Genes and Variants Underlying Human Congenital Lactic Acidosis—From Genetics to Personalized Treatment

 ,
, 
 ,
,  , ,
, ,  , add
Show full author list
, add
Show full author list
Abstract
1. Introduction
2. Experimental Section
2.1. Patients
2.2. Genetic Analysis
2.2.1. Clinical Exome Sequencing
2.2.2. Mitochondrial DNA Sequencing
2.2.3. Variant Prioritization and Pathogenicity Prediction of Nuclear DNA Variants
2.2.4. High-Density Genotyping
2.2.5. mRNA Studies
2.3. Cellular Studies
2.3.1. Cell Culture
2.3.2. CoQ10 Measurement
2.3.3. Cellular Oxygen Consumption
2.3.4. Mitochondrial Mass and Membrane Potential
2.3.5. Mitochondrial Isolation and Western Blotting
2.3.6. Transmission Electron Microscopy
2.3.7. Minigene Analysis and Morpholino Assay
2.3.8. Statistical Analysis
3. Results
3.1. Biochemical Profile
3.2. Genetic Analysis
3.2.1. DNA Sequencing of Nuclear Genes
3.2.2. Whole Mitochondrial DNA Analysis
3.2.3. RNA Analysis Helped Diagnose Patients and Can Be Used as Proof of Concept of Personalized Therapies
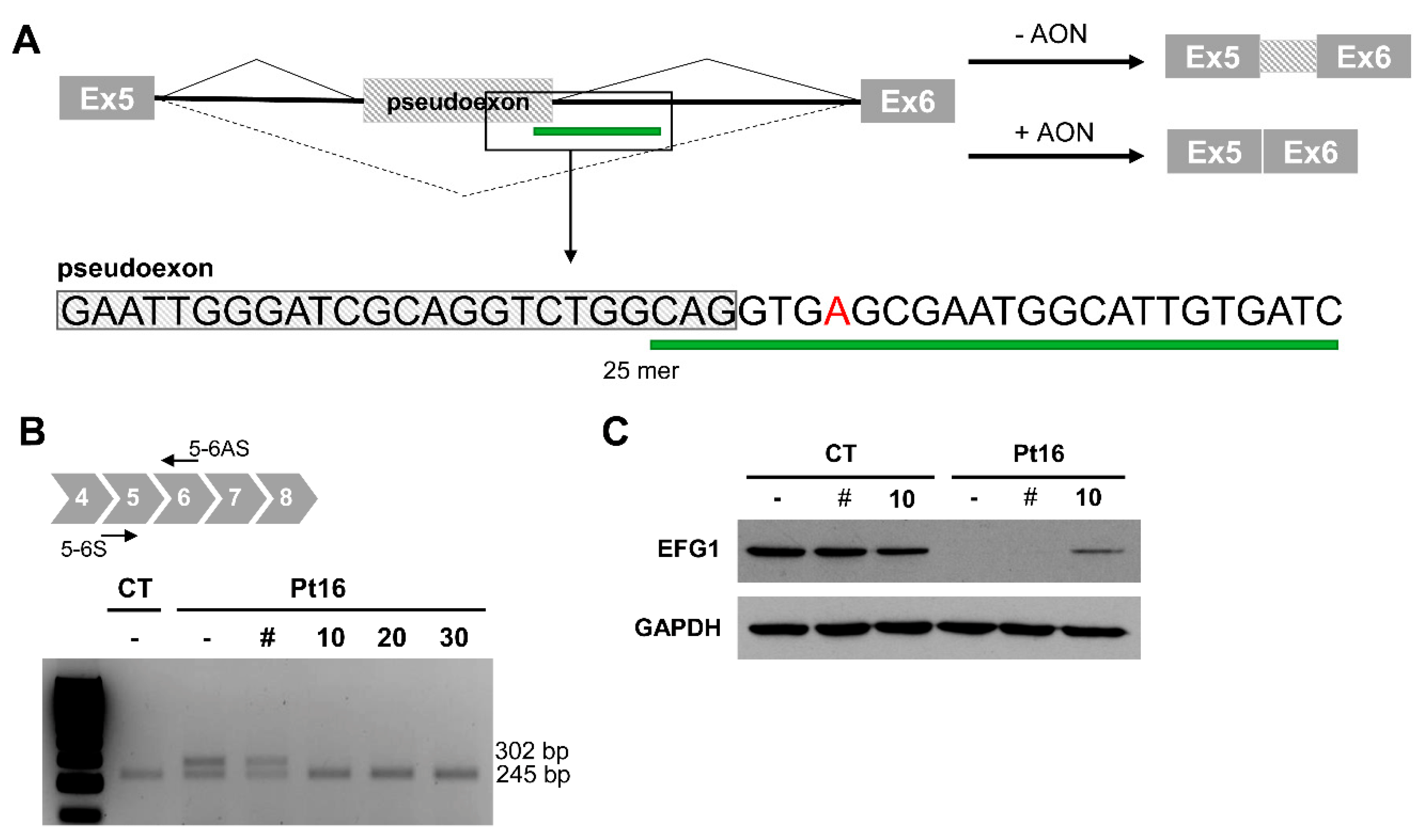
3.2.4. Antisense Oligonucleotide Treatment Rescues the Aberrant Splicing Event Caused by the Intronic Variant GFM1 c.689+908G>A
3.3. Functional Studies in Patients with Novel Genotypes
3.3.1. Biochemical Confirmation of Genetic Data
3.3.2. Mitochondrial Respiration and OxPhos Protein Analysis Confirmed Mitochondrial Dysfunction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kraut, J.A.; Madias, N.E. Lactic acidosis. N. Engl. J. Med. 2014, 371, 2309–2319. [Google Scholar] [CrossRef] [PubMed]
- Morava, E.; van den Heuvel, L.; Hol, F.; de Vries, M.C.; Hogeveen, M.; Rodenburg, R.J.; Smeitink, J.A. Mitochondrial disease criteria: Diagnostic applications in children. Neurology 2006, 67, 1823–1826. [Google Scholar] [CrossRef] [PubMed]
- Legati, A.; Reyes, A.; Nasca, A.; Invernizzi, F.; Lamantea, E.; Tiranti, V.; Garavaglia, B.; Lamperti, C.; Ardissone, A.; Moroni, I.; et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochim. Biophys. Acta 2016, 1857, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Parikh, S.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Anselm, I.; Brunel-Guitton, C.; Christodoulou, J.; Cohen, B.H.; Dimmock, D.; Enns, G.M.; et al. Patient care standards for primary mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2017, 19, 1380. [Google Scholar] [CrossRef]
- Thompson Legault, J.; Strittmatter, L.; Tardif, J.; Sharma, R.; Tremblay-Vaillancourt, V.; Aubut, C.; Boucher, G.; Clish, C.B.; Cyr, D.; Daneault, C.; et al. A Metabolic Signature of Mitochondrial Dysfunction Revealed through a Monogenic Form of Leigh Syndrome. Cell Rep. 2015, 13, 981–989. [Google Scholar] [CrossRef]
