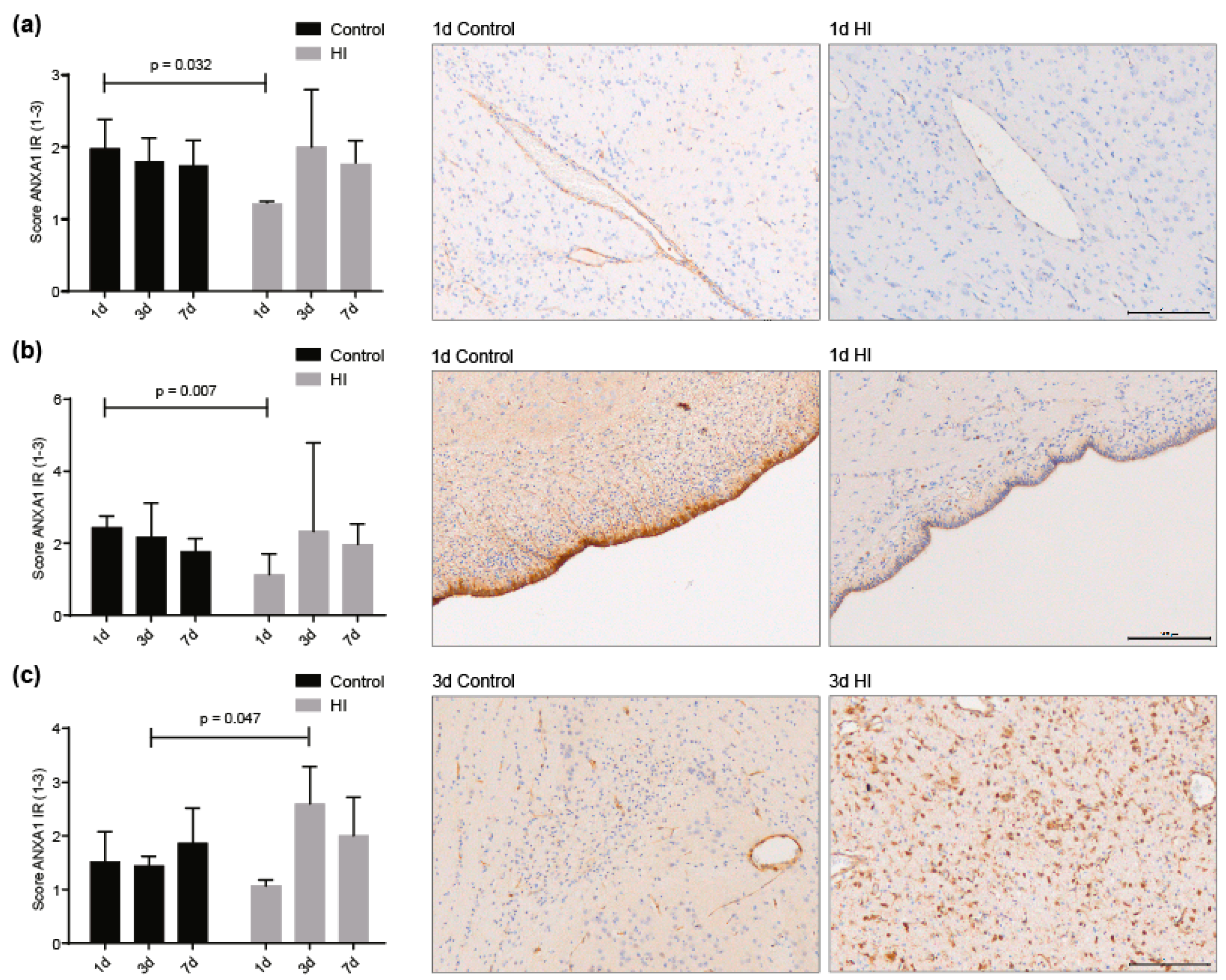
3.3. ANXA1 is Widely Expressed in the Preterm Ovine Brain and the Expression Decreases Acutely after Global Hypoxia-Ischemia
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allen, K.A.; Brandon, D.H. Hypoxic ischemic encephalopathy: Pathophysiology and experimental treatments. Newborn Infant Nurs. Rev. 2011, 11, 125–133. [Google Scholar] [CrossRef]
- Pathirana, J.; Muñoz, F.M.; Abbing-Karahagopian, V.; Bhat, N.; Harris, T.; Kapoor, A.; Keene, D.L.; Mangili, A.; Padula, M.A.; Pande, S.L.; et al. Neonatal death: Case definition and guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 2016, 34, 6027–6037. [Google Scholar] [CrossRef]
- Gopagondanahalli, K.R.; Li, J.; Fahey, M.C.; Hunt, R.W.; Jenkin, G.; Miller, S.L.; Malhotra, A. Preterm hypoxic-ischemic encephalopathy. Front. Pediatr. 2016, 4, 114. [Google Scholar] [CrossRef]
- Davidson, J.O.; Wassink, G.; van den Heuij, L.G.; Bennet, L.; Gunn, A.J. Therapeutic hypothermia for neonatal hypoxic-ischemic encephalopathy—Where to from here? Front. Neurol. 2015, 6, 198. [Google Scholar] [CrossRef]
- Rao, R.; Trivedi, S.; Vesoulis, Z.; Liao, S.M.; Smyser, C.D.; Mathur, A.M. Safety and short-term outcomes of therapeutic hypothermia in preterm neonates 34-35 weeks gestational age with hypoxic-ischemic encephalopathy. J. Pediatr. 2017, 183, 37–42. [Google Scholar] [CrossRef]
- Cotten, C.M.; Shankaran, S. Hypothermia for hypoxic-ischemic encephalopathy. Expert Rev. Obstet. Gynecol. 2010, 5, 227–239. [Google Scholar] [CrossRef]
- Kumar, A.; Mittal, R.; Khanna, H.D.; Basu, S. Free radical injury and blood-brain barrier permeability in hypoxic-ischemic encephalopathy. Pediatrics 2008, 122, e722–e727. [Google Scholar] [CrossRef]
- Lee, W.L.A.; Michael-Titus, A.T.; Shah, D.K. Hypoxic-ischaemic encephalopathy and the blood-brain barrier in neonates. Dev. Neurosci. 2017, 39, 49–58. [Google Scholar] [CrossRef]
- Chen, X.; Threlkeld, S.W.; Cummings, E.E.; Juan, I.; Makeyev, O.; Besio, W.G.; Gaitanis, J.; Banks, W.A.; Sadowska, G.B.; Stonestreet, B.S. Ischemia-reperfusion impairs blood-brain barrier function and alters tight junction protein expression in the ovine fetus. Neuroscience 2012, 226, 89–100. [Google Scholar] [CrossRef]
- Baburamani, A.A.; Castillo-Melendez, M.; Walker, D.W. VEGF expression and microvascular responses to severe transient hypoxia in the fetal sheep brain. Pediatr. Res. 2013, 73, 310–316. [Google Scholar] [CrossRef]
- Moretti, R.; Pansiot, J.; Bettati, D.; Strazielle, N.; Ghersi-Egea, J.F.; Damante, G.; Fleiss, B.; Titomanlio, L.; Gressens, P. Blood-brain barrier dysfunction in disorders of the developing brain. Front. Neurosci. 2015, 9, 40. [Google Scholar] [CrossRef]
- McArthur, S.; Loiola, R.A.; Maggioli, E.; Errede, M.; Virgintino, D.; Solito, E. The restorative role of annexin A1 at the blood-brain barrier. Fluids Barriers CNS 2016, 13, 17. [Google Scholar] [CrossRef]
