Immune-Inflammatory Responses in Atherosclerosis: The Role of Myeloid Cells

 , , and
, , and 
{kind=link}
Abstract
1. Introduction
2. Myeloid Immune Cells Involved in Atherosclerosis
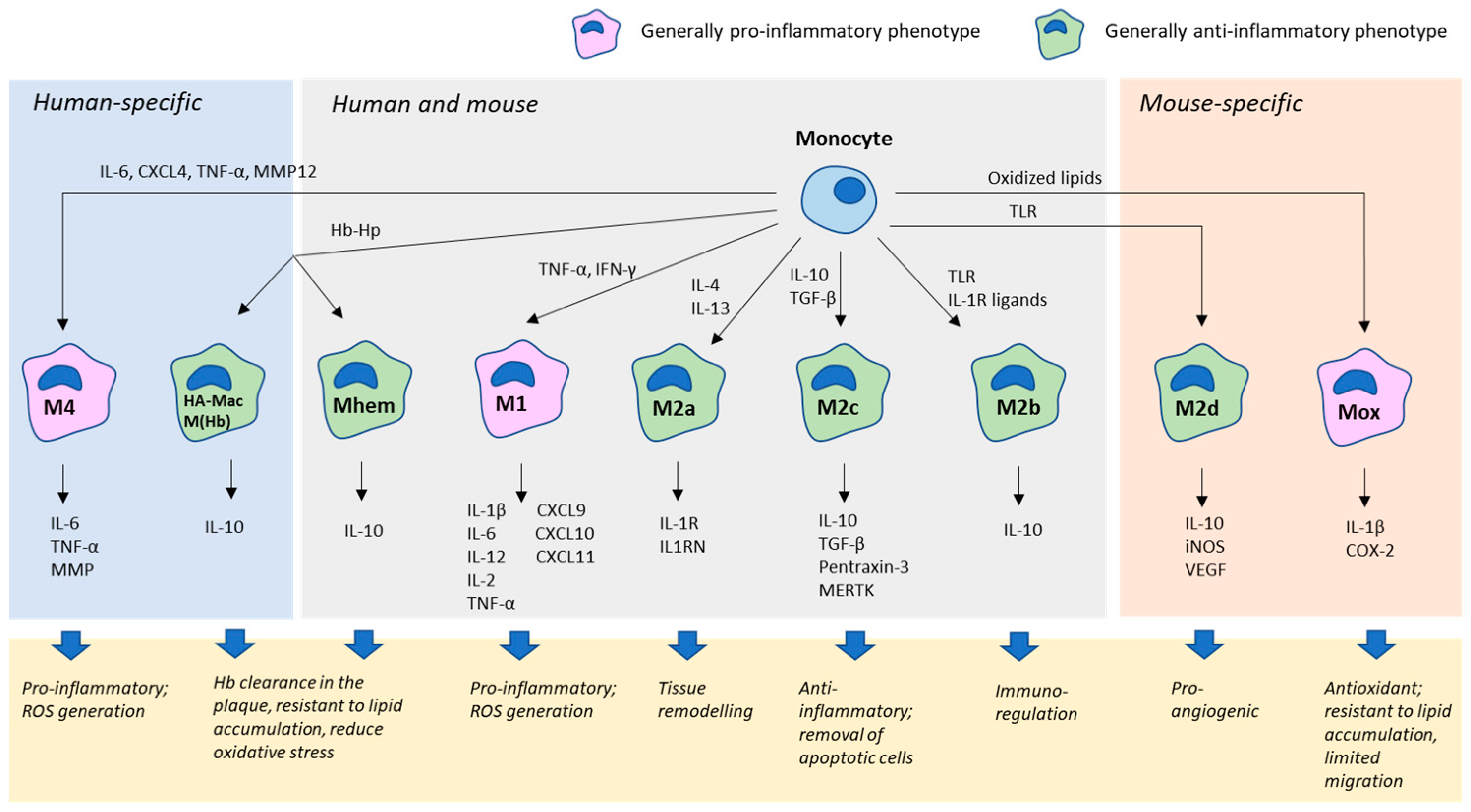
2.1. Monocytes
2.2. Macrophages
2.3. Dendritic Cells
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Alipov, V.I.; Sukhorukov, V.N.; Karagodin, V.P.; Grechko, A.V.; Orekhov, A.N. Chemical composition of circulating native and desialylated low density lipoprotein: What is the difference? Vessel Plus 2017, 1, 107–115. [Google Scholar] [CrossRef]
- Wong, B.W.; Meredith, A.; Lin, D.; McManus, B.M. The biological role of inflammation in atherosclerosis. Can. J. Cardiol. 2012, 28, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc. Pathol. 2013, 22, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Kasikara, C.; Doran, A.C.; Cai, B.; Tabas, I. The role of non-resolving inflammation in atherosclerosis. J. Clin. Investig. 2018, 128, 2713–2723. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef]
- Gerrity, R.G.; Naito, H.K.; Richardson, M.; Schwartz, C.J. Dietary induced atherogenesis in swine. Morphology of the intima in prelesion stages. Am. J. Pathol. 1979, 95, 775–792. [Google Scholar]
- Galkina, E.; Ley, K. Leukocyte influx in atherosclerosis. Curr. Drug Targets 2007, 8, 1239–1248. [Google Scholar] [CrossRef]
- Jongstra-Bilen, J.; Haidari, M.; Zhu, S.N.; Chen, M.; Guha, D.; Cybulsky, M.I. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J. Exp. Med. 2006, 203, 2073–2083. [Google Scholar] [CrossRef]
- Lessner, S.M.; Prado, H.L.; Waller, E.K.; Galis, Z.S. Atherosclerotic lesions grow through recruitment and proliferation of circulating monocytes in a murine model. Am. J. Pathol. 2002, 160, 2145–2155. [Google Scholar] [CrossRef]
- Lord, R.S.; Bobryshev, Y.V. Clustering of dendritic cells in athero-prone areas of the aorta. Atherosclerosis 1999, 146, 197–198. [Google Scholar] [PubMed]
- Strieter, R.M.; Wiggins, R.; Phan, S.H.; Wharram, B.L.; Showell, H.J.; Remich, D.G.; Chensue, S.W.; Kunkel, S.L. Monocyte chemotactic protein gene expression by cytokine-treated human fibroblasts and endothelial cells. Biochem. Biophys. Res. Commun. 1989, 162, 694–700. [Google Scholar] [CrossRef]
- Pober, J.S.; Gimbrone, M.A., Jr.; Collins, T.; Cotran, R.S.; Ault, K.A.; Fiers, W.; Krensky, A.M.; Clayberger, C.; Reiss, C.S.; Burakoff, S.J. Interactions of T lymphocytes with human vascular endothelial cells: Role of endothelial cells surface antigens. Immunobiology 1984, 168, 483–494. [Google Scholar] [CrossRef]
- Bobryshev, Y.V. Monocyte recruitment and foam cell formation in atherosclerosis. Micron 2006, 37, 208–222. [Google Scholar] [CrossRef]