- Ortigoza-Escobar, J.D.; Molero-Luis, M.; Arias, A.; Oyarzabal, A.; Darin, N.; Serrano, M.; Garcia-Cazorla, A.; Tondo, M.; Hernandez, M.; Garcia-Villoria, J.; et al. Free-thiamine is a potential biomarker of thiamine transporter-2 deficiency: A treatable cause of Leigh syndrome. Brain 2016, 139, 31–38. [Google Scholar] [CrossRef]
- Desbats, M.A.; Lunardi, G.; Doimo, M.; Trevisson, E.; Salviati, L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J. Inherit. Metab. Dis. 2015, 38, 145–156. [Google Scholar] [CrossRef]
- Witters, P.; Saada, A.; Honzik, T.; Tesarova, M.; Kleinle, S.; Horvath, R.; Goldstein, A.; Morava, E. Revisiting mitochondrial diagnostic criteria in the new era of genomics. Genet. Med. 2018, 20, 444–451. [Google Scholar] [CrossRef]
- Rahman, J.; Noronha, A.; Thiele, I.; Rahman, S. Leigh map: A novel computational diagnostic resource for mitochondrial disease. Ann. Neurol. 2017, 81, 9–16. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Koolen, D.A.; Smeitink, J.A.; van den Heuvel, L.; Rodenburg, R.J. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J. Inherit. Metab. Dis. 2015, 38, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef] [PubMed]
- Puusepp, S.; Reinson, K.; Pajusalu, S.; Murumets, U.; Oiglane-Shlik, E.; Rein, R.; Talvik, I.; Rodenburg, R.J.; Ounap, K. Effectiveness of whole exome sequencing in unsolved patients with a clinical suspicion of a mitochondrial disorder in Estonia. Mol. Genet. Metab. Rep. 2018, 15, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef]
- Raymond, F.L.; Horvath, R.; Chinnery, P.F. First-line genomic diagnosis of mitochondrial disorders. Nat. Rev. Genet. 2018, 19, 399–400. [Google Scholar] [CrossRef]
- Chalmers, R.A.; Lawson, A.M. Gas Chomatography-Mass spectometry. In Organic Acids in Man. Analytical Chemistry, Biochemistry and Diagnosis of the Organic Acidurias; Chalmers, R.A., Lawson, A.M., Eds.; Chapman and Hall: London, UK; New York, NY, USA, 1982; pp. 81–127. [Google Scholar]
- Ferrer, I.; Ruiz-Sala, P.; Vicente, Y.; Merinero, B.; Perez-Cerda, C.; Ugarte, M. Separation and identification of plasma short-chain acylcarnitine isomers by HPLC/MS/MS for the differential diagnosis of fatty acid oxidation defects and organic acidemias. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 860, 121–126. [Google Scholar] [CrossRef]
- Vega, A.I.; Medrano, C.; Navarrete, R.; Desviat, L.R.; Merinero, B.; Rodriguez-Pombo, P.; Vitoria, I.; Ugarte, M.; Perez-Cerda, C.; Perez, B. Molecular diagnosis of glycogen storage disease and disorders with overlapping clinical symptoms by massive parallel sequencing. Genet. Med. 2016, 18, 1037–1043. [Google Scholar] [CrossRef]
- Ruiz-Pesini, E.; Lott, M.T.; Procaccio, V.; Poole, J.C.; Brandon, M.C.; Mishmar, D.; Yi, C.; Kreuziger, J.; Baldi, P.; Wallace, D.C. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007, 35, D823–D828. [Google Scholar] [CrossRef]
