Brain-Immune Alterations and Mitochondrial Dysfunctions in a Mouse Model of Paediatric Autoimmune Disorder Associated with Streptococcus: Exacerbation by Chronic Psychosocial Stress

 and
and 
Abstract
1. Introduction
2. Experimental Section
2.1. Animals and Rearing
2.2. Immunization Protocol
2.3. Chronic Psychosocial Stress
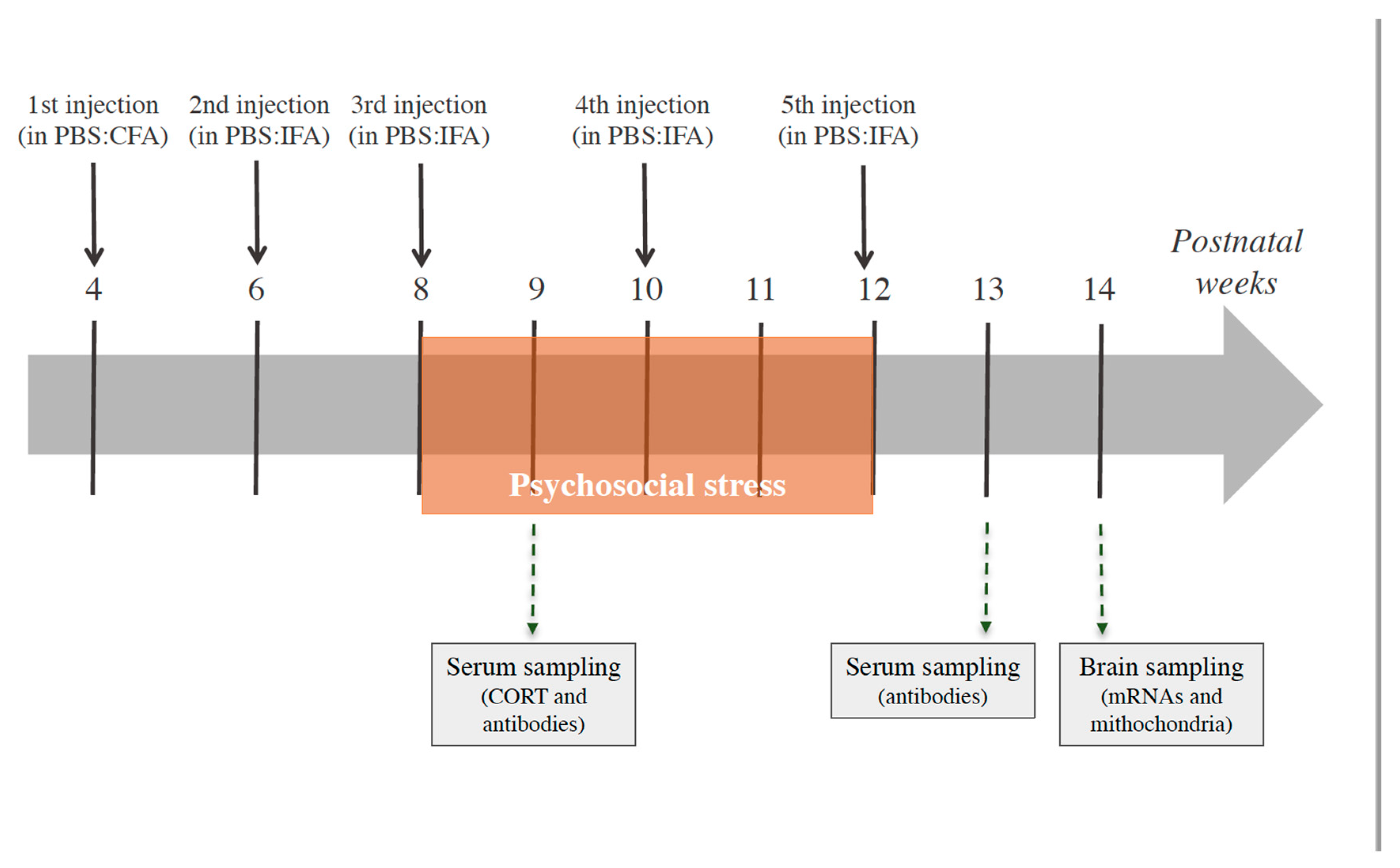
2.4. Experimental Design
2.5. Blood Serum Sampling and Corticosterone Determination
2.6. Analysis of Anti-Group A Streptococcal Antibodies in Serum Samples
2.7. Brain Sampling
2.8. Real-Time Quantitative Polymerase Chain Reaction (RT-PCR)
- HPRT (NM_013556): forward 5′-CAGGCCAGACTTTG-TTGGAT-3′; reverse 5′-TTGCGCTCATC-TTAGGCTTT-3′;
- IL-1β (NM_008361): forward 5′-CGACAAAATACCTGTGGCCT-3′, reverse 5′-TTCTTTGGGTATTCCTTGGG-3′;
- TNF-α (NM_013693.3): forward 5′-AGCCCCCAGTCTGTATCCTT-3′, reverse 5′-ACAGTCCAGGTCACTGTCCC-3′;
- IL-10 (NM_010548): forward 5′-TTAAGCTGTTTCCATTGGGG-3′, reverse 5′-AAGTGTGGCCAGCCTTAGAA-3′;
- iNOS (NM_010927): forward 5′-CAGCTGGGCTGTACAAACCTT-3′, reverse 5′-CATTGGAAGTGAAGCGTTTCG-3′;
- Arg-1 (NM_007482): forward 5′-GGAAAGCCAATGAAGAGCTG-3′, reverse 5′-AACACTCCCCTGACAACCAG-3′;
- MnSOD (NC_000083.6): forward 5′-GCTCTGGCCAAGGGAGATGT-3′, reverse, 5′-GGGCTCAGGTTTGTCCAGAAA-3′;
- GR (NM_008173.3): forward 5′-CGCCAAGTGATTGCCGC-3′, reverse 5′-TGTAGAAGGGTCATTTGGTCATCCA-3′.