- Huo, Y.; Xia, L. P-selectin glycoprotein ligand-1 plays a crucial role in the selective recruitment of leukocytes into the atherosclerotic arterial wall. Trends Cardiovasc. Med. 2009, 19, 140–145. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Randolph, G.J.; Jakubzick, C.; Qu, C. Antigen presentation by monocytes and monocyte-derived cells. Curr. Opin. Immunol. 2008, 20, 52–60. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Iwabuchi, K.; Ogasawara, K.; Good, R.A.; Onoé, K. The murine c-fgr gene product associated with Ly6C and p70 integral membrane protein is expressed in cells of a monocyte/macrophage lineage. Proc. Natl. Acad. Sci. USA 1994, 91, 3458–3462. [Google Scholar] [CrossRef]
- Lauvau, G.; Chorro, L.; Spaulding, E.; Soudja, S.M. Inflammatory monocyte effector mechanisms. Cell. Immunol. 2014, 291, 32–40. [Google Scholar] [CrossRef]
- Fong, A.M.; Robinson, L.A.; Steeber, D.A.; Tedder, T.F.; Yoshie, O.; Imai, T.; Patel, D.D. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J. Exp. Med. 1998, 188, 1413–1419. [Google Scholar] [CrossRef]
- Tacke, F.; Alvarez, D.; Kaplan, T.J.; Jakubzick, C.; Spanbroek, R.; Llodra, J.; Garin, A.; Liu, J.; Mack, M.; van Rooijen, N.; et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Investig. 2007, 117, 185–194. [Google Scholar] [CrossRef]
- Auffray, C.; Fogg, D.; Garfa, M.; Elain, G.; Join-Lambert, O.; Kayal, S.; Sarnacki, S.; Cumano, A.; Lauvau, G.; Geissmann, F. Monitoring of blood vessels and tissues by a population of monocytes withpatrolling behavior. Science 2007, 317, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.; Tacke, R.; Hedrick, C.C.; Hanna, R.N. Nonclassical patrolling monocyte function in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Combadière, C.; Potteaux, S.; Rodero, M.; Simon, T.; Pezard, A.; Esposito, B.; Merval, R.; Proudfoot, A.; Tedgui, A.; Mallat, Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6Chi and Ly6Clo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 2008, 117, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Ilhan, F.; Kalkanli, S.T. Atherosclerosis and the role of immune cells. World J. Clin. Cases 2015, 3, 345–352. [Google Scholar] [CrossRef]
- Funk, J.L.; Feingold, K.R.; Moser, A.H.; Grunfeld, C. Lipopolysaccharide stimulation of RAW 264.7 macrophages induces lipid accumulation and foam cell formation. Atherosclerosis 1993, 98, 67–82. [Google Scholar] [CrossRef]
- Lee, J.G.; Lim, E.J.; Park, D.W.; Lee, S.H.; Kim, J.R.; Baek, S.H. A combination of Lox-1 and Nox1 regulates TLR9-mediated foam cell formation. Cell. Signal. 2008, 20, 2266–2275. [Google Scholar] [CrossRef]
- Higashimori, M.; Tatro, J.B.; Moore, K.J.; Mendelsohn, M.E.; Galper, J.B.; Beasley, D. Role of toll-like receptor 4 in intimal foam cell accumulation in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 50–57. [Google Scholar] [CrossRef]
- Cole, J.E.; Georgiou, E.; Monaco, C. The expression and functions of toll-like receptors in atherosclerosis. Mediat. Inflamm. 2010, 2010, 393946. [Google Scholar] [CrossRef]
