Oral Anticoagulant Therapy—When Art Meets Science

 , ,
, ,  ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
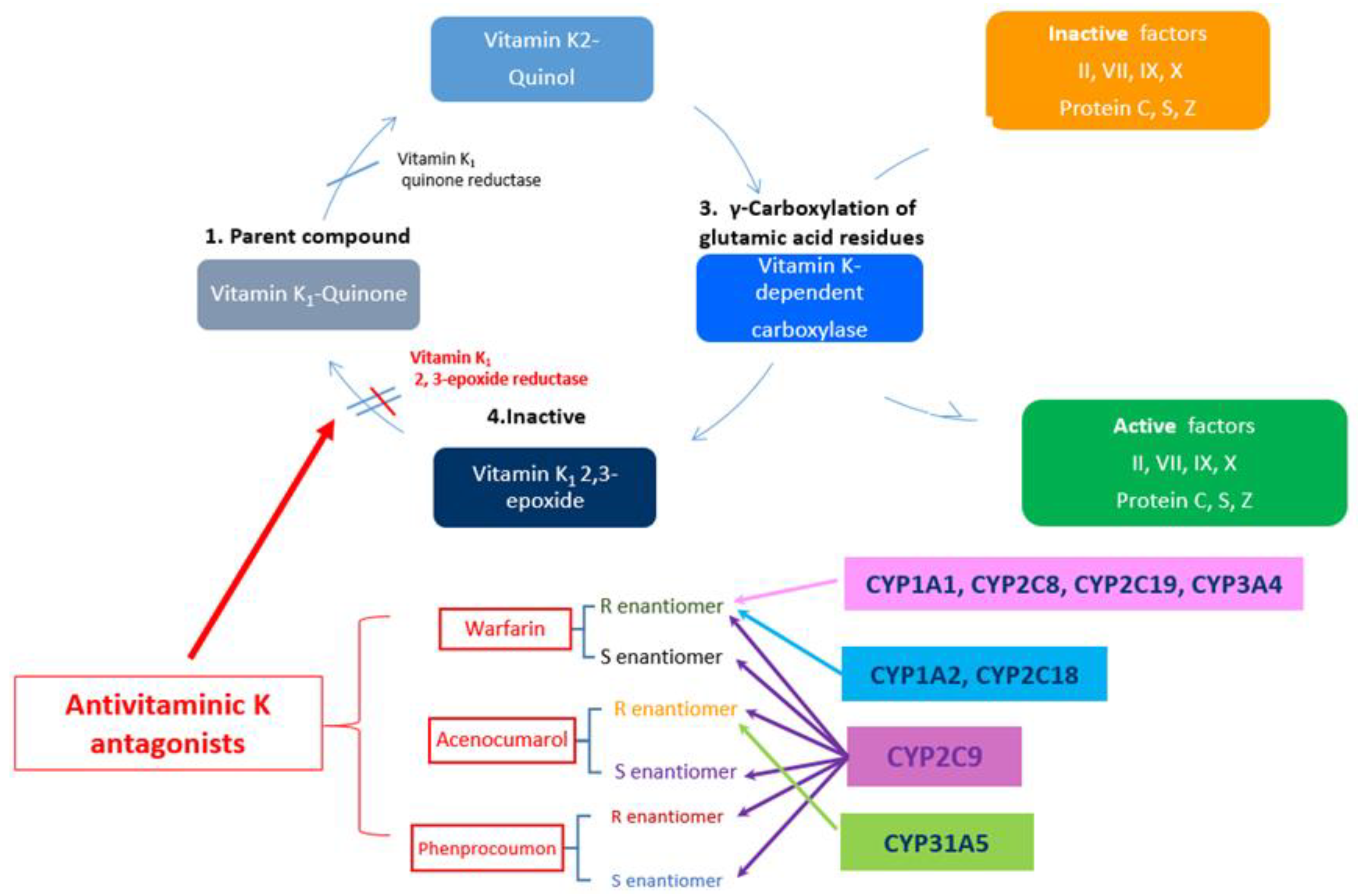
2. General Consideration on VKA
3. Genetic Determinism of the Response to VKA