- Weissensteiner, H.; Forer, L.; Fuchsberger, C.; Schopf, B.; Kloss-Brandstatter, A.; Specht, G.; Kronenberg, F.; Schonherr, S. mtDNA-Server: Next-generation sequencing data analysis of human mitochondrial DNA in the cloud. Nucleic Acids Res. 2016, 44, W64–W69. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2018, 35, 1978–1980. [Google Scholar] [CrossRef]
- Li, B.; Krishnan, V.G.; Mort, M.E.; Xin, F.; Kamati, K.K.; Cooper, D.N.; Mooney, S.D.; Radivojac, P. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 2009, 25, 2744–2750. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Thomas, P.D. PANTHER-PSEP: Predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics 2016, 32, 2230–2232. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Alonso, I.; Navarrete, R.; Arribas-Carreira, L.; Perona, A.; Abia, D.; Couce, M.L.; Garcia-Cazorla, A.; Morais, A.; Domingo, R.; Ramos, M.A.; et al. Nonketotic hyperglycinemia: Functional assessment of missense variants in GLDC to understand phenotypes of the disease. Hum. Mutat. 2017, 38, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Oyarzabal, A.; Martinez-Pardo, M.; Merinero, B.; Navarrete, R.; Desviat, L.R.; Ugarte, M.; Rodriguez-Pombo, P. A novel regulatory defect in the branched-chain alpha-keto acid dehydrogenase complex due to a mutation in the PPM1K gene causes a mild variant phenotype of maple syrup urine disease. Hum. Mutat. 2013, 34, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Alonso, I.; Oyarzabal, A.; Sanchez-Arago, M.; Rejas, M.T.; Merinero, B.; Garcia-Cazorla, A.; Artuch, R.; Ugarte, M.; Rodriguez-Pombo, P. Dataset reporting BCKDK interference in a BCAA-catabolism restricted environment. Data Br. 2016, 7, 755–759. [Google Scholar] [CrossRef]
- Mohanraj, K.; Wasilewski, M.; Beninca, C.; Cysewski, D.; Poznanski, J.; Sakowska, P.; Bugajska, Z.; Deckers, M.; Dennerlein, S.; Fernandez-Vizarra, E.; et al. Inhibition of proteasome rescues a pathogenic variant of respiratory chain assembly factor COA7. EMBO Mol. Med. 2019, 11, e9561. [Google Scholar] [CrossRef]
- Garcia-Cazorla, A.; Oyarzabal, A.; Fort, J.; Robles, C.; Castejon, E.; Ruiz-Sala, P.; Bodoy, S.; Merinero, B.; Lopez-Sala, A.; Dopazo, J.; et al. Two novel mutations in the BCKDK (branched-chain keto-acid dehydrogenase kinase) gene are responsible for a neurobehavioral deficit in two pediatric unrelated patients. Hum. Mutat. 2014, 35, 470–477. [Google Scholar] [CrossRef]
- Oyarzabal, A.; Bravo-Alonso, I.; Sanchez-Arago, M.; Rejas, M.T.; Merinero, B.; Garcia-Cazorla, A.; Artuch, R.; Ugarte, M.; Rodriguez-Pombo, P. Mitochondrial response to the BCKDK-deficiency: Some clues to understand the positive dietary response in this form of autism. Biochim. Biophys. Acta 2016, 1862, 592–600. [Google Scholar] [CrossRef]