- Seneviratne, A.N.; Sivagurunathan, B.; Monaco, C. Toll-like receptors and macrophage activation in atherosclerosis. Clin. Chim. Acta 2012, 413, 3–14. [Google Scholar] [CrossRef]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef]
- Moore, K.J.; Kunjathoor, V.V.; Koehn, S.L.; Manning, J.J.; Tseng, A.A.; Silver, J.M.; McKee, M.; Freeman, M.W. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J. Clin. Investig. 2005, 115, 2192–2201. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, S.; Vanegas, D.; Kennedy, D.J.; Guy, E.; Nimako, G.; Morton, R.E.; Febbraio, M. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc. Res. 2008, 78, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Kume, N.; Miyamoto, S.; Minami, M.; Moriwaki, H.; Murase, T.; Sawamura, T.; Masaki, T.; Hashimoto, N.; Kita, T. Expression of lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation 1999, 99, 3110–3117. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kondratenko, N.; Green, S.; Steinberg, D.; Quehenberger, O. Identification of the lectin-like receptor for oxidized low-density lipoprotein in human macrophages and its potential role as a scavenger receptor. Biochem. J. 1998, 334, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Kume, N.; Kita, T. Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) in atherogenesis. Trends Cardiovasc. Med. 2001, 11, 22–25. [Google Scholar] [CrossRef]
- Schaeffer, D.F.; Riazy, M.; Parhar, K.S.; Chen, J.H.; Duronio, V.; Sawamura, T.; Steinbrecher, U.P. LOX-1 augments oxLDL uptake by lysoPC-stimulated murine macrophages but is not required for oxLDL clearance from plasma. J. Lipid. Res. 2009, 50, 1676–1684. [Google Scholar] [CrossRef]
- Inoue, K.; Arai, Y.; Kurihara, H.; Kita, T.; Sawamura, T. Overexpression of lectin-like oxidized low-density lipoprotein receptor-1 induces intramyocardial vasculopathy in apolipoprotein E-null mice. Circ. Res. 2005, 97, 176–184. [Google Scholar] [CrossRef]
- Mehta, J.L.; Sanada, N.; Hu, C.P.; Chen, J.; Dandapat, A.; Sugawara, F.; Satoh, H.; Inoue, K.; Kawase, Y.; Jishage, K.; et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ. Res. 2007, 100, 1634–1642. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Bobryshev, Y.V.; Nikiforov, N.G.; Elizova, N.V.; Sobenin, I.A.; Orekhov, A.N. Macrophage phenotypic plasticity in atherosclerosis: The associated features and the peculiarities of the expression of inflammatory genes. Int. J. Cardiol. 2015, 184, 436–445. [Google Scholar] [CrossRef]
- Taghavie-Moghadam, P.L.; Butcher, M.J.; Galkina, E.V. The dynamic lives of macrophage and dendritic cell subsets in atherosclerosis. Ann. N. Y. Acad. Sci. 2014, 1319, 19–37. [Google Scholar] [CrossRef]
- Zhang, S.; Kim, C.C.; Batra, S.; McKerrow, J.H.; Loke, P. Delineation of diverse macrophage activation programs in response to intracellular parasites and cytokines. PLoS Negl. Trop. Dis. 2010, 4, e648. [Google Scholar] [CrossRef] [PubMed]