3.1. What Does CYP2C9 Means?
3.2. What Does VKORC1 Mean?
4. Practical Issues to Be Solved
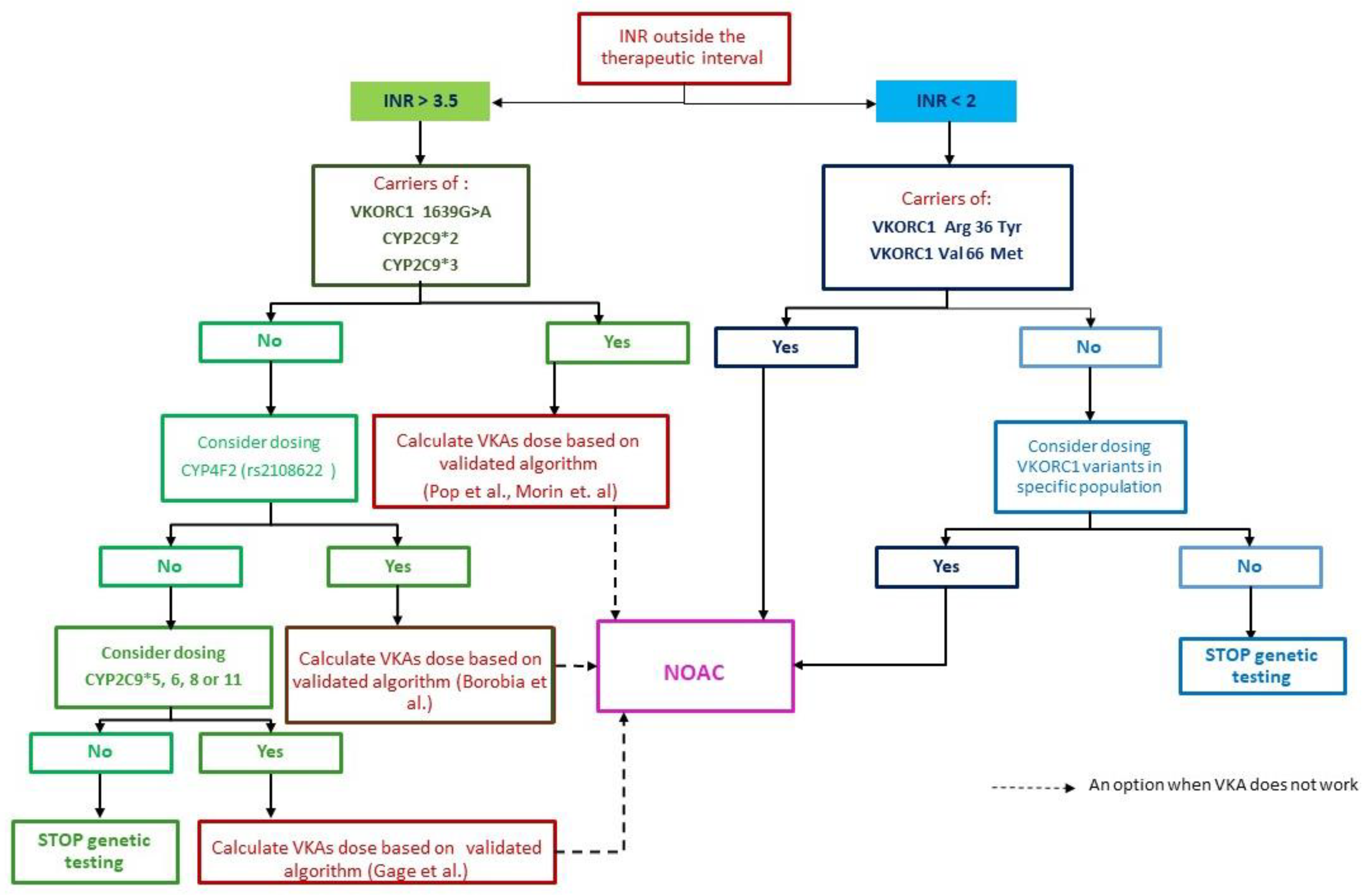
4.1. How to Diagnose and Treat Patient in the Presence of the CYP2C9*2 Mutation (Heterozygote) and the VKORC C1173T Variant