- De Vos, K.J.; Allan, V.J.; Grierson, A.J.; Sheetz, M.P. Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Curr. Biol. 2005, 15, 678–683. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Simon, M.T.; Ng, B.G.; Friederich, M.W.; Wang, R.Y.; Boyer, M.; Kircher, M.; Collard, R.; Buckingham, K.J.; Chang, R.; Shendure, J.; et al. Activation of a cryptic splice site in the mitochondrial elongation factor GFM1 causes combined OXPHOS deficiency. Mitochondrion 2017, 34, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Emperador, S.; Bayona-Bafaluy, M.P.; Fernandez-Marmiesse, A.; Pineda, M.; Felgueroso, B.; Lopez-Gallardo, E.; Artuch, R.; Roca, I.; Ruiz-Pesini, E.; Couce, M.L.; et al. Molecular-genetic characterization and rescue of a TSFM mutation causing childhood-onset ataxia and nonobstructive cardiomyopathy. Eur. J. Hum. Genet. 2017, 25, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, J.; Zhang, V.W.; Li, F.Y.; Landsverk, M.; Cui, H.; Truong, C.K.; Wang, G.; Chen, L.C.; Graham, B.; et al. Transition to next generation analysis of the whole mitochondrial genome: A summary of molecular defects. Hum. Mutat. 2013, 34, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Swalwell, H.; Kirby, D.M.; Blakely, E.L.; Mitchell, A.; Salemi, R.; Sugiana, C.; Compton, A.G.; Tucker, E.J.; Ke, B.X.; Lamont, P.J.; et al. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. Eur. J. Hum. Genet. 2011, 19, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Boczonadi, V.; Ricci, G.; Horvath, R. Mitochondrial DNA transcription and translation: Clinical syndromes. Essays Biochem. 2018, 62, 321–340. [Google Scholar] [PubMed]
- Ghezzi, D.; Zeviani, M. Human diseases associated with defects in assembly of OXPHOS complexes. Essays Biochem. 2018, 62, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Catteruccia, M.; Verrigni, D.; Martinelli, D.; Torraco, A.; Agovino, T.; Bonafe, L.; D'Amico, A.; Donati, M.A.; Adorisio, R.; Santorelli, F.M.; et al. Persistent pulmonary arterial hypertension in the newborn (PPHN): A frequent manifestation of TMEM70 defective patients. Mol. Genet. Metab. 2014, 111, 353–359. [Google Scholar] [CrossRef]
- Brito, S.; Thompson, K.; Campistol, J.; Colomer, J.; Hardy, S.A.; He, L.; Fernandez-Marmiesse, A.; Palacios, L.; Jou, C.; Jimenez-Mallebrera, C.; et al. Long-term survival in a child with severe encephalopathy, multiple respiratory chain deficiency and GFM1 mutations. Front. Genet. 2015, 6, 102. [Google Scholar]
- Salviati, L.; Sacconi, S.; Murer, L.; Zacchello, G.; Franceschini, L.; Laverda, A.M.; Basso, G.; Quinzii, C.; Angelini, C.; Hirano, M.; et al. Infantile encephalomyopathy and nephropathy with CoQ10 deficiency: A CoQ10-responsive condition. Neurology 2005, 65, 606–608. [Google Scholar] [CrossRef]