- Lacavé-Lapalun, J.V.; Benderitter, M.; Linard, C. Flagellin or lipopolysaccharide treatment modified macrophage populations after colorectal radiation of rats. J. Pharmacol. Exp. Ther. 2013, 346, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef]
- Pinhal-Enfield, G.; Ramanathan, M.; Hasko, G.; Vogel, S.N.; Salzman, A.L.; Boons, G.J.; Leibovich, S.J. An angiogenic switch in macrophages involving synergy between Toll-like receptors 2, 4, 7, and 9 and adenosine A(2A) receptors. Am. J. Pathol. 2003, 163, 711–721. [Google Scholar] [CrossRef]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovitch, S.J. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin- 4 receptor alpha (IL-4Ralpha) signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef]
- Gleissner, C.A.; Shaked, I.; Little, K.M.; Ley, K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J. Immunol. 2010, 184, 4810–4818. [Google Scholar] [CrossRef]
- Erbel, C.; Tyka, M.; Helmes, C.M.; Akhavanpoor, M.; Rupp, G.; Domschke, G.; Linden, F.; Wolf, A.; Doesch, A.; Lasitschka, F.; et al. CXCL4-induced plaque macrophages can be specifically identified by co-expression of MMP7+S100A8+ in vitro and in vivo. Innate Immun. 2015, 21, 255–265. [Google Scholar] [CrossRef]
- Gleissner, C.A. Macrophage phenotype modulation by CXCL4 in atherosclerosis. Front. Physiol. 2012, 3, 1. [Google Scholar] [CrossRef]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.J.; Harrington, H.A.; Piper, E.; Elderfield, K.; Stark, J.; Landis, R.C.; Haskard, D.O. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am. J. Pathol. 2009, 174, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Finn, A.V.; Nakano, M.; Polavarapu, R.; Karmali, V.; Saeed, O.; Zhao, X.; Yazdani, S.; Otsuka, F.; Davis, T.; Habib, A.; et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J. Am. Coll. Cardiol. 2012, 59, 166–177. [Google Scholar] [CrossRef]
- Boyle, J.J.; Johns, M.; Kampfer, T.; Nguyen, A.T.; Game, L.; Schaer, D.J.; Mason, J.C.; Haskard, D.O. Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ. Res. 2012, 110, 20–33. [Google Scholar] [CrossRef]
- Philippidis, P.; Mason, J.C.; Evans, B.J.; Nadra, I.; Taylor, K.M.; Haskard, D.O.; Landis, R.C. Hemoglobin scavenger receptor CD163 mediates interleukin-10 release and heme oxygenase-1 synthesis: Antiinflammatory monocyte-macrophage responses in vitro, in resolving skin blisters in vivo, and after cardiopulmonary bypass surgery. Circ. Res. 2004, 94, 119–126. [Google Scholar] [CrossRef]
- Groh, L.; Keating, S.T.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P. Monocyte and macropjhage immunometabolism in atherosclerosis. Semin. Immunopathol. 2018, 40, 203–214. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signaling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Bonacina, F.; Guinamard, R.R.; Norata, G.D. Immunometabolic function of cholesterol in cardiovascular disease and beyond. Cardiovasc. Res. 2019, 115, 1393–1407. [Google Scholar] [CrossRef]