4.2. Genetic Testing—How Should We Do It, How Far Should We Go?
5. NOAC the Practical Solution
6. Conclusions
Supplementary Materials
Supplementary File 1Author Contributions
Acknowledgments
Conflicts of Interest
References
- Stehle, S.; Kirchheiner, J.; Lazar, A.; Fuhr, U. Pharmacogenetics of oral anticoagulants: A basis for dose individualization. Clin. Pharmacokinet. 2008, 47, 565–594. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; McEwan, P.; Morgan, C.L.; Peters, J.R.; Goodfellow, J.; Currie, C.J. Evaluation of the pattern of treatment, level of anticoagulation control, and outcome of treatment with warfarin in patients with non-valvar atrial fibrillation: A record linkage study in a large British population. Heart 2005, 91, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Markatos, C.N.; Grouzi, E.; Politou, M.; Gialeraki, A.; Merkouri, E.; Panagou, I.; Spiliotopoulou, I.; Travlou, A. VKORC1 and CYP2C9 allelic variants influence acenocoumarol dose requirements in Greek patients. Pharmacogenomics 2008, 9, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Steffel, J.; Verhamme, P.; Potpara, T.S.; Albaladejo, P.; Antz, M.; Desteghe, L.; Haeusler, K.G.; Oldgren, J.; Reinecke, H.; Roldan-Schilling, V.; et al. The 2018 European Heart Rhythm Association Practical Guide on the use ofnon-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation. Eur. Heart. J. 2018, 60, 1–64. [Google Scholar]
- Hansson, N.C.; Grove, E.L.; Andersen, H.R.; Leipsic, J.; Mathiassen, O.N.; Jensen, J.M.; Jensen, K.T.; Blanke, P.; Leetmaa, T.; Tang, M.; et al. Transcatheter aortic valve thrombosis: Incidence, predisposing factors, and clinical implications. J. Am. Coll. Cardiol. 2016, 68, 2059–2069. [Google Scholar] [CrossRef]
- Dangas, G.D.; Weitz, J.I.; Giustino, G.; Makkar, R.; Mehran, R. Prosthetic heart valve thrombosis. J. Am. Coll. Cardiol. 2016, 68, 2670–2689. [Google Scholar] [CrossRef]
- Sachdev, S.; Bardia, N.; Nguyen, L.; Omar, B. Bioprosthetic valve thrombosis. Cardio. Res. 2018, 9, 335–342. [Google Scholar] [CrossRef]
- Sorrentino, S.; Giustino, G.; Moalem, K.; Indolfi, C.; Mehran, R.; Dangas, G.D. Antithrombotic treatment after transcatheter heart valves implant. Semin. Thromb. Hemost. 2018, 44, 38–45. [Google Scholar] [CrossRef]
- Baumgartner, H.; Falk, V.; Bax, J.J.; De Bonis, M.; Hamm, C.; Holm, P.J.; Iung, B.; Lancellotti, P.; Lansac, E.; Munoz, D.R.; et al. 2017 ESC/EACTS guidelines for the management of valvular heart disease. The task force for the management of valvular heart disease of the European Society of Cardiology (ESC) and the European. Eur. Heart J. 2017, 38, 2739–2791. [Google Scholar] [CrossRef]
- Cohen, H.; Efthymiou, M.; Isenberg, D.A. Use of direct oral anticoagulants in antiphospholipid syndrome. J. Thromb. Haemost. 2018, 16, 1028–1039. [Google Scholar] [CrossRef]
- Garcia, D.; Erkan, D. Diagnosis and management of the antiphospholipid syndrome. N. Engl. J. Med. 2018, 378, 2010–2110. [Google Scholar] [CrossRef] [PubMed]
- Sciascia, S.; Lopez-Pedrera, C.; Cecchi, I.; Pecoraro, C.; Roccatello, D.; Cuadrado, M.J. Non-vitamin K antagonist oral anticoagulants and antiphospholipid syndrome. Rheumatology (Oxford) 2016, 55, 1726–1735. [Google Scholar] [CrossRef] [PubMed]
- Andrade, D.; Cervera, R.; Cohen, H.; Al, E. Antiphospholipid Syndrome: Current Research Highlights and Clinical Insights; Erkan, D., Lockshin, M.D., Eds.; Springer: New York, NY, USA, 2017. [Google Scholar]
- Elsebaie, M.A.T.; van Es, N.; Langston, A.; Büller, H.R.; Gaddh, M. Direct oral anticoagulants in patients with venous thromboembolism and thrombophilia: a systematic review and meta-analysis. J. Thromb. Haemost. 2019, 17, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Serrao, A.; Lucan, B.; Mansou, D.; Ferrett, A.; Baldacc, E.; Santor, C.; Foà, R.; Chistolini, A. Direct oral anticoagulants in patients affected by major congenital thrombophilia. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019044. [Google Scholar] [CrossRef]