2.9. Measurement of Mitochondrial Respiratory Chain Complex (MRC) Activities
2.10. Measurement of Mitochondrial ATP Production Rate
2.11. Measurement of Mouse Brain ATP Levels
2.12. T-Maze
2.13. Statistical Analyses
2.14. Data Statement
3. Results
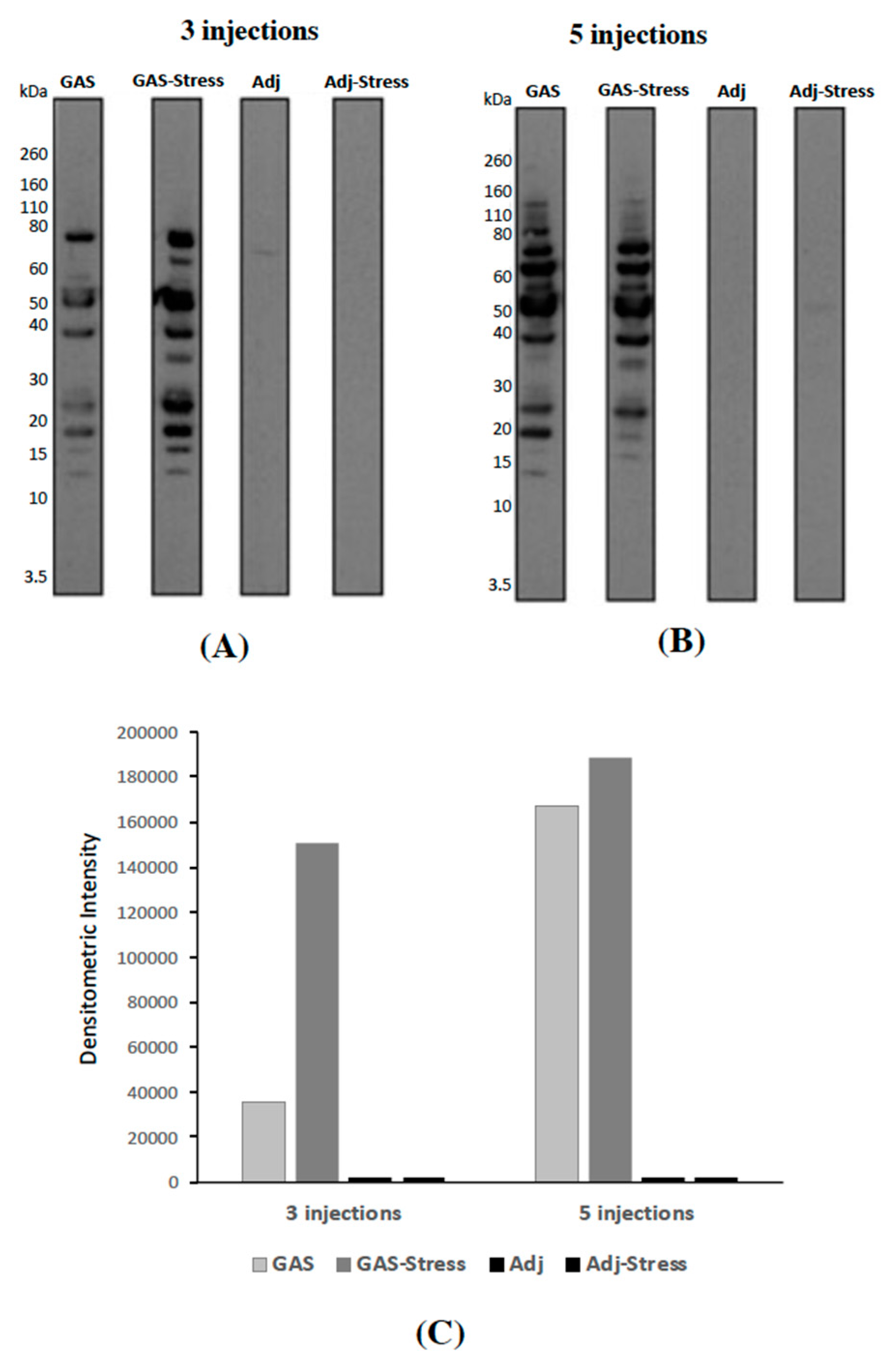
3.1. Evaluation of Anti-GAS Antibody Responses in Sera from Mice Injected with GAS Homogenates and Exposed (or not) to Chronic Psychosocial Stress
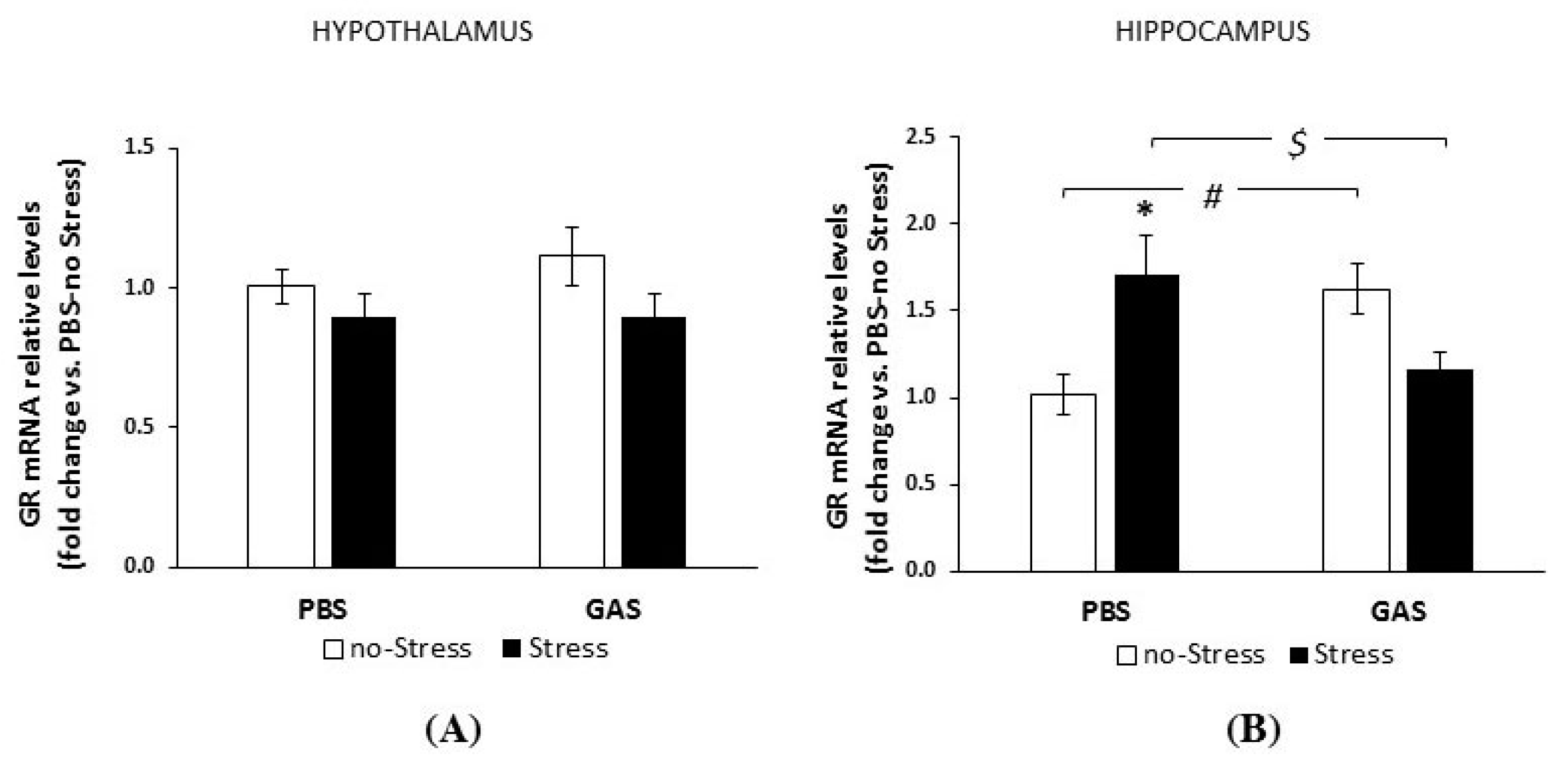
3.2. Consequences of Psychosocial Stress on Serum Corticosterone Concentrations and Glucocorticoid Receptor mRNA Levels in Hypothalamus and Hippocampus
3.3. Behavioural Profile Exhibited during the First Active Social Confrontation
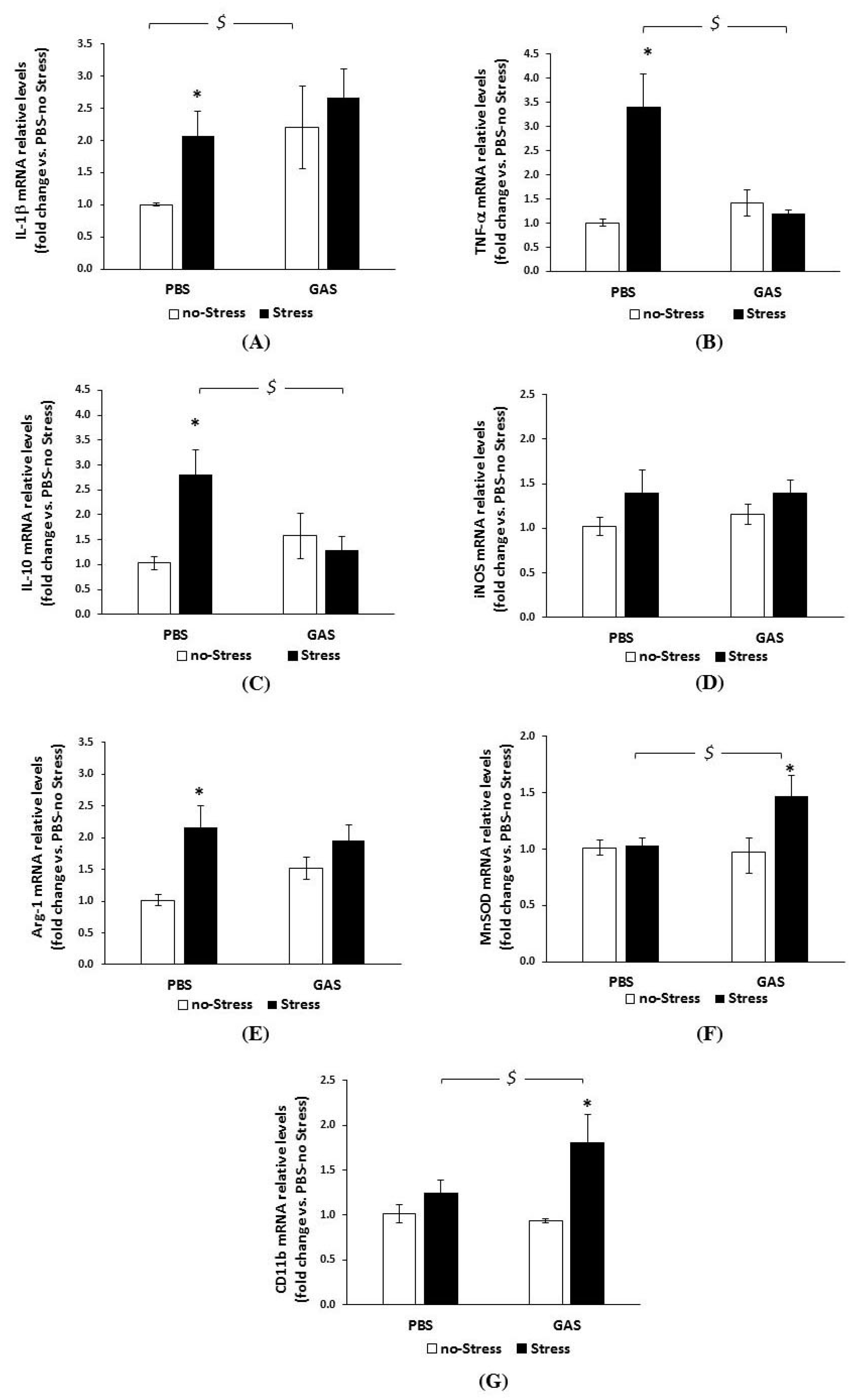
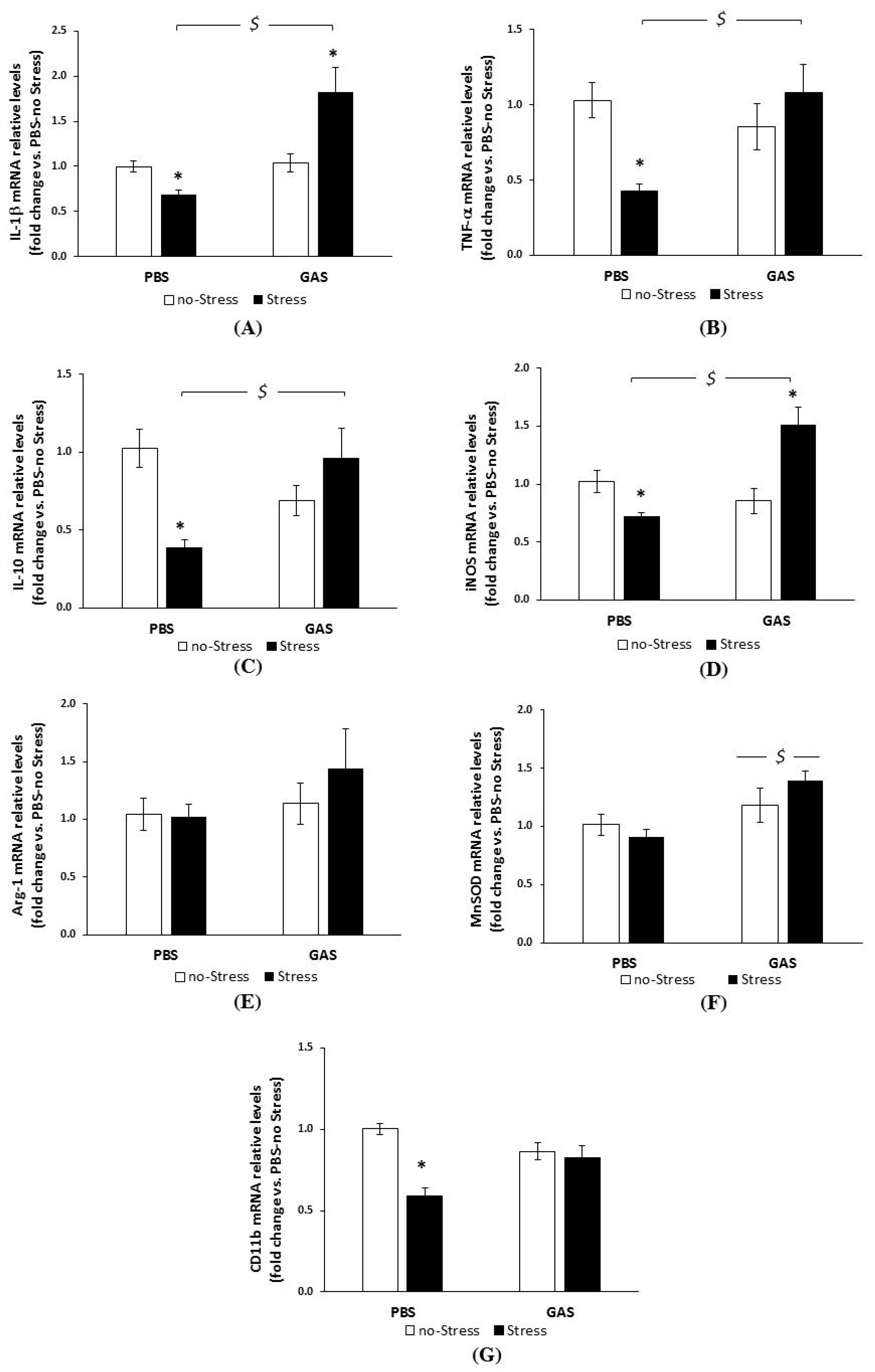
3.4. Chronic Psychosocial Stress Increased Inflammatory Genes Expression in the Brain of GAS Mice
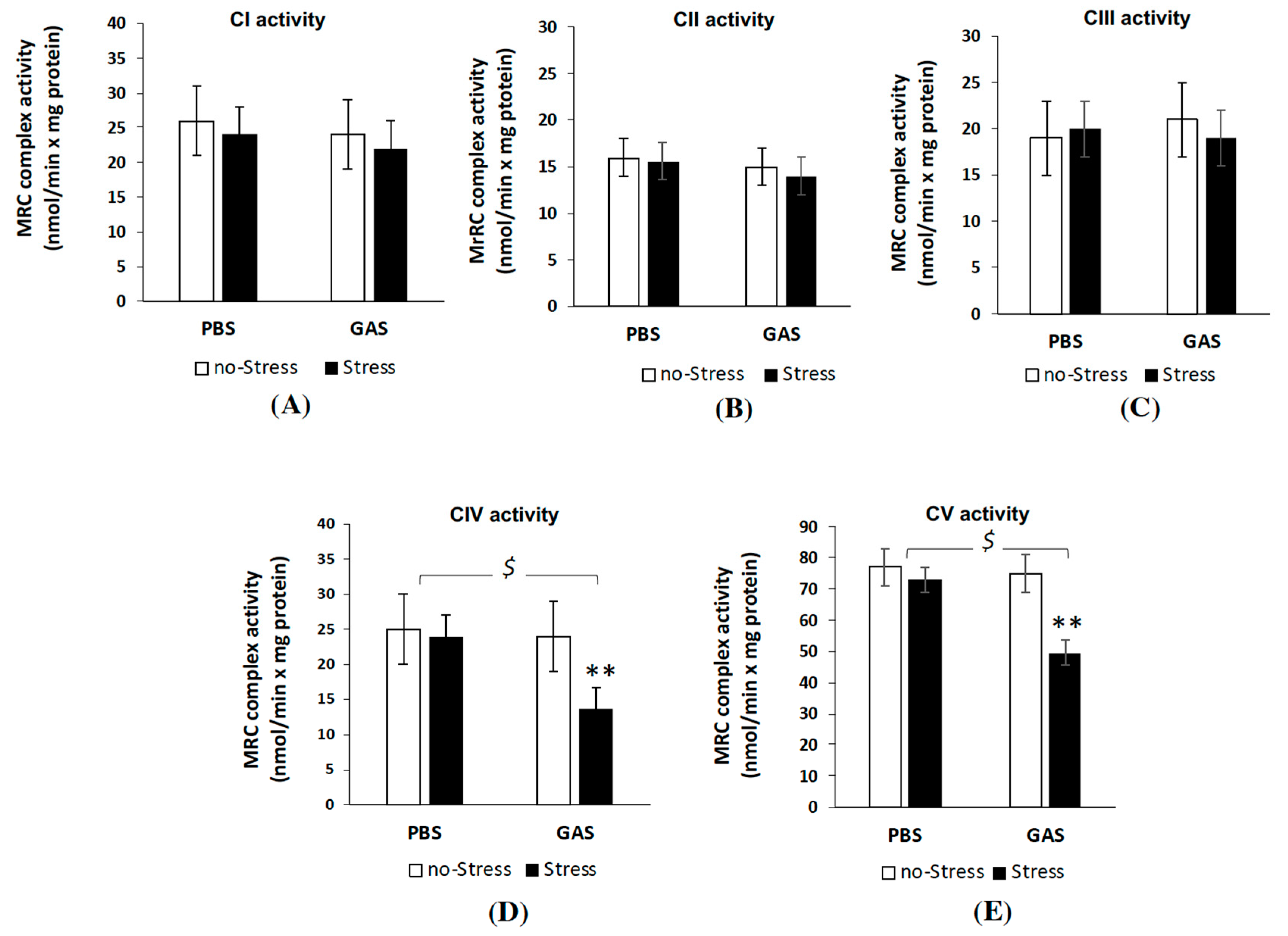
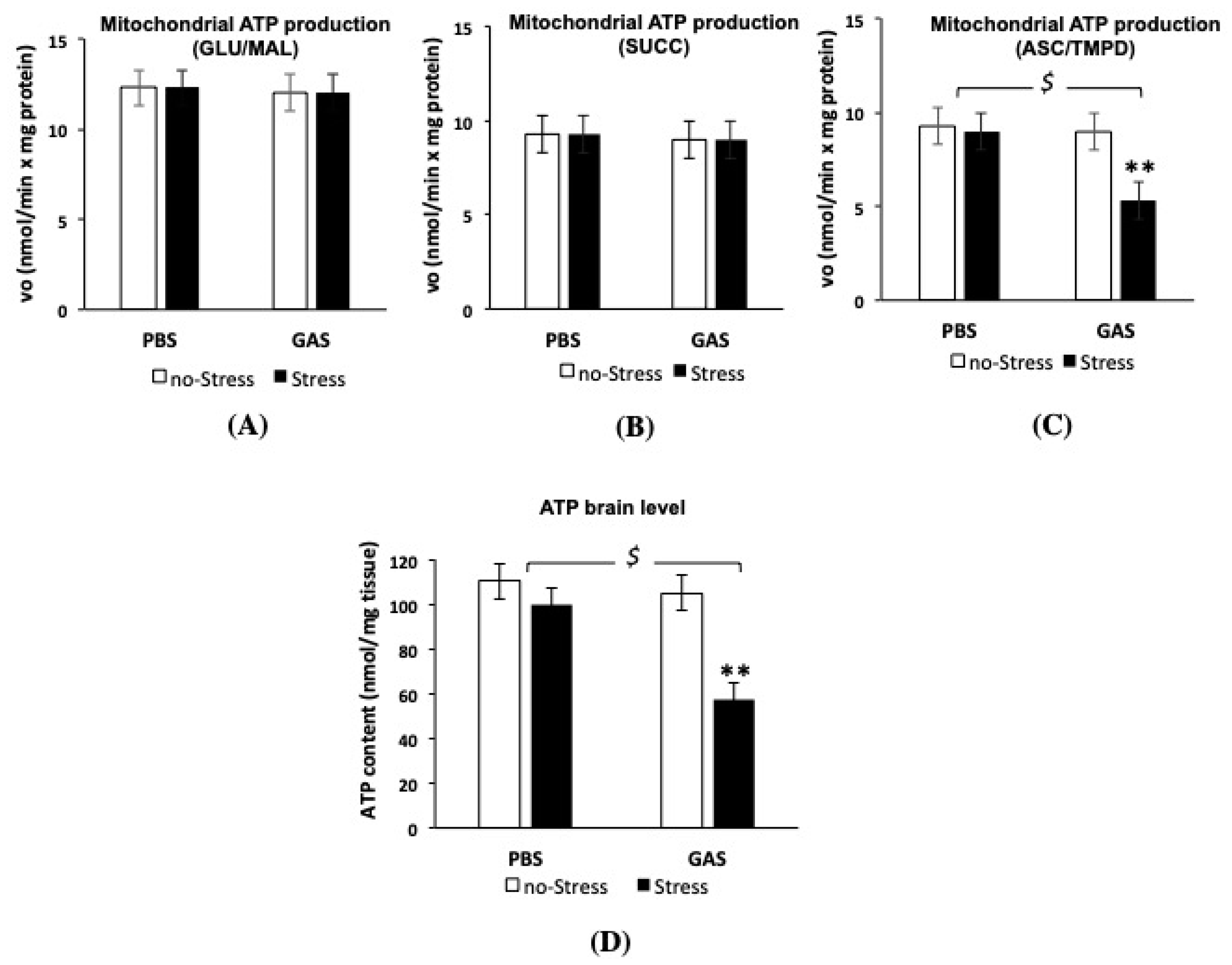
3.5. Psychosocial Stress Affects Mitochondrial OXPHOS Machinery and Reduces Energy Status in the Brain of GAS Immunized Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Benros, M.E.; Waltoft, B.L.; Nordentoft, M.; Østergaard, S.D.; Eaton, W.W.; Krogh, J.; Mortensen, P.B. Autoimmune Diseases and Severe Infections as Risk Factors for Mood Disorders. JAMA Psychiatry 2013, 70, 812. [Google Scholar] [CrossRef] [PubMed]
- Köhler-Forsberg, O.; Petersen, L.; Gasse, C.; Mortensen, P.B.; Dalsgaard, S.; Yolken, R.H.; Mors, O.; Benros, M.E. A Nationwide Study in Denmark of the Association between Treated Infections and the Subsequent Risk of Treated Mental Disorders in Children and Adolescents. JAMA Psychiatry 2018. [Google Scholar] [CrossRef] [PubMed]
- Sperner-Unterweger, B. Immunological aetiology of major psychiatric disorders: Evidence and therapeutic implications. Drugs 2005, 65, 1493–1520. [Google Scholar] [CrossRef] [PubMed]
- Tylee, D.S.; Sun, J.; Hess, J.L.; Tahir, M.A.; Sharma, E.; Malik, R.; Worrall, B.B.; Levine, A.J.; Martinson, J.J.; Nejentsev, S.; et al. Genetic correlations among psychiatric and immune-related phenotypes based on genome-wide association data. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2018, 177, 641–657. [Google Scholar] [CrossRef] [PubMed]
- Khandaker, G.M.; Dantzer, R.; Jones, P.B. Immunopsychiatry: Important facts. Psychol. Med. 2017, 47, 2229–2237. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.C.; Lee, S.M.; Kalume, F.; Morcos, Y.; Dohan, F.C.; Hasty, K.A.; Callaway, J.C.; Zunt, J.; Desiderio, D.M.; Stuart, J.M. Autoimmunity due to molecular mimicry as a cause of neurological disease. Nat. Med. 2002, 8, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Brimberg, L.; Benhar, I.; Mascaro-Blanco, A.; Alvarez, K.; Lotan, D.; Winter, C.; Klein, J.; Moses, A.E.; Somnier, F.E.; Leckman, J.F.; et al. Behavioral, pharmacological, and immunological abnormalities after streptococcal exposure: A novel rat model of Sydenham chorea and related neuropsychiatric disorders. Neuropsychopharmacology 2012, 37, 2076–2087. [Google Scholar] [CrossRef] [PubMed]
- Garvey, M.A.; Giedd, J.; Swedo, S.E. PANDAS: The search for environmental triggers of pediatric neuropsychiatric disorders. Lessons from rheumatic fever. J. Child Neurol. 1998, 13, 413–423. [Google Scholar] [CrossRef]
- Hoffman, K.L.; Hornig, M.; Yaddanapudi, K.; Jabado, O.; Lipkin, W.I. A murine model for neuropsychiatric disorders associated with group A beta-hemolytic streptococcal infection. J. Neurosci. 2004, 24, 1780–1791. [Google Scholar] [CrossRef]
- Snider, L.A.; Swedo, S.E. PANDAS: Current status and directions for research. Mol. Psychiatry 2004, 9, 900–907. [Google Scholar] [CrossRef]