- Saunders, N.R.; Daneman, R.; Dziegielewska, K.M.; Liddelow, S.A. Transporters of the blood-brain and blood-CSF interfaces in development and in the adult. Mol. Asp. Med. 2013, 34, 742–752. [Google Scholar] [CrossRef]
- Smyth, L.C.D.; Rustenhoven, J.; Park, T.I.; Schweder, P.; Jansson, D.; Heppner, P.A.; O’Carroll, S.J.; Mee, E.W.; Faull, R.L.M.; Curtis, M.; et al. Unique and shared inflammatory profiles of human brain endothelia and pericytes. J. Neuroinflamm. 2018, 15, 138. [Google Scholar] [CrossRef]
- Da Fonseca, A.C.; Matias, D.; Garcia, C.; Amaral, R.; Geraldo, L.H.; Freitas, C.; Lima, F.R. The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 2014, 8, 362. [Google Scholar] [CrossRef]
- Jellema, R.K.; Lima Passos, V.; Zwanenburg, A.; Ophelders, D.R.M.G.; De Munter, S.; Vanderlocht, J.; Germeraad, W.T.V.; Kuypers, E.; Collins, J.J.P.; Cleutjens, J.P.M.; et al. Cerebral inflammation and mobilization of the peripheral immune system following global hypoxia-ischemia in preterm sheep. J. Neuroinflamm. 2013, 10, 807. [Google Scholar] [CrossRef]
- Jellema, R.K.; Wolfs, T.G.A.M.; Lima Passos, V.; Zwanenburg, A.; Ophelders, D.R.M.G.; Kuypers, E.; Hopman, A.H.N.; Dudink, J.; Steinbusch, H.W.; Andriessen, P.; et al. Mesenchymal stem cells induce T-cell tolerance and protect the preterm brain after global hypoxia-ischemia. PLoS ONE 2013, 8, e73031. [Google Scholar] [CrossRef]
- Ek, C.J.; D’Angelo, B.; Baburamani, A.A.; Lehner, C.; Leverin, A.L.; Smith, P.L.; Nilsson, H.; Svedin, P.; Hagberg, H.; Mallard, C. Brain barrier properties and cerebral blood flow in neonatal mice exposed to cerebral hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 818–827. [Google Scholar] [CrossRef]
- Ophelders, D.R.; Wolfs, T.G.; Jellema, R.K.; Zwanenburg, A.; Andriessen, P.; Delhaas, T.; Ludwig, A.K.; Radtke, S.; Peters, V.; Janssen, L.; et al. Mesenchymal stromal cell-derived extracellular vesicles protect the fetal brain after hypoxia-ischemia. Stem Cells Transl. Med. 2016, 5, 754–763. [Google Scholar] [CrossRef]
- Cristante, E.; McArthur, S.; Mauro, C.; Maggioli, E.; Romero, I.A.; Wylezinska-Arridge, M.; Couraud, P.O.; Lopez-Tremoleda, J.; Christian, H.C.; Weksler, B.B.; et al. Identification of an essential endogenous regulator of blood-brain barrier integrity, and its pathological and therapeutic implications. Proc. Natl. Acad. Sci. USA 2013, 110, 832–841. [Google Scholar] [CrossRef]
- Perretti, M.; D’Acquisto, F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 2009, 9, 62. [Google Scholar] [CrossRef]
- Chen, K.; Bao, Z.; Gong, W.; Tang, P.; Yoshimura, T.; Wang, J.M. Regulation of inflammation by members of the formyl-peptide receptor family. J. Autoimmun. 2017, 85, 64–77. [Google Scholar] [CrossRef]
- He, H.Q.; Ye, R.D. The formyl peptide receptors: Diversity of ligands and mechanism for recognition. Molecules 2017, 22. [Google Scholar] [CrossRef]