- Shiga, T.; Naganuma, M.; Nagao, T.; Maruyam, K.; Suzuk, A.; Murasak, K.; Hagiwara, N. Persistence of non-vitamin K antagonist oral anticoagulant use in Japanese patients with atrial fibrillation: A single-center observational study. J. Arrhythm. 2015, 31, 339. [Google Scholar] [CrossRef]
- Pirmohamed, M. Warfarin: Almost 60 years old and still causing problems. Br. J. Clin. Pharmacol. 2006, 62, 509–511. [Google Scholar] [CrossRef]
- Last, J.A. The missing link: The story of Karl Paul Link. Toxicol. Sci. 2002, 66, 4–6. [Google Scholar] [CrossRef]
- Gómez-Outes, A.; Suárez-Gea, M.L.; Calvo-Rojas, G.; Lecumberri, R.; Rocha, E.; Pozo-Hernández, C.; Terleira-Fernández, A.I.; Vargas-Castrillón, E. Discovery of anticoagulant drugs: A historical perspective. Curr. Drug Discov. Technol. 2012, 9, 83–104. [Google Scholar] [CrossRef]
- Wardrop, D.; Keeling, D. The story of the discovery of heparin and warfarin. Br. J. Haematol. 2008, 141, 757–763. [Google Scholar] [CrossRef]
- Schalekamp, T.; de Boer, A. Pharmacogenetics of oral anticoagulant therapy. Curr. Pharm. Des. 2010, 16, 187–203. [Google Scholar] [CrossRef]
- Kaminsky, L.S.; Zhang, Z.Y. Human P450 metabolism of warfarin. Pharmacol. Ther. 1997, 73, 67–74. [Google Scholar] [CrossRef]
- Takahashi, H.; Echizen, H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin. Pharmacokinet. 2001, 40, 587–603. [Google Scholar] [CrossRef] [PubMed]
- Ufer, M. Comparative pharmacokinetics of vitamin K antagonists. Clin. Pharmacokinet. 2005, 44, 1227–1246. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, T.I.; Redekop, W.K.; Daly, A.K.; Schie, R.M.F.; de Boer, A.; van der Zee, A.M.H. Pharmacogenetic-guided dosing of coumarin anticoagulants: Algorithms for warfarin, acenocoumarol and phenprocoumon. Br. J. Clin. Pharmacol. 2014, 77, 626–641. [Google Scholar] [CrossRef] [PubMed]
- Mili, F.D.; Allen, T.; Wadell, P.W.; Hooper, W.C.; Staercke, D.; Bean, C.J.; Lally, C.; Austin, H.; Wenger, N.K. VKORC1-1639A allele influences warfarin maintenance dosage among Blacks receiving warfarin anticoagulation: A retrospective cohort study. Future Cardiol. 2018, 14, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Schalekamp, T.; Brasse, B.; Roijers, J.F.; Chahid, Y.; van Geest-Daalderop, J.H.; de Vries-Goldschmeding, H.; van Wijk, E.M.; Egberts, A.C.; de Boer, A. VKORC1 and CYP2C9 genotypes and acenocoumarol anticoagulation status: Interaction between both genotypes affects overanticoagulation. Clin. Pharmacol. Ther. 2006, 80, 13–22. [Google Scholar] [CrossRef]
- Jia, L.; Wang, Z.; Men, J.; Cai, H.; Wei, M. Polymorphisms of VKORC1 and CYP2C9 are associated with warfarin sensitivity in Chinese population. Ther. Clin. Risk Manag. 2017, 13, 421–425. [Google Scholar] [CrossRef]
- Daly, A.K.; King, B.P. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003, 13, 247–252. [Google Scholar] [CrossRef]
- Franchini, M.; Castaman, G.; Coppola, A.; Santoro, C.; Zanon, E.; Di Minno, G.; Morfini, M.; Santagostino, E.; Rocino, A. AICE Working Group. Acquired inhibitors of clotting factors: AICE recommendations for diagnosis and management. Blood Transfus. 2015, 13, 498–513. [Google Scholar]
- Paulus, E.; Komperda, K.; Park, G.; Fusco, J. Anticoagulation therapy considerations in factor VII deficiency. Drug Saf. Case Rep. 2016, 3, 8. [Google Scholar] [CrossRef]
- DeStefano, C.; Sylvester, K.; Connors, J.; Sheikh, F.; Catlett, J. Warfarin management in the setting of FVII deficiency and mechanical circulatory support. Vasc. Med. 2017, 22, 345–346. [Google Scholar] [CrossRef] [PubMed]