- Swedo, S.E.; Leonard, H.L.; Rapoport, J.L. The pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection (PANDAS) subgroup: Separating fact from fiction. Pediatrics 2004, 113, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Yaddanapudi, K.; Hornig, M.; Serge, R.; De Miranda, J.; Baghban, A.; Villar, G.; Lipkin, W.I. Passive transfer of streptococcus-induced antibodies reproduces behavioral disturbances in a mouse model of pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection. Mol. Psychiatry 2010, 15, 712–726. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, P.J.; Dietrich, A.; Edwards, M.J.; Elamin, I.; Martino, D. Environmental factors in Tourette syndrome. Neurosci. Biobehav. Rev. 2013, 37, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Leonard, H.L.; Swedo, S.E. Paediatric autoimmune neuropsychiatric disorders associated with streptococcal infection (PANDAS). Int. J. Neuropsychopharmacol. 2001, 4, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Cardona, F.; Orefici, G. Group A streptococcal infections and tic disorders in an Italian pediatric population. J. Pediatr. 2001, 138, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, R.; Gulisano, M.; Pavone, P.; Fogliani, F.; Robertson, M.M. Increased antistreptococcal antibody titers and anti-basal ganglia antibodies in patients with Tourette syndrome: Controlled cross-sectional study. J. Child Neurol. 2006, 21, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Orlovska, S.; Vestergaard, C.H.; Bech, B.H.; Nordentoft, M.; Vestergaard, M.; Benros, M.E. Association of Streptococcal Throat Infection with Mental Disorders: Testing Key Aspects of the PANDAS Hypothesis in a Nationwide Study. JAMA Psychiatry 2017, 74, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Macrì, S.; Ceci, C.; Onori, M.P.; Invernizzi, R.W.; Bartolini, E.; Altabella, L.; Canese, R.; Imperi, M.; Orefici, G.; Creti, R.; et al. Mice repeatedly exposed to Group-A β-Haemolytic Streptococcus show perseverative behaviors, impaired sensorimotor gating, and immune activation in rostral diencephalon. Sci. Rep. 2015, 5, 13257. [Google Scholar] [CrossRef] [PubMed]
- Macrì, S.; Spinello, C.; Widomska, J.; Magliozzi, R.; Poelmans, G.; Invernizzi, R.W.; Creti, R.; Roessner, V.; Bartolini, E.; Margarit, I.; et al. Neonatal corticosterone mitigates autoimmune neuropsychiatric disorders associated with streptococcus in mice. Sci. Rep. 2018, 8, 10188. [Google Scholar] [CrossRef] [PubMed]
- Conelea, C.A.; Woods, D.W. The influence of contextual factors on tic expression in Tourette’s syndrome: A review. J. Psychosom. Res. 2008, 65, 487–496. [Google Scholar] [CrossRef]
- Buse, J.; Kirschbaum, C.; Leckman, J.F.; Münchau, A.; Roessner, V. The Modulating Role of Stress in the Onset and Course of Tourette’s Syndrome: A Review. Behav. Modif. 2014, 38, 184–216. [Google Scholar] [CrossRef] [PubMed]
- Godar, S.C.; Bortolato, M. What makes you tic? Translational approaches to study the role of stress and contextual triggers in Tourette syndrome. Neurosci. Biobehav. Rev. 2017, 76, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Leckman, J.F. Tourette’s syndrome. Lancet 2002, 360, 1577–1586. [Google Scholar] [CrossRef]
- Motlagh, M.G.; Katsovich, L.; Thompson, N.; Lin, H.; Kim, Y.-S.; Scahill, L.; Lombroso, P.J.; King, R.A.; Peterson, B.S.; Leckman, J.F. Severe psychosocial stress and heavy cigarette smoking during pregnancy: An examination of the pre- and perinatal risk factors associated with ADHD and Tourette syndrome. Eur. Child. Adolesc. Psychiatry 2010, 19, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Saccomani, L.; Fabiana, V.; Manuela, B.; Giambattista, R. Tourette syndrome and chronic tics in a sample of children and adolescents. Brain Dev. 2005, 27, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Conelea, C.A.; Woods, D.W.; Brandt, B.C. The impact of a stress induction task on tic frequencies in youth with Tourette Syndrome. Behav. Res. Ther. 2011, 49, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Katsovich, L.; Ghebremichael, M.; Findley, D.B.; Grantz, H.; Lombroso, P.J.; King, R.A.; Zhang, H.; Leckman, J.F. Psychosocial stress predicts future symptom severities in children and adolescents with Tourette syndrome and/or obsessive-compulsive disorder. J. Child Psychol. Psychiatry 2007, 48, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Bateson, P.; Barker, D.; Clutton-Brock, T.; Deb, D.; D’Udine, B.; Foley, R.A.; Gluckman, P.; Godfrey, K.; Kirkwood, T.; Lahr, M.M.; et al. Developmental plasticity and human health. Nature 2004, 430, 419–421. [Google Scholar] [CrossRef]
- Bale, T.L.; Epperson, C.N. Sex differences and stress across the lifespan. Nat. Neurosci. 2015, 18, 1413–1420. [Google Scholar] [CrossRef]
- Bartolomucci, A. Social stress, immune functions and disease in rodents. Front. Neuroendocrinol. 2007, 28, 28–49. [Google Scholar] [CrossRef]