- Persidsky, Y.; Ramirez, S.H.; Haorah, J.; Kanmogne, G.D. Blood-brain barrier: Structural components and function under physiologic and pathologic conditions. J. Neuroimmune Pharmacol. 2006, 1, 223–236. [Google Scholar] [CrossRef]
- McArthur, S.; Cristante, E.; Paterno, M.; Christian, H.; Roncaroli, F.; Gillies, G.E.; Solito, E. Annexin A1: A central player in the anti-inflammatory and neuroprotective role of microglia. J. Immunol. 2010, 185, 6317–6328. [Google Scholar] [CrossRef]
- Probst-Cousin, S.; Kowolik, D.; Kuchelmeister, K.; Kayser, C.; Neundörfer, B.; Heuss, D. Expression of annexin-1 in multiple sclerosis plaques. Neuropathol. Appl. Neurobiol. 2002, 28, 292–300. [Google Scholar] [CrossRef]
- Knott, C.; Stern, G.; Wilkin, G.P. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol Cell Neurosci 2000, 16, 724–739. [Google Scholar] [CrossRef]
- Maia, L.; de Moraes, C.N.; Dias, M.C.; Martinez, J.B.; Caballol, A.O.; Testoni, G.; de Queiroz, C.M.; Peña, R.D.; Landim-Alvarenga, F.C.; de Oliveira, E. A proteomic study of mesenchymal stem cells from equine umbilical cord. Theriogenology 2017, 100, 8–15. [Google Scholar] [CrossRef]
- De Moraes, C.N.; Maia, L.; de Oliveira, E.; de Paula Freitas Dell’Aqua, C.; Chapwanya, A.; da Cruz Landim-Alvarenga, F.; Oba, E. Shotgun proteomic analysis of the secretome of bovine endometrial mesenchymal progenitor/stem cells challenged or not with bacterial lipopolysaccharide. Vet. Immunol. Immunopathol. 2017, 187, 42–47. [Google Scholar] [CrossRef]
- Rackham, C.L.; Vargas, A.E.; Hawkes, R.G.; Amisten, S.; Persaud, S.J.; Austin, A.L.; King, A.J.; Jones, P.M. Annexin A1 Is a Key Modulator of Mesenchymal Stromal Cell-Mediated Improvements in Islet Function. Diabetes 2016, 65, 129–139. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef]
- Back, S.A.; Riddle, A.; Hohimer, A.R. Role of instrumented fetal sheep preparations in defining the pathogenesis of human periventricular white-matter injury. J. Child Neurol. 2006, 21, 582–589. [Google Scholar] [CrossRef]
- Kordelas, L.; Rebmann, V.; Ludwig, A.K.; Radtke, S.; Ruesing, J.; Doeppner, T.R.; Epple, M.; Horn, P.A.; Beelen, D.W.; Giebel, B. MSC-derived exosomes: A novel tool to treat therapy-refractory graft-versus-host disease. Leukemia 2014, 28, 970–973. [Google Scholar] [CrossRef]
- Sokolova, V.; Ludwig, A.-K.; Hornung, S.; Rotan, O.; Horn, P.A.; Epple, M.; Giebel, B. Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids Surf. B Biointerfaces 2011, 87, 146–150. [Google Scholar] [CrossRef]
- Kusters, D.H.M.; Chatrou, M.L.; Willems, B.A.G.; De Saint-Hubert, M.; Bauwens, M.; van der Vorst, E.; Bena, S.; Biessen, E.A.L.; Perretti, M.; Schurgers, L.J.; et al. Pharmacological treatment with annexin a1 reduces atherosclerotic plaque burden in LDLR-/-mice on western type diet. PLoS ONE 2015, 10, e0130484. [Google Scholar] [CrossRef]