- Montini, G.; Malaventura, C.; Salviati, L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 2008, 358, 2849–2850. [Google Scholar] [CrossRef]
- Vantroys, E.; Larson, A.; Friederich, M.; Knight, K.; Swanson, M.A.; Powell, C.A.; Smet, J.; Vergult, S.; De Paepe, B.; Seneca, S.; et al. New insights into the phenotype of FARS2 deficiency. Mol. Genet. Metab. 2017, 122, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Friederich, M.W.; Timal, S.; Powell, C.A.; Dallabona, C.; Kurolap, A.; Palacios-Zambrano, S.; Bratkovic, D.; Derks, T.G.J.; Bick, D.; Bouman, K.; et al. Pathogenic variants in glutamyl-tRNA(Gln) amidotransferase subunits cause a lethal mitochondrial cardiomyopathy disorder. Nat. Commun. 2018, 9, 4065. [Google Scholar] [CrossRef] [PubMed]
- Formosa, L.E.; Mimaki, M.; Frazier, A.E.; McKenzie, M.; Stait, T.L.; Thorburn, D.R.; Stroud, D.A.; Ryan, M.T. Characterization of mitochondrial FOXRED1 in the assembly of respiratory chain complex I. Hum. Mol. Genet. 2015, 24, 2952–2965. [Google Scholar] [CrossRef] [PubMed]
- Galmiche, L.; Serre, V.; Beinat, M.; Zossou, R.; Assouline, Z.; Lebre, A.S.; Chretien, F.; Shenhav, R.; Zeharia, A.; Saada, A.; et al. Toward genotype phenotype correlations in GFM1 mutations. Mitochondrion 2012, 12, 242–247. [Google Scholar] [CrossRef]
- Pagani, F.; Baralle, F.E. Genomic variants in exons and introns: Identifying the splicing spoilers. Nat. Rev. Genet. 2004, 5, 389–396. [Google Scholar] [CrossRef]
- Perez, B.; Vilageliu, L.; Grinberg, D.; Desviat, L.R. Antisense mediated splicing modulation for inherited metabolic diseases: Challenges for delivery. Nucleic Acid Ther. 2014, 24, 48–56. [Google Scholar] [CrossRef]
- Holmes-Hampton, G.P.; Crooks, D.R.; Haller, R.G.; Guo, S.; Freier, S.M.; Monia, B.P.; Rouault, T.A. Use of antisense oligonucleotides to correct the splicing error in ISCU myopathy patient cell lines. Hum. Mol. Genet. 2016, 25, 5178–5187. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | MDC | Inheritance | Gene | Phenotype MIM Number | RefSeq/Variant 1 | RefSeq/Variant 2 |
|---|---|---|---|---|---|---|
| Pt1.1 * Pt1.2 | Definite | AR | ACAD9 | # 611126 Mitochondrial Complex I Deficiency Due To ACAD9 Deficiency | NM_014049.4: c.359delT (p.Phe120Serfs*9) | NM_014049.4: c.473C>T (p.Thr158Ile) |
| Pt2 * | Definite | AR | FOXRED1 | # 256000 Leigh Syndrome Due To Mitochondrial Complex I Deficiency | NM_017547.3: c.628T>G (p.Tyr210Asp) | NM_017547.3: c.1273C>T (p.His425Tyr) |
| Pt3 | Definite | Mit | MT-ATP6 | # 516060 Mitochondrial Complex V(ATP Synthase) Deficiency | NC_012920.1: m.G8719A (p.Gly65 *) | |
| Pt4 * | Definite | AR | TMEM70 | # 614052 Mitochondrial Complex V (ATPSynthase) Deficiency, Nuclear Type 2 | NM_017866.5: c.317-2A>G | NM_017866.5: c.317-2A>G |