- Krishnan, V.; Han, M.-H.; Graham, D.L.; Berton, O.; Renthal, W.; Russo, S.J.; LaPlant, Q.; Graham, A.; Lutter, M.; Lagace, D.C.; et al. Molecular Adaptations Underlying Susceptibility and Resistance to Social Defeat in Brain Reward Regions. Cell 2007, 131, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Lassance-Soares, R.M.; Sood, S.; Chakraborty, N.; Jhamnani, S.; Aghili, N.; Nashin, H.; Hammamieh, R.; Jett, M.; Epstein, S.E.; Burnett, M.S. Chronic stress impairs collateral blood flow recovery in aged mice. J. Cardiovasc. Transl. Res. 2014, 7, 749–755. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Physiology and neurobiology of stress and adaptation: Central role of the brain. Physiol. Rev. 2007, 87, 873–904. [Google Scholar] [CrossRef] [PubMed]
- Pryce, C.R.; Fuchs, E. Chronic psychosocial stressors in adulthood: Studies in mice, rats and tree shrews. Neurobiol. Stress 2017, 6, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Scharf, S.H.; Sterlemann, V.; Liebl, C.; Müller, M.B.; Schmidt, M.V. Chronic social stress during adolescence: Interplay of paroxetine treatment and ageing. Neuropharmacology 2013, 72, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Goshen, I.; Yirmiya, R. Interleukin-1 (IL-1): A central regulator of stress responses. Front. Neuroendocrinol. 2009, 30, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Wilcockson, D.C.; Campion, S.; Lunnon, K.; Perry, V.H. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J. Neurosci. 2005, 25, 9275–9284. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Stress-induced neuroinflammatory priming: A liability factor in the etiology of psychiatric disorders. Neurobiol. Stress 2016, 4, 62–70. [Google Scholar] [CrossRef]
- Johnson, J.; O’Connor, K.; Watkins, L.; Maier, S. The role of IL-1β in stress-induced sensitization of proinflammatory cytokine and corticosterone responses. Neuroscience 2004, 127, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Newman, T.A.; Cunningham, C. The impact of systemic infection on the progression of neurodegenerative disease. Nat. Rev. Neurosci. 2003, 4, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Barron, H.; Hafizi, S.; Andreazza, A.; Mizrahi, R. Neuroinflammation and Oxidative Stress in Psychosis and Psychosis Risk. Int. J. Mol. Sci. 2017, 18, 651. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.; Wallace, D.C. Mitochondrial Etiology of Neuropsychiatric Disorders. Biol. Psychiatry 2018, 83, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46 Pt 2, 202–217. [Google Scholar] [CrossRef]
- Vacca, R.A.; Bawari, S.; Valenti, D.; Tewari, D.; Nabavi, S.F.; Shirooie, S.; Sah, A.N.; Volpicella, M.; Braidy, N.; Nabavi, S.M. Down syndrome: Neurobiological alterations and therapeutic targets. Neurosci. Biobehav. Rev. 2019, 98, 234–255. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; Braidy, N.; De Rasmo, D.; Signorile, A.; Rossi, L.; Atanasov, A.G.; Volpicella, M.; Henrion-Caude, A.; Nabavi, S.M.; Vacca, R.A. Mitochondria as pharmacological targets in Down syndrome. Free Radic. Biol. Med. 2018, 114, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Stubbs, B.; Köhler, C.A.; Walder, K.; Slyepchenko, A.; Berk, M.; Carvalho, A.F. The putative role of oxidative stress and inflammation in the pathophysiology of sleep dysfunction across neuropsychiatric disorders: Focus on chronic fatigue syndrome, bipolar disorder and multiple sclerosis. Sleep Med. Rev. 2018, 41, 255–265. [Google Scholar] [CrossRef] [PubMed]
- De Simone, R.; Ajmone-Cat, M.A.; Pandolfi, M.; Bernardo, A.; De Nuccio, C.; Minghetti, L.; Visentin, S. The mitochondrial uncoupling protein-2 is a master regulator of both M1 and M2 microglial responses. J. Neurochem. 2015, 135, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Ferger, A.I.; Campanelli, L.; Reimer, V.; Muth, K.N.; Merdian, I.; Ludolph, A.C.; Witting, A. Effects of mitochondrial dysfunction on the immunological properties of microglia. J. Neuroinflamm. 2010, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, H.-M. The Role of Manganese Superoxide Dismutase in Inflammation Defense. Enzym. Res. 2011, 2011, 387176. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Suliman, H.B. Transcriptional control of mitochondrial biogenesis and its interface with inflammatory processes. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 532–541. [Google Scholar] [CrossRef]