- Bernas, M.J.; Cardoso, F.L.; Daley, S.K.; Weinand, M.E.; Campos, A.R.; Ferreira, A.J.; Hoying, J.B.; Witte, M.H.; Brites, D.; Persidsky, Y.; et al. Establishment of primary cultures of human brain microvascular endothelial cells to provide an in vitro cellular model of the blood-brain barrier. Nat. Protoc. 2010, 5, 1265–1272. [Google Scholar] [CrossRef]
- Kinney, H.C.; Volpe, J.J. Modeling the encephalopathy of prematurity in animals: The important role of translational research. Neurol. Res. Int. 2012, 2012, 295389. [Google Scholar] [CrossRef]
- Galinsky, R.; Draghi, V.; Wassink, G.; Davidson, J.O.; Drury, P.P.; Lear, C.A.; Gunn, A.J.; Bennet, L. Magnesium sulfate reduces EEG activity but is not neuroprotective after asphyxia in preterm fetal sheep. J. Cereb. Blood Flow Metab. 2017, 37, 1362–1373. [Google Scholar] [CrossRef]
- Wassink, G.; Davidson, J.O.; Dhillon, S.K.; Fraser, M.; Galinsky, R.; Bennet, L.; Gunn, A.J. Partial white and grey matter protection with prolonged infusion of recombinant human erythropoietin after asphyxia in preterm fetal sheep. J. Cereb. Blood Flow Metab. 2017, 37, 1080–1094. [Google Scholar] [CrossRef]
- Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P.O. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta 2009, 1788, 842–857. [Google Scholar] [CrossRef]
- Engelhardt, S.; Huang, S.-F.; Patkar, S.; Gassmann, M.; Ogunshola, O.O. Differential responses of blood-brain barrier associated cells to hypoxia and ischemia: A comparative study. Fluids Barriers CNS 2015, 12, 4. [Google Scholar] [CrossRef]
- Nishioku, T.; Matsumoto, J.; Dohgu, S.; Sumi, N.; Miyao, K.; Takata, F.; Shuto, H.; Yamauchi, A.; Kataoka, Y. Tumor necrosis factor-alpha mediates the blood-brain barrier dysfunction induced by activated microglia in mouse brain microvascular endothelial cells. J. Pharmacol. Sci. 2010, 112, 251–254. [Google Scholar] [CrossRef]
- Ries, M.; Loiola, R.; Shah, U.N.; Gentleman, S.M.; Solito, E.; Sastre, M. The anti-inflammatory Annexin A1 induces the clearance and degradation of the amyloid-beta peptide. J. Neuroinflamm. 2016, 13, 234. [Google Scholar] [CrossRef]
- Tang, G.; Liu, Y.; Zhang, Z.; Lu, Y.; Wang, Y.; Huang, J.; Li, Y.; Chen, X.; Gu, X.; Wang, Y.; et al. Mesenchymal stem cells maintain blood-brain barrier integrity by inhibiting aquaporin-4 upregulation after cerebral ischemia. Stem Cells 2014, 32, 3150–3162. [Google Scholar] [CrossRef]
- Bedi, S.S.; Aertker, B.M.; Liao, G.P.; Caplan, H.W.; Bhattarai, D.; Mandy, F.; Mandy, F.; Fernandez, L.G.; Zelnick, P.; Mitchell, M.B.; et al. Therapeutic time window of multipotent adult progenitor therapy after traumatic brain injury. J. Neuroinflamm. 2018, 15, 84. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Z.; Yang, J.; Yang, Z.; Yin, J.; Zuo, G.; Duan, X.; Shen, H.; Li, H.; Chen, G. Identification of two phosphorylation sites essential for annexin A1 in blood-brain barrier protection after experimental intracerebral hemorrhage in rats. J. Cereb. Blood Flow Metab. 2017, 37, 2509–2525. [Google Scholar] [CrossRef]