| Pt5 | Definite | AR | DGUOK | # 251880 Mitochondrial DNA Depletion Syndrome 3 (Hepato-cerebral Type) | NM_080916.2: c.763_766dupGATT (p.Phe256*) | NM_080916.2: c.763_766dupGATT (p.Phe256*) |
| Pt6 | Definite | AR | MRPS22 | # 611719 Combined Oxidative Phosphorylation Deficiency 5 | NM_020191.2: c.509G>A (p.Arg170His) | NM_020191.2: c.1032_1035dupAACA (p.Leu346Asnfs*21) |
| Pt7 * | Definite | AR | TRMU | # 613070 Liver Failure, Infantile, Transient | NM_018006.4: c.680G>C (p.Arg227Thr) | NM_018006.4: c.1041_1044dupTCAA (p.Asp349Serfs*58) |
| Pt8 | Definite | AR | TSFM | # 610505 Combined Oxidative Phosphorylation Deficiency 3 | NM_001172696.1: c.782G>C (p.Cys261Ser) | NM_001172696.1: c.848G>A (p.Gly283Asp) |
| Pt9 * | Definite | AR | COQ2 | # 607426 Coenzyme Q10 Deficiency, Primary, 1 | NM_015697.7: c.163C>T (p.Arg55 *) | NM_015697.7: c.1197delT (p.Asn401Ilefs*15) |
| Pt10 * | Definite | AR | PDSS1 | # 614651 Coenzyme Q10 Deficiency, Primary, 2 | NM_014317.3: c.716T>G (p.Val239Gly) | NM_014317.3: c.1183C>T (p.Arg395*) |
| Pt11 * | Definite | AR | NPHS2 | # 600995 Nephrotic Syndrome, Type 2 | NM_014625.3: c.413G>A (p.Arg138Gln) | NM_014625.3: c.413G>A (p.Arg138Gln) |
| Pt12 | Definite | XL | PDHA1 | # 312170 Pyruvate Dehydrogenase E1-Alpha Deficiency | NM_000284.3: c.787C>G (p.Arg263Gly) | |
| Pt15 | Probable | AR | FARS2 | # 614946 Combined Oxidative Phosphorylation Deficiency 14 | NM_006567.3: c.737C>T (p.Thr246Met) | NM_006567.3: c.1082C>T (p.Pro361Leu) |
| Pt16 * | Probable | AR | GFM1 | # 609060 Combined Oxidative Phosphorylation Deficiency 1 | NM_024996.5: c.2011C>T (p.Arg671Cys) | a.NM_024996.5: c.689+908G>A r.(689_690ins57) (p.Gly230_231Glnins19) |
| Pt17 * | Probable | AR | GFM1 | # 609060 Combined Oxidative Phosphorylation Deficiency 1 | NM_024996.5: c.2011C>T (p.Arg671Cys) | NM_024996.5: c.1404delA (p.Gly469Valfs*84) |
| Pt18 | Probable | AR | DLD | # 246900 Dihydrolipoamide Dehydrogenase Deficiency | NM_000108.4: c.259C>T (p.Pro87Ser) | NM_000108.4: c.946C>T (p.Arg316*) |
| Pt19 | Probable | AR | DLD | # 246900 Dihydrolipoamide Dehydrogenase Deficiency | NM_000108.4: c.788G>A (p.Arg263His) | not found |
| Pt20 ** | Probable | XL | PDHA1 | # 312170 Pyruvate Dehydrogenase E1-Alpha Deficiency | NM_000284.3: c.506C>T (p.Ala169Val) | = |
| Pt21 | Probable | AR | PDHX | # 245349 Pyruvate Dehydrogenase E3-Binding Protein Deficiency | NM_003477.2: c.965-1G>A (p.Asp322Alafs*6) | b.NG_013368.1: g.34984192_34988219del r.642_816del (p.Asp215Alafs*19) |
| Pt22 * | Probable | AR | SLC16A1 | # 616095 Monocarboxylate Transporter 1 Deficiency | NM_003051.3: c.747_750delTAAT (p.Asn250Serfs*5) | NM_003051.3: c.747_750delTAAT (p.Asn250Serfs*5) |