- Van Tuijl, J.; Joosten, L.A.B.; Netea, M.G.; Bekkering, S.; Riksen, N.P. Immunometabolism orchestrates training of innate immunity in atherosclerosis. Cardiovasc. Res. 2019, 115, 1416–1424. [Google Scholar] [CrossRef]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Lord, R.S. Ultrastructural recognition of cells with dendritic cell morphology in human aortic intima. Contacting interactions of Vascular Dendritic Cells in athero-resistant and athero-prone areas of the normal aorta. Arch. Histol. Cytol. 1995, 58, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Wigren, M.; Nilsson, J.; Kolbus, D. Lymphocytes in atherosclerosis. Clin. Chim. Acta 2012, 413, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Myeloid dendritic cells: Development, functions, and role in atherosclerotic inflammation. Immunobiology 2015, 220, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, N.; Ho, S.; Antonenko, S.; Malefyt, R.W.; Kastelein, R.A.; Bazan, F.; Liu, Y.J. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 2001, 194, 863–869. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Sobenin, I.A.; Bobryshev, Y.V. Plasmacytoid dendritic cells: Development, functions, and role in atherosclerotic inflammation. Front. Physiol. 2014, 5, 279. [Google Scholar] [CrossRef] [PubMed]
- Penna, G.; Vulcano, M.; Sozzani, S.; Adorini, L. Differential migration behavior and chemokine production by myeloid and plasmacytoid dendritic cells. Hum. Immunol. 2002, 63, 1164–1171. [Google Scholar] [CrossRef]
- Dzionek, A.; Fuchs, A.; Schmidt, P.; Cremer, S.; Zysk, M.; Miltenyi, S.; Buck, D.W.; Schmitz, J. BDCA-2, BDCA-3, and BDCA-4: Three markers for distinct subsets of dendritic cells in human peripheral blood. J. Immunol. 2000, 165, 6037–6046. [Google Scholar] [CrossRef]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+(BDCA-3) + dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef]
- Legein, B.; Temmerman, L.; Biessen, E.A.; Lutgens, E. Inflammation and immune system interactions in atherosclerosis. Cell. Mol. Life Sci. 2013, 70, 3847–3869. [Google Scholar] [CrossRef]
- Dopheide, J.F.; Sester, U.; Schlitt, A.; Horstick, G.; Rupprecht, H.J.; Munzel, T.; Blankenberg, S. Monocyte-derived dendritic cells of patients with coronary artery disease show an increased expression of costimulatory molecules CD40, CD80 and CD86 in vitro. Coron. Artery Dis. 2007, 18, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, D.; Yang, K.; Hu, Y.; Zeng, Q. Toll-like receptor-4 and mitogen-activated protein kinase signal system are involved in activation of dendritic cells in patients with acute coronary syndrome. Immunology 2008, 125, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Ghattas, A.; Griffiths, H.R.; Devitt, A.; Lip, G.Y.; Shantsila, E. Monocytes in coronary artery disease and atherosclerosis: Where are we now? J. Am. Coll. Cardiol. 2013, 62, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Sage, A.P.; Mallat, Z.; Tedgui, A. Adaptive (T and B cells) immunity and control by dendritic cells in atherosclerosis. Circ. Res. 2014, 114, 1640–1660. [Google Scholar] [CrossRef]