- Sheikh, M.H.; Solito, E. Annexin A1: Uncovering the many talents of an old protein. Int. J. Mol. Sci. 2018, 19, 1045. [Google Scholar] [CrossRef]
- Park, J.C.; Baik, S.H.; Han, S.H.; Cho, H.J.; Choi, H.; Kim, H.J.; Choi, H.; Lee, W.; Kim, D.K.; Mook-Jung, I. Annexin A1 restores Aβ(1-42)-induced blood–brain barrier disruption through the inhibition of RhoA-ROCK signaling pathway. Aging Cell 2017, 16, 149–161. [Google Scholar] [CrossRef]
- Maggioli, E.; McArthur, S.; Mauro, C.; Kieswich, J.; Kusters, D.H.; Reutelingsperger, C.P.; Yaqoob, M.; Solito, E. Estrogen protects the blood-brain barrier from inflammation-induced disruption and increased lymphocyte trafficking. Brain Behav. Immun. 2016, 51, 212–222. [Google Scholar] [CrossRef]
- Johnston, M.V.; Hagberg, H. Sex and the pathogenesis of cerebral palsy. Dev. Med. Child Neurol. 2007, 49, 74–78. [Google Scholar] [CrossRef]
- Mayoral, S.R.; Omar, G.; Penn, A.A. Sex differences in a hypoxia model of preterm brain damage. Pediatr. Res. 2009, 66, 248–253. [Google Scholar] [CrossRef]
- Al Mamun, A.; Yu, H.; Romana, S.; Liu, F. Inflammatory Responses are Sex Specific in Chronic Hypoxic-Ischemic Encephalopathy. Cell Transplant. 2018, 27, 1328–1339. [Google Scholar] [CrossRef]
- Morris, J.; Christian, H.; Flower, R. 19: Annexin 1 distribution in the CNS: An association with stem cells? J. Anat. 2004, 205, 525. [Google Scholar]
- Solito, E.; McArthur, S.; Christian, H.; Gavins, F.; Buckingham, J.C.; Gillies, G.E. Annexin A1 in the brain – undiscovered roles? Trends Pharmacol. Sci. 2008, 29, 135–142. [Google Scholar] [CrossRef]
- Saunders, N.R.; Liddelow, S.A.; Dziegielewska, K.M. Barrier mechanisms in the developing brain. Front. Pharmacol. 2012, 3, 46. [Google Scholar] [CrossRef]
- Whish, S.; Dziegielewska, K.M.; Mollgard, K.; Noor, N.M.; Liddelow, S.A.; Habgood, M.D.; Richardson, S.J.; Saunders, N.R. The inner CSF-brain barrier: Developmentally controlled access to the brain via intercellular junctions. Front. Neurosci. 2015, 9, 16. [Google Scholar] [CrossRef]
- Fossan, G.; Cavanagh, M.; Evans, C.; Malinowska, D.; Møllgård, K.; Reynolds, M.; Saunders, N.J.D.B.R. CSF-brain permeability in the immature sheep fetus: A CSF-brain barrier. Dev. Brain Res. 1985, 18, 113–124. [Google Scholar] [CrossRef]
- Liu, J.H.; Feng, D.; Zhang, Y.F.; Shang, Y.; Wu, Y.; Li, X.F.; Pei, L. Chloral hydrate preconditioning protects against ischemic stroke via upregulating Annexin A1. CNS Neurosci. Ther. 2015, 21, 718–726. [Google Scholar] [CrossRef]
- Locatelli, I.; Sutti, S.; Jindal, A.; Vacchiano, M.; Bozzola, C.; Reutelingsperger, C.; Kusters, D.; Bena, S.; Parola, M.; Paternostro, C.; et al. Endogenous annexin A1 is a novel protective determinant in nonalcoholic steatohepatitis in mice. Hepatology 2014, 60, 531–544. [Google Scholar] [CrossRef]