- Gulati, G.; Hevelow, M.; George, M.; Behling, E.; Siegel, J. International normalized ratio versus plasma levels of coagulation factors in patients on vitamin K antagonist therapy. Arch. Pathol. Lab. Med. 2011, 135, 490–494. [Google Scholar] [PubMed]
- Rettie, A.E.; Korzekwa, K.R.; Kunze, K.L.; Lawrence, R.F.; Eddy, A.C.; Aoyama, T.; Gelboin, H.V.; Gonzalez, F.J.; Trager, W.F. Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: A role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem. Res. Toxicol. 1992, 5, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chang, C.-Y.; Jin, D.-Y.; Lin, P.-J.; Khvorova, A.; Stafford, D.W. Identification of the gene for vitamin K epoxide reductase. Nature 2004, 427, 541–544. [Google Scholar] [CrossRef]
- Rost, S.; Fregin, A.; Ivaskevicius, V.; Conzelmann, E.; Hörtnagel, K.; Pelz, H.J.; Lappegard, K.; Seifried, E.; Scharrer, I.; Tuddenham, E.G.; et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004, 427, 537–541. [Google Scholar] [CrossRef]
- Geisen, C.; Spohn, G.; Sittinger, K.; Rost, S.; Watzka, M.; Lages, P.; Huth-Kühne, A.; Zimmermann, R.; Müller, C.R.; Seifried, E.; et al. A novel mutation (Asp36Tyr) in the Vitamin K Epoxide Reductase Complex Subunit 1 Gene (VKORC1) causes increased phenprocoumon requirement. In 35th Hemophilia Symposium; Hamburg, 2004; Scharrer, I., Schramm, W., Eds.; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Osinbowale, O.; Al Malki, M.; Schade, A.; Bartholomew, J.R. An algorithm for managing warfarin resistance. Cleve. Clin. J. Med. 2009, 76, 724–730. [Google Scholar] [CrossRef]
- Jiménez-Var, E.; Cañadas-Garre, M.; Aguilera, M.; Callejas, D.G.; Ramirez, C.P.; Hernández, M.A.C. Omics for Personalized Medicine; Springer: New Delhi, India, 2013; pp. 435–467. [Google Scholar]
- Lee, C.R.; Goldstein, J.A.; Pieper, J.A. Cytochrome P450 2C9 polymorphisms: A comprehensive review of the in-vitro and human data. Pharmacogenetics 2002, 12, 251–263. [Google Scholar] [CrossRef]
- Sanderson, S.; Emery, J.; Higgins, J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: A HuGEnet systematic review and meta-analysis. Genet. Med. 2005, 7, 97–104. [Google Scholar] [CrossRef]
- Buzoianu, A.D.; Trifa, A.P.; Muresanu, D.F.; Crisan, S. Analysis of CYP2C9*2, CYP2C9*3 and VKORC1-1639 G>A polymorphisms in a population from South-Eastern Europe. J. Cell. Mol. Med. 2012, 16, 2919–2924. [Google Scholar] [CrossRef]
- Scordo, M.; Pengo, V.; Spina, E.; Dahl, M.L.; Gusella, M.; Padrin, R. Influence of CYP2C9 and CYP2C19 please genetic polymorphisms on warfarin maintenance dose and metabolic clearance. Clin. Pharmacol. Ther. 2002, 72, 702–710. [Google Scholar] [CrossRef]
- Takahashi, H.; Kashima, T.; Nomoto, S.; Iwade, K.; Tainaka, H.; Shimizu, T.; Nomizo, Y.; Muramoto, N.; Kimura, S.; Echizen, H. Comparisons between in-vitro and in-vivo metabolism of (S)-warfarin: Catalytic activities of cDNA-expressed CYP2C9, its Leu359 variant and their mixture versus unbound clearance in patients with the corresponding CYP2C9 genotypes. Pharmacogenetics 1998, 8, 365–373. [Google Scholar] [CrossRef] [PubMed]
- LaSala, A.; Bowerm, B.; Windemuth, A.; White, C.M.; Kocherla, M.; Seip, R.; Duconge, J.; Ruaño, G. Integrating genomic based information into clinical Warfarin (Coumadin®) management: An illustrative case report. Conn. Med. 2008, 72, 399–403. [Google Scholar] [PubMed]
- Pop, T.R.; Vesa, Ş.C.; Trifa, A.P.; Crişan, S.; Buzoianu, A.D. An acenocoumarol dose algorithm based on a south-eastern European population. Eur. J. Clin. Pharmacol. 2013, 69, 1901–1907. [Google Scholar] [CrossRef] [PubMed]
- Van Schie, R.M.; el Khedr, N.; Verhoef, T.I.; Teichert, M.; Stricker, B.H.; Hofman, A.; Buhre, P.N.; Wessels, J.A.; Schalekamp, T.; le Cessie, S.; et al. Validation of the acenocoumarol EU-PACT algorithms: Similar performance in the Rotterdam Study cohort as in the original study. Pharmacogenomics 2012, 13, 1239–1245. [Google Scholar] [CrossRef]