- Döring, Y.; Manthey, H.D.; Drechsler, M.; Lievens, D.; Megens, R.T.; Soehnlein, O.; Busch, M.; Manca, M.; Koenen, R.R.; Pelisek, J.; et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 2012, 125, 1673–1683. [Google Scholar] [CrossRef]
- Niessner, A.; Sato, K.; Chaikof, E.L.; Colmegna, I.; Goronzy, J.J.; Weyand, C.M. Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-alpha. Circulation 2006, 114, 2482–2489. [Google Scholar] [CrossRef]
- Anand, P.K.; Malireddi, R.K.; Kanneganti, T.D. Role of the nlrp3 inflammasome in microbial infection. Front. Microbiol. 2011, 2, 12. [Google Scholar] [CrossRef]
- Sozzani, S.; Vermi, W.; Del Prete, A.; Facchetti, F. Trafficking properties of plasmacytoid dendritic cells in health and disease. Trends Immunol. 2010, 31, 270–277. [Google Scholar] [CrossRef]
- Butcher, M.J.; Galkina, E.V. Phenotypic and functional heterogeneity of macrophages and dendritic cell subsets in the healthy and atherosclerosis-prone aorta. Front. Physiol. 2012, 3, 44. [Google Scholar] [CrossRef]
- Bobryshev, Y.V.; Lord, R.S.A. Mapping of vascular dendritic cells in atherosclerotic arteries suggests their involvement in local immune inflammatory reaction. Cardiovasc. Res. 1998, 37, 799–810. [Google Scholar] [CrossRef]
- Koltsova, E.K.; Garcia, Z.; Chodaczek, G.; Landau, M.; McArdle, S.; Scott, S.R.; von Vietinghoff, S.; Galkina, E.; Miller, Y.I.; Acton, S.T.; et al. Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis. J. Clin. Investig. 2012, 122, 3114–3126. [Google Scholar] [CrossRef] [PubMed]
- Perrin-Cocon, L.; Coutant, F.; Agaugué, S.; Deforges, S.; André, P.; Lotteau, V. Oxidized low-density lipoprotein promotes mature dendritic cell transition from differentiating monocyte. J. Immunol. 2001, 167, 3785–3791. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.E.; Zhu, S.N.; Chen, M.; Nurmohamed, S.; Jongstra-Bilen, J.; Cybulsky, M.I. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ. Res. 2010, 106, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Meiler, S.; Döring, Y.; Koch, M.; Drechsler, M.; Megens, R.T.; Rowinska, Z.; Bidzhekov, K.; Fecher, C.; Ribechini, E.; et al. CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J. Clin. Investig. 2011, 121, 2898–2910. [Google Scholar] [CrossRef]
- Maldonado, R.A.; von Andrian, U.H. How tolerogenic dendritic cells induce regulatory T cells. Adv. Immunol. 2010, 108, 111–165. [Google Scholar]
- Choi, J.H.; Cheong, C.; Dandamudi, D.B.; Park, C.G.; Rodriguez, A.; Mehandry, S.; Velinzon, K.; Jung, I.H.; Yoo, J.Y.; Oh, G.T.; et al. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity 2011, 35, 819–831. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N. Regulatory T cells in atherosclerosis and strategies to induce the endogenous atheroprotective immune response. Immunol Lett. 2013, 151, 10–22. [Google Scholar] [CrossRef]