- Luo, Z.Z.; Gao, Y.; Sun, N.; Zhao, Y.; Wang, J.; Tian, B.; Shi, J. Enhancing the interaction between annexin-1 and formyl peptide receptors regulates microglial activation to protect neurons from ischemia-like injury. J. Neuroimmunol. 2014, 276, 24–36. [Google Scholar] [CrossRef]
- McArthur, S.; Gobbetti, T.; Juban, G.; Desgeorges, T.; Theret, M.; Gondin, J.; Toller-Kawahisa, J.; Reutelingsperger, C.; Perretti, M.; Mounier, R. Annexin A1 drives macrophage skewing towards a resolving phenotype to accelerate the regeneration of muscle injury through AMPK activation. bioRxiv 2018. [Google Scholar] [CrossRef]
- Plendl, J.; Neumüller, C.; Vollmar, A.; Auerbach, R.; Sinowatz, F. Isolation and characterization of endothelial cells from different organs of fetal pigs. Anat. Embryol. 1996, 194, 445–456. [Google Scholar] [CrossRef]
- Plendl, J.; Sinowatz, F. Fetal versus adult brain endothelium in vitro: Characterization and reaction to tumor-conditioned media. Electr. J. Pathol. Histol. 1995, 1, 28–39. [Google Scholar]
- Takata, F.; Dohgu, S.; Yamauchi, A.; Matsumoto, J.; Machida, T.; Fujishita, K.; Shibata, K.; Shinozaki, Y.; Sato, K.; Kataoka, Y.; et al. In vitro blood-brain barrier models using brain capillary endothelial cells isolated from neonatal and adult rats retain age-related barrier properties. PLoS ONE 2013, 8, e55166. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gussenhoven, R.; Klein, L.; Ophelders, D.R.M.G.; Habets, D.H.J.; Giebel, B.; Kramer, B.W.; Schurgers, L.J.; Reutelingsperger, C.P.M.; Wolfs, T.G.A.M. Annexin A1 as Neuroprotective Determinant for Blood-Brain Barrier Integrity in Neonatal Hypoxic-Ischemic Encephalopathy. J. Clin. Med. 2019, 8, 137. https://doi.org/10.3390/jcm8020137
Gussenhoven R, Klein L, Ophelders DRMG, Habets DHJ, Giebel B, Kramer BW, Schurgers LJ, Reutelingsperger CPM, Wolfs TGAM. Annexin A1 as Neuroprotective Determinant for Blood-Brain Barrier Integrity in Neonatal Hypoxic-Ischemic Encephalopathy. Journal of Clinical Medicine. 2019; 8(2):137. https://doi.org/10.3390/jcm8020137
Chicago/Turabian StyleGussenhoven, Ruth, Luise Klein, Daan R. M. G. Ophelders, Denise H. J. Habets, Bernd Giebel, Boris W. Kramer, Leon J. Schurgers, Chris P. M. Reutelingsperger, and Tim G. A. M. Wolfs. 2019. "Annexin A1 as Neuroprotective Determinant for Blood-Brain Barrier Integrity in Neonatal Hypoxic-Ischemic Encephalopathy" Journal of Clinical Medicine 8, no. 2: 137. https://doi.org/10.3390/jcm8020137
APA StyleGussenhoven, R., Klein, L., Ophelders, D. R. M. G., Habets, D. H. J., Giebel, B., Kramer, B. W., Schurgers, L. J., Reutelingsperger, C. P. M., & Wolfs, T. G. A. M. (2019). Annexin A1 as Neuroprotective Determinant for Blood-Brain Barrier Integrity in Neonatal Hypoxic-Ischemic Encephalopathy. Journal of Clinical Medicine, 8(2), 137. https://doi.org/10.3390/jcm8020137