| Pt23 | Probable | XLR | PHKA2 | # 306000 Glycogen Storage Disease IXA1 | NM_000292.2: c.1246G>A (p.Gly416Arg) | |
| Pt33 | Possible | AR | BCS1L | # 124000 Mitochondrial Complex III Deficiency, Nuclear Type 1 | NM_004328.4 c.166C>T (p.Arg56*) | NM_004328.4 c.-147A>G (p.?) |
| Pt34 | Possible | Mit | MT-ND5 | # 540000 Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis and Stroke-Like Episodes | NC_012920.1: m.13513G>A (p.Asp393Asn) | |
| Pt35 * | Possible | AR | DLD | # 246900 Dihydrolipoamide Dehydrogenase Deficiency | NM_000108.4 c.647T>C (p.Met216Thr) | NM_000108.4 c.647T>C (p.Met216Thr) |
| Pt36 * | Possible | AR | SLC19A3 | # 607483 Thiamine Metabolism Dysfunction Syndrome 2 (Biotin- Or Thiamine-Responsive Type) | NM_025243.3: c.20C>A (p.Ser7*) | NM_025243.3: c.20C>A (p.Ser7*) |
| Pt39 * | AR | SLC19A3 | # 607483 Thiamine Metabolism Dysfunction Syndrome 2 (Biotin- Or Thiamine-Responsive Type) | NM_025243.3: c.20C>A (p.Ser7*) | NM_025243.3: c.20C>A (p.Ser7*) |
| Gene | Variant/ Consequence | ACMG Tags | Classification | HGMD Accession | gnomaD |
|---|---|---|---|---|---|
| ACAD9 | c.359delT (p.Phe120Serfs*9) | PVS1, PM2, PP1, PP5 | Pathogenic | CD153914 | 0.0001042 |
| ACAD9 | c.473C>T (p.Thr158Ile) | PM2, PM3, PP1, PP3 | Likely pathogenic | - | 0 |
| BCS1L | c.166C>T (p.Arg56Ter) | PS3, PM4, PP3, PP5 | Likely pathogenic | CM022763 | 0.0001626 |
| BCS1L | c.-147A>G (p.?) | PM2 PM3, PP5 | VUS | CS098028 | 0 |
| COQ2 | c.1197delT (p.Asn401Ilefs*15) | PM2, PM3, PM4, PP5 | Likely pathogenic | CD071308 | 1.217e-5 |
| COQ2 | c.163C>T (p.Arg55Ter) | PM2, PM3, PM4, PP1 PS3 | Likely pathogenic | - | 0 |
| DGUOK | c.763_766dupGATT (p.Phe256Ter) | PM4, PM2, PM5, PS3, PP5 | Likely pathogenic | CI034484 | 2.031e-5 |
| DLD | c. 259C>T (p.Pro87Ser) | PM2, PM3, PP2, PP3, PP4 | VUS | - | 0 |
| DLD | c.647T>C (p.Met216Thr) | PM2, PP2, PP3 | VUS | - | 0 |
| DLD | C.788G>A (p.Arg263Hys) | PM3, PP3 | VUS | - | 0.000817 |
| DLD | c.946C>T (p.Arg316Ter) | PM2, PM4, PP3, PP4 | Likely pathogenic | - | 1.63e-05 |
| FARS2 | c.737C>T (p.Thr246Met) | BS2, BP6 | Likely benign | - | 0.004064 |
| FARS2 | c.1082C>T (p.Pro361Leu) | PP3 | VUS | CM1718796 | 0.0001339 |
| FOXRED1 | c.628T>G (p.Tyr210Asp) | PM2, PP3 | VUS | - | 0 |
| FOXRED1 | c.1273C>T (p.His425Tyr) | PM2, PP3 | VUS | - | 0 |
| GFM1 | c.1404delA (p.Gly469Valfs*84) | PVS1, PM2, PP5 | Pathogenic | CD154422 | 1.635e-5 |
| GFM1 | c.2011C>T (p.Arg671Cys) | PM2, PM3, PP2, PP3, PP5 | Likely pathogenic | CM11881 | 7.216e-5 |
| MRPS22 | c.1032_1035dupAACA (p.Leu346Asnfs*21) | PVS1, PM3, PP5 | Pathogenic | CI152171 | 9.028e-5 |
| MRPS22 | c.509G>A (p.Arg170His) | PM2, PM3, PP3, PP5 | Likely pathogenic | CM076316 | 7.221e-5 |
| NPHS2 | c.413G>A (p.Arg138Gln) | PP2, PP3, PP5 | VUS | CM000581 | 0.0005739 |