- Kim, E.K.; Kwon, J.-E.; Lee, S.-Y.; Lee, E.-J.; Kim, D.S.; Moon, S.-J.; Lee, J.; Kwok, S.-K.; Park, S.-H.; Cho, M.-L. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis. 2017, 8, e2565. [Google Scholar] [CrossRef] [PubMed]
- Flies, D.B.; Chen, L. A simple and rapid vortex method for preparing antigen/adjuvant emulsions for immunization. J. Immunol. Methods 2003, 276, 239–242. [Google Scholar] [CrossRef]
- Bartolomucci, A.; Cabassi, A.; Govoni, P.; Ceresini, G.; Cero, C.; Berra, D.; Dadomo, H.; Franceschini, P.; Dell’Omo, G.; Parmigiani, S.; et al. Metabolic consequences and vulnerability to diet-induced obesity in male mice under chronic social stress. PLoS ONE 2009, 4, e4331. [Google Scholar] [CrossRef] [PubMed]
- Sanghez, V.; Razzoli, M.; Carobbio, S.; Campbell, M.; McCallum, J.; Cero, C.; Ceresini, G.; Cabassi, A.; Govoni, P.; Franceschini, P.; et al. Psychosocial stress induces hyperphagia and exacerbates diet-induced insulin resistance and the manifestations of the Metabolic Syndrome. Psychoneuroendocrinology 2013, 38, 2933–2942. [Google Scholar] [CrossRef] [PubMed]
- Bartolomucci, A.; Carola, V.; Pascucci, T.; Puglisi-Allegra, S.; Cabib, S.; Lesch, K.-P.; Parmigiani, S.; Palanza, P.; Gross, C. Increased vulnerability to psychosocial stress in heterozygous serotonin transporter knockout mice. Dis. Model. Mech. 2010, 3, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Dadomo, H.; Sanghez, V.; Di Cristo, L.; Lori, A.; Ceresini, G.; Malinge, I.; Parmigiani, S.; Palanza, P.; Sheardown, M.; Bartolomucci, A. Vulnerability to chronic subordination stress-induced depression-like disorders in adult 129SvEv male mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Zoratto, F.; Sbriccoli, M.; Martinelli, A.; Glennon, J.C.; Macrì, S.; Laviola, G. Intranasal oxytocin administration promotes emotional contagion and reduces aggression in a mouse model of callousness. Neuropharmacology 2018, 143, 250–267. [Google Scholar] [CrossRef] [PubMed]
- Bartolomucci, A.; Pederzani, T.; Sacerdote, P.; Panerai, A.E.; Parmigiani, S.; Palanza, P. Behavioral and physiological characterization of male mice under chronic psychosocial stress. Psychoneuroendocrinology 2004, 29, 899–910. [Google Scholar] [CrossRef]
- Macrì, S.; Pasquali, P.; Bonsignore, L.T.; Pieretti, S.; Cirulli, F.; Chiarotti, F.; Laviola, G. Moderate neonatal stress decreases within-group variation in behavioral, immune and HPA responses in adult mice. PLoS ONE 2007, 2, e1015. [Google Scholar] [CrossRef]
- Macrì, S.; Granstrem, O.; Shumilina, M.; Antunes Gomes dos Santos, F.J.; Berry, A.; Saso, L.; Laviola, G. Resilience and vulnerability are dose-dependently related to neonatal stressors in mice. Horm. Behav. 2009, 56, 391–398. [Google Scholar] [CrossRef]
- Gao, W.; Stalder, T.; Foley, P.; Rauh, M.; Deng, H.; Kirschbaum, C. Quantitative analysis of steroid hormones in human hair using a column-switching LC-APCI-MS/MS assay. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 928, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; de Bari, L.; De Filippis, B.; Ricceri, L.; Vacca, R.A. Preservation of mitochondrial functional integrity in mitochondria isolated from small cryopreserved mouse brain areas. Anal. Biochem. 2014, 444, 25–31. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Valenti, D.; Chiodi, V.; Ferrante, A.; de Bari, L.; Fiorentini, C.; Domenici, M.R.; Ricceri, L.; Vacca, R.A.; Fabbri, A.; et al. Modulation of Rho GTPases rescues brain mitochondrial dysfunction, cognitive deficits and aberrant synaptic plasticity in female mice modeling Rett syndrome. Eur. Neuropsychopharmacol. 2015, 25, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Manente, A.G.; Valenti, D.; Pinton, G.; Jithesh, P.V.; Daga, A.; Rossi, L.; Gray, S.G.; O’Byrne, K.J.; Fennell, D.A.; Vacca, R.A.; et al. Estrogen receptor β activation impairs mitochondrial oxidative metabolism and affects malignant mesothelioma cell growth in vitro and in vivo. Oncogenesis 2013, 2, e72. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Valenti, D.; de Bari, L.; De Rasmo, D.; Musto, M.; Fabbri, A.; Ricceri, L.; Fiorentini, C.; Laviola, G.; Vacca, R.A. Mitochondrial free radical overproduction due to respiratory chain impairment in the brain of a mouse model of Rett syndrome: Protective effect of CNF1. Free Radic. Biol. Med. 2015, 83, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.A. Bioluminometric assay of ATP in mouse brain: Determinant factors for enhanced test sensitivity. J. Biosci. 