- Morin, S.; Bodin, L.; Loriot, M.A.; Thijssen, H.H.W.; Robert, A.; Strabach, S.; Verstuyft, C.; Tregouet, D.-A.; Dubert, L.; Laurent-Puig, P.; et al. Pharmacogenetics of acenocoumarol pharmacodynamics. Clin. Pharmacol. Ther. 2004, 75, 403–414. [Google Scholar] [CrossRef]
- Borobia, A.M.; Lubomirov, R.; Ramírez, E.; Lorenzo, A.; Campos, A.; Muñoz-Romo, R.; Fernández-Capitán, C.; Frías, J.; Carcas, A.J. An Acenocoumarol dosing algorithm using clinical and pharmacogenetic data in Spanish patients with thromboembolic disease. PLoS ONE 2012, 7, e41360. [Google Scholar] [CrossRef]
- Sconce, E.A.; Kamali, F. Appraisal of current vitamin K dosing algorithms for the reversal of over-anticoagulation with warfarin: The need for a more tailored dosing regimen. Eur. J. Haematol. 2006, 77, 457–462. [Google Scholar] [CrossRef]
- Johnson, J.A.; Gong, L.; Whirl-Carrillo, M.; Gage, B.F.; Scott, S.A.; Stein, C.M.; Anderson, J.L.; Kimmel, S.E.; Lee, M.T.; Pirmohamed, M.; et al. Clinical pharmacogenetics implementation consortium guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin. Pharmacol. Ther. 2011, 90, 625–629. [Google Scholar] [CrossRef]
- Aithal, G.P.; Day, C.P.; Kesteven, P.J.; Daly, A.K. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet 1999, 353, 717–719. [Google Scholar] [CrossRef]
- Peyvandi, F.; Spreafico, M.; Siboni, S.M.; Moia, M.; Mannucci, P.M. CYP2C9 genotypes and dose requirements during the induction phase of oral anticoagulant therapy. Clin. Pharmacol. Ther. 2004, 75, 198–203. [Google Scholar] [CrossRef]
- Schalekamp, T.; Van Geest-Daalderop, J.H.H.; De Vries-Goldschmeding, H.; Conemans, J.; Bernesen, M.J.; De Boer, A. Acenocoumarol stabilization is delayed in CYP2C9*3 carriers. Clin. Pharmacol. Ther. 2004, 75, 294–402. [Google Scholar] [CrossRef] [PubMed]
- Visser, L.E.; van Vliet, M.; van Schaik, R.H.N.; Kasbergen, A.A.H.; De Smet, P.A.G.M.; Vulto, A.G.; Hofman, A.; van Duijn, C.M.; Stricker, B.H. The risk of overanticoagulation in patients with cytochrome P450 CYP2C9*2 or CYP2C9*3 alleles on acenocoumarol or phenprocoumon. Pharmacogenetics 2004, 14, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Tàssies, D.; Freire, C.; Pijoan, J.; Maragall, S.; Monteagudo, J.; Ordinas, A.; Reverter, J.C. Pharmacogenetics of Acenocoumarol: Cytochrome P450 CYP2C9 polymorphisms influence dose requirements and stability of anticoagulation. Haematologica 2002, 87, 1185–1191. [Google Scholar] [PubMed]
- Mark, L.; Marki-Zay, J.; Fodor, L.; Hajdara, I.; Paragh, G.; Katona, A.C. Cytochrome P450 2C9 polymorphism and acenocoumarol therapy. Kardiol. Pol. 2006, 64, 397–402. [Google Scholar] [PubMed]
- Hermida, J.; Zarza, J.; Alberca, I.; Montes, R.; López, M.L.; Molina, E.; Rocha, E. Differential effects of 2C9*3 and 2C9*2 variants of cytochrome P-450 CYP2C9 on sensitivity to acenocoumarol. Blood 2002, 99, 4237–4239. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, H.H.; Verkooijen, I.W.; Frank, H.L. The possession of the CYP2C9*3 allele is associated with low dose requirement of acenocoumarol. Pharmacogenetics 2000, 10, 757–760. [Google Scholar] [CrossRef]
- Chaidaroglou, A.; Kanellopoulou, T.; Panopoulos, G.; Stavridis, G.; Degiannis, D. Extremely low therapeutic doses of acenocoumarol in a patient with CYP2C9*3/*3 and VKORC1-1639A/A genotype. Pharmacogenomics 2019, 20, 311–317. [Google Scholar] [CrossRef]
- Cavallari, L.H.; Perera, M.A. The future of warfarin pharmacogenetics in under-represented minority groups. Future Cardiol. 2012, 8, 563–576. [Google Scholar] [CrossRef]
- Rieder, M.J.; Reiner, A.P.; Gage, B.F.; Nickerson, D.A.; Eby, C.S.; McLeod, H.L.; Blough, D.K.; Thummel, K.E.; Veenstra, D.L.; Rettie, A.E. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N. Engl. J. Med. 2005, 352, 2285–2293. [Google Scholar] [CrossRef]