- Lievens, D.; Habets, K.L.; Robertson, A.K.; Laouar, Y.; Winkels, H.; Rademakers, T.; Beckers, L.; Wijnands, E.; Boon, L.; Mosaheb, M.; et al. Abrogated transforming growth factor beta receptor II (TGFβRII) signalling in dendritic cells promotes immune reactivity of T cells resulting in enhanced atherosclerosis. Eur. Heart J. 2013, 34, 3717–3727. [Google Scholar] [CrossRef]
- Fife, B.T.; Pauken, K.E.; Eagar, T.N.; Obu, T.; Wu, J.; Tang, Q.; Azuma, M.; Krummel, M.F.; Bluestone, J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 2009, 10, 1185–1192. [Google Scholar] [CrossRef]
- Heitger, A. Regulation of expression and function of IDO in human dendritic cells. Curr. Med. Chem. 2011, 18, 2222–2233. [Google Scholar] [CrossRef]
- Fallarino, F.; Orabona, C.; Vacca, C.; Bianchi, R.; Gizzi, S.; Asselin-Paturel, C.; Fioretti, M.C.; Trincheri, G.; Grohmann, U.; Puccetti, P. Ligand and cytokine dependence of the immunosuppressive pathway of tryptophan catabolism in plasmacytoid dendritic cells. Int. Immunol. 2005, 17, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Daissormont, I.T.; Christ, A.; Temmerman, L.; Sampedro Millares, S.; Seijkens, T.; Manca, M.; Rousch, M.; Poggi, M.; Boon, L.; van der Loos, C.; et al. Plasmacytoid dendritic cells protect against atherosclerosis by tuning T-cell proliferation and activity. Circ. Res. 2011, 109, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikova, O.A.; Berge, N.; Kang, C.; Urien, C.; Ketelhuth, D.F.; Pottier, J.; Drouet, L.; Hansson, G.K.; Marchal, G.; Back, M.; et al. Mycobacterium bovis BCG killed by extended freeze-drying induces an immunoregulatory profile and protects against atherosclerosis. J. Intern. Med. 2014, 275, 49–58. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chistiakov, D.A.; Kashirskikh, D.A.; Khotina, V.A.; Grechko, A.V.; Orekhov, A.N. Immune-Inflammatory Responses in Atherosclerosis: The Role of Myeloid Cells. J. Clin. Med. 2019, 8, 1798. https://doi.org/10.3390/jcm8111798
Chistiakov DA, Kashirskikh DA, Khotina VA, Grechko AV, Orekhov AN. Immune-Inflammatory Responses in Atherosclerosis: The Role of Myeloid Cells. Journal of Clinical Medicine. 2019; 8(11):1798. https://doi.org/10.3390/jcm8111798
Chicago/Turabian StyleChistiakov, Dimitry A., Dmitry A. Kashirskikh, Victoriya A. Khotina, Andrey V. Grechko, and Alexander N. Orekhov. 2019. "Immune-Inflammatory Responses in Atherosclerosis: The Role of Myeloid Cells" Journal of Clinical Medicine 8, no. 11: 1798. https://doi.org/10.3390/jcm8111798
APA StyleChistiakov, D. A., Kashirskikh, D. A., Khotina, V. A., Grechko, A. V., & Orekhov, A. N. (2019). Immune-Inflammatory Responses in Atherosclerosis: The Role of Myeloid Cells. Journal of Clinical Medicine, 8(11), 1798. https://doi.org/10.3390/jcm8111798