2003, 28, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Vigli, D.; Rusconi, L.; Valenti, D.; La Montanara, P.; Cosentino, L.; Lacivita, E.; Leopoldo, M.; Amendola, E.; Gross, C.; Landsberger, N.; et al. Rescue of prepulse inhibition deficit and brain mitochondrial dysfunction by pharmacological stimulation of the central serotonin receptor 7 in a mouse model of CDKL5 Deficiency Disorder. Neuropharmacology 2019, 144, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M.J.; Rawlins, J.N.P. T-maze alternation in the rodent. Nat. Protoc. 2006, 1, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Storm, D.R.; Xia, Z. Conditional deletion of ERK5 MAP kinase in the nervous system impairs pheromone information processing and pheromone-evoked behaviors. PLoS ONE 2013, 8, e76901. [Google Scholar] [CrossRef] [PubMed]
- Razzoli, M.; Nyuyki-Dufe, K.; Gurney, A.; Erickson, C.; McCallum, J.; Spielman, N.; Marzullo, M.; Patricelli, J.; Kurata, M.; Pope, E.A.; et al. Social stress shortens lifespan in mice. Aging Cell 2018, 17, e12778. [Google Scholar] [CrossRef]
- Spinello, C.; Laviola, G.; Macrì, S. Pediatric Autoimmune Disorders Associated with Streptococcal Infections and Tourette’s Syndrome in Preclinical Studies. Front. Neurosci. 2016, 10, 310. [Google Scholar] [CrossRef] [PubMed]
- Swedo, S.E.; Grant, P.J. Annotation: PANDAS: A model for human autoimmune disease. J. Child Psychol. Psychiatry 2005, 46, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Trifiletti, R.R.; Packard, A.M. Immune mechanisms in pediatric neuropsychiatric disorders. Tourette’s syndrome, OCD, and PANDAS. Child. Adolesc. Psychiatr. Clin. N. Am. 1999, 8, 767–775. [Google Scholar] [CrossRef]
- Lalonde, R. The neurobiological basis of spontaneous alternation. Neurosci. Biobehav. Rev. 2002, 26, 91–104. [Google Scholar] [CrossRef]
- Irwin, J.; Tombaugh, T.N.; Zacharko, R.M.; Anisman, H. Alteration of exploration and the response to food associated cues after treatment with pimozide. Pharmacol. Biochem. Behav. 1983, 18, 235–246. [Google Scholar] [CrossRef]
- Jaffard, R.; Mocaer, E.; Poignant, J.-C.; Micheau, J.; Marighetto, A.; Meunier, M.; Béracochéa, D. Effects of tianeptine on spontaneous alternation, simple and concurrent spatial discrimination learning and on alcohol-induced alternation deficits in mice. Behav. Pharmacol. 1991, 2, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Bartolomucci, A.; Palanza, P.; Parmigiani, S.; Pederzani, T.; Merlot, E.; Neveu, P.J.; Dantzer, R. Chronic psychosocial stress down-regulates central cytokines mRNA. Brain Res. Bull. 2003, 62, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Ajmone-Cat, M.A.; D’Urso, M.C.; di Blasio, G.; Brignone, M.S.; De Simone, R.; Minghetti, L. Glycogen synthase kinase 3 is part of the molecular machinery regulating the adaptive response to LPS stimulation in microglial cells. Brain Behav. Immun. 2016, 55, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Ajmone-Cat, M.A.; Mancini, M.; De Simone, R.; Cilli, P.; Minghetti, L. Microglial polarization and plasticity: Evidence from organotypic hippocampal slice cultures. Glia 2013, 61, 1698–1711. [Google Scholar] [CrossRef] [PubMed]
- Alboni, S.; Maggi, L. Editorial: Cytokines as Players of Neuronal Plasticity and Sensitivity to Environment in Healthy and Pathological Brain. Front. Cell. Neurosci. 2016, 9, 508. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Oliva, A.M.; de Pablos, R.M.; Villarán, R.F.; Argüelles, S.; Venero, J.L.; Machado, A.; Cano, J. Stress is critical for LPS-induced activation of microglia and damage in the rat hippocampus. Neurobiol. Aging 2011, 32, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.-A.; Eu, M.Y.; Kang, S.-H.; Gwag, B.J.; Jou, I.; Joe, E.-H. Differential neutrophil infiltration contributes to regional differences in brain inflammation in the substantia nigra pars compacta and cortex. Glia 2008, 56, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Kipp, M.; Norkute, A.; Johann, S.; Lorenz, L.; Braun, A.; Hieble, A.; Gingele, S.; Pott, F.; Richter, J.; Beyer, C. Brain-region-specific astroglial responses in vitro after LPS exposure. J. Mol. Neurosci. 2008, 35, 235–243. [Google Scholar] [CrossRef]