- D’Andrea, G.; D’Ambrosio, R.L.; Di Perna, P.; Chetta, M.; Santacroce, R.; Brancaccio, V.; Grandone, E.; Margaglione, M. A polymorphism in the VKORC1 gene is associated with an interindividual variability in the dose-anticoagulant effect of warfarin. Blood 2005, 105, 645–649. [Google Scholar] [CrossRef]
- Shuen, A.Y.; Wong, B.Y.L.; Fu, L.; Selby, R.; Cole, D.E.C. Evaluation of the warfarin-resistance polymorphism, VKORC1 Asp36Tyr, and its effect on dosage algorithms in a genetically heterogeneous anticoagulant clinic. Clin. Biochem. 2012, 45, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Geisen, C.; Watzka, M.; Sittinger, K.; Steffens, M.; Daugela, L.; Seifried, E.; Müller, C.R.; Wienker, T.F.; Oldenburg, J. VKORC1 haplotypes and their impact on the inter-individual and inter-ethnical variability of oral anticoagulation. Thromb. Haemost. 2005, 94, 773–779. [Google Scholar] [PubMed]
- Oldenburg, J.; Bevans, C.; Fregin, A.; Geisen, C.; Müller-Reible, C.; Watzka, M. Current pharmacogenetic developments in oral anticoagulation therapy: The influence of variant VKORC1 and CYP2C9 alleles. Thromb. Haemost. 2007, 98, 570–578. [Google Scholar] [PubMed]
- Cooper, G.M.; Johnson, J.A.; Langaee, T.Y.; Feng, H.; Stanaway, I.B.; Schwarz, U.I.; Ritchie, M.D.; Stein, C.M.; Roden, D.M.; Smith, J.D.; et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood 2008, 112, 1022–1027. [Google Scholar] [CrossRef]
- Takeuchi, F.; McGinnis, R.; Bourgeois, S.; Barnes, C.; Eriksson, N.; Soranzo, N.; Whittaker, P.; Ranganath, V.; Kumanduri, V.; McLaren, W.; et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 2009, 5, e1000433. [Google Scholar] [CrossRef]
- Limdi, N.A.; Wadelius, M.; Cavallari, L.; Eriksson, N.; Crawford, D.C.; Lee, M.T.; Chen, C.H.; Motsinger-Reif, A.; Sagreiya, H.; Liu, N.; et al. Warfarin pharmacogenetics: A single VKORC1 polymorphism is predictive of dose across 3 racial groups. Blood 2010, 115, 3827–3834. [Google Scholar] [CrossRef]
- Limdi, N.; McGwin, G.; Goldstein, J.; Beasley, T.; Arnett, D.; Adler, B.; Baird, M.F.; Acton, R.T. Influence of CYP2C9 and VKORC1 1173C/T genotype on the risk of hemorrhagic complications in African-American and European-American patients on Warfarin. Clin. Pharmacol. Ther. 2008, 83, 312–321. [Google Scholar] [CrossRef]
- Van Schie, R.M.; Wessels, J.A.; le Cessie, S.; de Boer, A.; Schalekamp, T.; van der Meer, F.J.; Verhoef, T.I.; van Meegen, E.; Rosendaal, F.R.; Maitland-van der Zee, A.H.; et al. Loading and maintenance dose algorithms for phenprocoumon and acenocoumarol using patient characteristics and pharmacogenetic data. Eur. Heart J. 2011, 32, 1909–1917. [Google Scholar] [CrossRef]
- Wadelius, M.; Chen, L.Y.; Lindh, J.D.; Eriksson, N.; Ghori, M.J.R.; Bumpstead, S.; Holm, L.; McGinnis, R.; Rane, A.; Deloukas, P. The largest prospective warfarin-treated cohort supports genetic forecasting. Blood 2009, 113, 784–792. [Google Scholar] [CrossRef]
- Ragia, G.; Kolovou, V.; Kolovou, G.; Konstantinides, S.; Maltezos, E.; Tavridou, A.; Tziakas, D.; van der Maitland-Zee, A.H.; Manolopoulos, V.G. A novel acenocoumarol pharmacogenomic dosing algorithm for the Greek population of EU-PACT trial. Pharmacogenomics 2017, 18, 23–34. [Google Scholar] [CrossRef]
- Anderson, J.L.; Horne, B.D.; Stevens, S.M.; Grove, A.S.; Barton, S.; Nicholas, Z.P.; Kahn, S.F.; May, H.T.; Samuelson, K.M.; Muhlestein, J.B.; et al. Randomized trial of genotype-guied versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation 2007, 116, 2563–2570. [Google Scholar] [CrossRef] [PubMed]
- Horne, B.; Lenzini, P.; Wadelius, M.; Jorgensen, A.; Kimmel, S.; Ridker, P.; Eriksson, N.; Anderson, J.; Pirmohamed, M.; Limdi, N.; et al. Pharmacogenetic warfarin dose refinements remain significantly influenced by genetic factors after one week of therapy. Thromb. Haemost. 2012, 107, 232–240. [Google Scholar] [PubMed]