| PDHA1 | c.506C>T (p.Ala169Val) | PM2, PP2, PP3, PP5 | VUS | CM091028 | 0 |
| PDHA1 | c.787C>G (p.Arg263Gly) | PM2, PM5, PS3, PS4, PP2, PP3, PP5 | Pathogenic | CM920573 | 0 |
| PDHX | c.965-1G>A (p.Asp322Alafs*6) | PVS1, PM2, PM3, PP3, PP5 | Pathogenic | CS024024 | 4.11e-6 |
| PDSS1 | c.716T>G (p.Val239Gly) | PM2, PP3 (PS3 - Likely path) | VUS | - | 0 |
| PDSS1 | c.1183C>T (p.Arg395Ter) | PM2, PM4, PP3 | Pathogenic | - | 4.064e-6 |
| PHKA2 | c.1246G>A (p. Gly416Arg) | PP3, BP6 | VUS | - | 0.00432 |
| SLC16A1 | c.747_750delTAAT (p.Asn250Serfs*5) | PVS1, PM2, PP5 | Pathogenic | CD1411339 | 8.123e-6 |
| SLC19A3 | c.20C>A (p.Ser7Ter) | PM2, PM3, PM4, PP4, PP5 | Likely pathogenic | CM131528 | 0 |
| TMEM70 | c.317-2A>G (p.?) | PVS1, PM2, PP1, PP5 | Pathogenic | CS084884 | 7.605e-5 |
| TRMU | c.1041_1044dupTCAA (p.Asp349Serfs*58) | PVS1, PM2, PP5 | Pathogenic | CD155923 | 1.219e-5 |
| TRMU | c.680G>C (p.Arg227Thr) | PM2, PM3, PP3 | VUS | - | 1.624e-5 |
| TSFM | c.782G>C (p. Cys261Ser) | PS3, PM2 | Likely pathogenic | CM170018 | 4.188e-6 |
| TSFM | c.848G>A (p. Gly283Asp) | PM2, PP3 | VUS | - | 5.889e-5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bravo-Alonso, I.; Navarrete, R.; Vega, A.I.; Ruíz-Sala, P.; García Silva, M.T.; Martín-Hernández, E.; Quijada-Fraile, P.; Belanger-Quintana, A.; Stanescu, S.; Bueno, M.; et al. Genes and Variants Underlying Human Congenital Lactic Acidosis—From Genetics to Personalized Treatment. J. Clin. Med. 2019, 8, 1811. https://doi.org/10.3390/jcm8111811
Bravo-Alonso I, Navarrete R, Vega AI, Ruíz-Sala P, García Silva MT, Martín-Hernández E, Quijada-Fraile P, Belanger-Quintana A, Stanescu S, Bueno M, et al. Genes and Variants Underlying Human Congenital Lactic Acidosis—From Genetics to Personalized Treatment. Journal of Clinical Medicine. 2019; 8(11):1811. https://doi.org/10.3390/jcm8111811
Chicago/Turabian StyleBravo-Alonso, Irene, Rosa Navarrete, Ana Isabel Vega, Pedro Ruíz-Sala, María Teresa García Silva, Elena Martín-Hernández, Pilar Quijada-Fraile, Amaya Belanger-Quintana, Sinziana Stanescu, María Bueno, and et al. 2019. "Genes and Variants Underlying Human Congenital Lactic Acidosis—From Genetics to Personalized Treatment" Journal of Clinical Medicine 8, no. 11: 1811. https://doi.org/10.3390/jcm8111811
APA StyleBravo-Alonso, I., Navarrete, R., Vega, A. I., Ruíz-Sala, P., García Silva, M. T., Martín-Hernández, E., Quijada-Fraile, P., Belanger-Quintana, A., Stanescu, S., Bueno, M., Vitoria, I., Toledo, L., Couce, M. L., García-Jiménez, I., Ramos-Ruiz, R., Martín, M. Á., Desviat, L. R., Ugarte, M., Pérez-Cerdá, C., ... Rodríguez-Pombo, P. (2019). Genes and Variants Underlying Human Congenital Lactic Acidosis—From Genetics to Personalized Treatment. Journal of Clinical Medicine, 8(11), 1811. https://doi.org/10.3390/jcm8111811