- Gądek-Michalska, A.; Spyrka, J.; Rachwalska, P.; Tadeusz, J.; Bugajski, J. Influence of chronic stress on brain corticosteroid receptors and HPA axis activity. Pharmacol. Rep. 2013, 65, 1163–1175. [Google Scholar] [CrossRef]
- Mizoguchi, K.; Ishige, A.; Aburada, M.; Tabira, T. Chronic stress attenuates glucocorticoid negative feedback: Involvement of the prefrontal cortex and hippocampus. Neuroscience 2003, 119, 887–897. [Google Scholar] [CrossRef]
- Levine, S.; Strebel, R.; Wenk, E.J.; Harman, P.J. Suppression of experimental allergic encephalomyelitis by stress. Proc. Soc. Exp. Biol. Med. 1962, 109, 294–298. [Google Scholar] [CrossRef]
- Levine, S.; Saltzman, A. Nonspecific stress prevents relapses of experimental allergic encephalomyelitis in rats. Brain Behav. Immun. 1987, 1, 336–341. [Google Scholar] [CrossRef]
- Levine, S.; Wenk, E.J.; Muldoon, T.N.; Cohen, S.G. Enhancement of experimental allergic encephalomyelitis by adrenalectomy. Proc. Soc. Exp. Biol. Med. 1962, 111, 383–385. [Google Scholar] [CrossRef]
- Newton, R.; Shah, S.; Altonsy, M.O.; Gerber, A.N. Glucocorticoid and cytokine crosstalk: Feedback, feedforward, and co-regulatory interactions determine repression or resistance. J. Biol. Chem. 2017, 292, 7163–7172. [Google Scholar] [CrossRef]
- Webster, J.C.; Oakley, R.H.; Jewell, C.M.; Cidlowski, J.A. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative isoform: A mechanism for the generation of glucocorticoid resistance. Proc. Natl. Acad. Sci. USA 2001, 98, 6865–6870. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Bonneau, R.H. Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J. Neuroimmunol. 2006, 171, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Kim, H.S.; Lee, J.; Chung, J.; Lee, J.S.; Choi, J.S.; Yoon, T.R.; Kim, H.K.; Chung, H.Y. Down-regulation of oxidative stress and COX-2 and iNOS expressions by dimethyl lithospermate in aged rat kidney. Arch. Pharm. Res. 2014, 37, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.-L.; Yang, C.-M. Role of Redox Signaling in Neuroinflammation and Neurodegenerative Diseases. Biomed. Res. Int. 2013, 2013, 484613. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, J.L.; Moro, M.A.; Lizasoain, I.; Lorenzo, P.; Castrillo, A.; Boscá, L.; Leza, J.C. Inducible nitric oxide synthase expression in brain cortex after acute restraint stress is regulated by nuclear factor kappaB-mediated mechanisms. J. Neurochem. 2001, 76, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Novaes, L.S.; Dos Santos, N.B.; Dragunas, G.; Perfetto, J.G.; Leza, J.C.; Scavone, C.; Munhoz, C.D. Repeated Restraint Stress Decreases Na,K-ATPase Activity via Oxidative and Nitrosative Damage in the Frontal Cortex of Rats. Neuroscience 2018, 393, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.H.; Goeddel, D.V. Induction of manganous superoxide dismutase by tumor necrosis factor: Possible protective mechanism. Science 1988, 242, 941–944. [Google Scholar] [CrossRef]
- Musatov, A.; Robinson, N.C. Susceptibility of mitochondrial electron-transport complexes to oxidative damage. Focus on cytochrome c oxidase. Free Radic. Res. 2012, 46, 1313–1326. [Google Scholar] [CrossRef]
- Hernández-Aguilera, A.; Rull, A.; Rodríguez-Gallego, E.; Riera-Borrull, M.; Luciano-Mateo, F.; Camps, J.; Menéndez, J.A.; Joven, J. Mitochondrial dysfunction: A basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediat. Inflamm. 2013, 2013, 135698. [Google Scholar] [CrossRef]
- Hoffmann, A.; Spengler, D. The Mitochondrion as Potential Interface in Early-Life Stress Brain Programming. Front. Behav. Neurosci. 2018, 12, 306. [Google Scholar] [CrossRef]
- Di Filippo, M.; Chiasserini, D.; Tozzi, A.; Picconi, B.; Calabresi, P. Mitochondria and the Link Between Neuroinflammation and Neurodegeneration. J. Alzheimer Dis. 2010, 20, S369–S379. [Google Scholar] [CrossRef]
- Witte, M.E.; Mahad, D.J.; Lassmann, H.; van Horssen, J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol. Med. 2014, 20, 179–187. [Google Scholar] [CrossRef]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef]
- Park, J.; Choi, H.; Min, J.-S.; Park, S.-J.; Kim, J.-H.; Park, H.-J.; Kim, B.; Chae, J.-I.; Yim, M.; Lee, D.-S. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J. Neurochem. 2013, 127, 221–232. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, X.; Shang, W.; Xu, J.; Wang, X.; Hu, X.; Ao, Y.; Cheng, H. Proinflammatory Cytokines Stimulate Mitochondrial Superoxide Flashes in Articular Chondrocytes In Vitro and In Situ. PLoS ONE 2013, 8, e66444. [Google Scholar] [CrossRef]
- Hahn, W.S.; Kuzmicic, J.; Burrill, J.S.; Donoghue, M.A.; Foncea, R.; Jensen, M.D.; Lavandero, S.; Arriaga, E.A.; Bernlohr, D.A. Proinflammatory cytokines differentially regulate adipocyte mitochondrial metabolism, oxidative stress, and dynamics. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1033–E1045. [Google Scholar] [CrossRef]
- Du, J.; McEwen, B.; Manji, H.K. Glucocorticoid receptors modulate mitochondrial function. Commun. Integr. Biol. 2009, 2, 350–352. [Google Scholar] [CrossRef]
- Hunter, R.G.; Seligsohn, M.; Rubin, T.G.; Griffiths, B.B.; Ozdemir, Y.; Pfaff, D.W.; Datson, N.A.; McEwen, B.S. Stress and corticosteroids regulate rat hippocampal mitochondrial DNA gene expression via the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2016, 113, 9099–9104. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Behaviour | Parameter | GAS-Stress | PBS-Stress | F (df) | p | Cohen’s d |
|---|---|---|---|---|---|---|
| Attack received | Duration | 6.559 ± 1.135 | 4.996 ± 0.957 | 3.578 (1,37) | 0.07 Treat × Time ^ | 1.489 |
| Frequency | 9.974 ± 1.866 | 7.175 ± 1.422 | 4.338 (1,37) | 0.04 Treat × Time * | 1.687 | |
| Defensive upright posture | Duration | 23.724 ± 3.616 | 16.925 ± 3.482 | 1.707 (1,38) | 0.20 | 1.915 |
| Frequency | 10.575 ± 1.729 | 6.079 ± 1.076 | 3.545 (1,37) | 0.07 Treat ^ | 3.122 | |
| Latency | 212.518 ± 36.059 | 337.022 ± 43.116 | 4.907 (1,38) | 0.03 Treat * | 3.132 | |
| Self-grooming | Duration | 13.048 ± 2.366 | 12.784 ± 2.318 | 0.007 (1,38) | 0.93 | 0.113 |
| Frequency | 3.350 ± 0.452 | 2.900 ± 0.477 | 0.707 (1,38) | 0.41 | 0.968 | |
| Immobility attack-related | Duration | 8.967 ± 2.156 | 2.923 ±.1.181 | 3.279 (1,36) | 0.08 Treat ^ | 3.477 |
| Frequency | 3.475 ± 0.791 | 2.350 ± 0.648 | 3.777 (1,38) | 0.06 Treat ^ | 1.556 | |
| Immobility contact-related | Duration | 13.3 ± 3.650 | 3.988 ± 1.022 | 5.402 (1,37) | 0.03 Treat * | 3.474 |
| Frequency | 2.950 ± 0.636 | 1.447 ± 0.375 | 3.982 (1,37) | 0.05 Treat ^ | 2.879 | |
| Submissive behaviours | Duration | 55.198 ± 6.601 | 45.790 ± 7.840 | 0.843 (1,38) | 0.36 | 1.298 |
| Frequency | 24.550 ± 3.890 | 17.700 ± 3.069 | 1.911 (1,38) | 0.17 | 1.955 | |
| Fleeing | Duration | 7.909 ± 1.452 | 6.480 ± 1.233 | 0.381 (1,37) | 0.54 | 1.061 |
| Frequency | 9.650 ± 1.743 | 6.350 ± 1.212 | 1.607 (1,38) | 0.21 | 2.198 | |
| Latency | 285.654 ± 39.534 | 349.358 ± 46.590 | 1.087 (1,38) | 0.30 | 1.474 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajmone-Cat, M.A.; Spinello, C.; Valenti, D.; Franchi, F.; Macrì, S.; Vacca, R.A.; Laviola, G. Brain-Immune Alterations and Mitochondrial Dysfunctions in a Mouse Model of Paediatric Autoimmune Disorder Associated with Streptococcus: Exacerbation by Chronic Psychosocial Stress. J. Clin. Med. 2019, 8, 1514. https://doi.org/10.3390/jcm8101514
Ajmone-Cat MA, Spinello C, Valenti D, Franchi F, Macrì S, Vacca RA, Laviola G. Brain-Immune Alterations and Mitochondrial Dysfunctions in a Mouse Model of Paediatric Autoimmune Disorder Associated with Streptococcus: Exacerbation by Chronic Psychosocial Stress. Journal of Clinical Medicine. 2019; 8(10):1514. https://doi.org/10.3390/jcm8101514
Chicago/Turabian StyleAjmone-Cat, Maria Antonietta, Chiara Spinello, Daniela Valenti, Francesca Franchi, Simone Macrì, Rosa Anna Vacca, and Giovanni Laviola. 2019. "Brain-Immune Alterations and Mitochondrial Dysfunctions in a Mouse Model of Paediatric Autoimmune Disorder Associated with Streptococcus: Exacerbation by Chronic Psychosocial Stress" Journal of Clinical Medicine 8, no. 10: 1514. https://doi.org/10.3390/jcm8101514
APA StyleAjmone-Cat, M. A., Spinello, C., Valenti, D., Franchi, F., Macrì, S., Vacca, R. A., & Laviola, G. (2019). Brain-Immune Alterations and Mitochondrial Dysfunctions in a Mouse Model of Paediatric Autoimmune Disorder Associated with Streptococcus: Exacerbation by Chronic Psychosocial Stress. Journal of Clinical Medicine, 8(10), 1514. https://doi.org/10.3390/jcm8101514