- Cho, S.M.; Lee, K.Y.; Choi, J.R.; Lee, K.A. Development and comparison of Warfarin dosing algorithms in stroke patients. Yonsei Med. J. 2016, 57, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Ye, F.; Xie, D.; Zhu, Y.; Zhu, J.; Tao, Y.; Feng, Y. A new algorithm to predict warfarin dose from polymorphisms of CYP4F2, CYP2C9 and VKORC1 and clinical variables: Derivation in Han Chinese patients with non valvular atrial fibrillation. Thromb. Haemost. 2012, 107, 1083–1091. [Google Scholar] [PubMed]
- Cho, H.J.; On, Y.K.; Bang, O.Y.; Kim, J.W.; Huh, W.; Ko, J.W.; Kim, J.S.; Le, S.Y. Development and comparison of a Warfarin-dosing algorithm for Korean patients with atrial fibrillation. Clin. Ther. 2011, 33, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Bernaitis, N.; Clark, G.; Kohja, S.; Leong, S.; Anoopkumar-Dukie, S. The SAMe-TT2R2 score predicts Warfarin control in an Australian population with atrial fibrillation. J. Clin. Med. 2019, 8, 882. [Google Scholar] [CrossRef]
- Rathore, S.S.; Agarwal, S.K.; Pande, S.; Singh, S.K.; Mittal, T.; Mittal, B. Therapeutic dosing of Acenocoumarol: Proposal of a population specific pharmacogenetic dosing algorithm and its validation in north Indians. PLoS ONE 2012, 7, 3–10. [Google Scholar] [CrossRef]
- Bodin, L.; Tregouet, D.; Robert, A.; Dubert, L.; Funck-brentano, C.; Jaillon, P.; Beaune, P.; Laurent-puig, P.; Becquemont, L.; Loriot, M. Genotypes as determinants of Acenocoumarol sensitivity. Pharmacogenetics 2005, 106, 135–140. [Google Scholar]
- Stack, G. Education Committee of the Academy of Clinical Laboratory Physicians and Scientists. Pathology consultation on Warfarin pharmacogenetic testing. Am. J. Clin. Pathol. 2011, 135, 13–19. [Google Scholar] [CrossRef]
- Kawai, V.K.; Cunningham, A.; Vear, S.I.; Van Driest, S.L.; Oginni, A.; Xu, H.; Min, J.; Chun, L.; Denny, J.C.; Shaffere, C.; et al. Genotype and risk of major bleeding during warfarin treatment. Pharmacogenomics 2014, 15, 1973–1983. [Google Scholar] [CrossRef]
- Lenzini, P.; Wadelius, M.; Kimmel, S.; Anderson, J.L.; Jorgensen, A.L.; Pirmohamed, M.; Caldwell, N.; Limdi, J.K.; Burmester, M.B.; Dowd, P.; et al. Integration of genetic, clinical, and INR data to refine Warfarin dosing. Clin. Pharmacol. Ther. 2010, 87, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Horne, B.; Stevens, S.; Woller, S.; Samuelson, K.; Mansfield, J.W.; Robinson, S.; Barton, K.; Brunisholz, C.P.; Mower, J.A.; et al. A randomized and clinical effectiveness trial comparing two pharmacogenetic algorithms and standard care for individualizing warfarin dosing (CoumaGen-II). Circulation 2012, 125, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, S.; French, B.; Kasner, S.; Johnson, J.; Anderson, J.; Gage, B.; Rosenberg, Y.D.; Eby, C.S.; Madigan, R.A.; McBane, R.B.; et al. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N. Engl. J. Med. 2013, 36, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Konstantinides, S.; Torbicki, A.; Agnelli, G.; Danchin, N.; Fitzmaurice, D.; Galiè, N.; Gibbs, J.S.; Huisman, M.V.; Humbert, M.; Kucher, N.; et al. 2014 ESC guidelines on the diagnosis and management of acute pulmonary embolism: The task force for the diagnosis and management of acute pulmonary embolism of the European Society of Cardiology (ESC) endorsed by the European Respiratory Society (ERS). Eur. Heart J. 2014, 35, 3033–3069. [Google Scholar]
- Kheiri, B.; Abdalla, A.; Haykal, T.; Osman, M.; Ahmed, S.; Hassan, M.; Bachuwa, G. Meta-analysis of genotype-guided versus standard dosing of vitamin K antagonists. Am. J. Cardiol. 2018, 121, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, N.; Wadelius, M. Prediction of warfarin dose: Why, when and how? Pharmacogenomics 2012, 13, 429–440. [Google Scholar] [CrossRef]
- Reitsma, P.H.; van der Heijden, J.F.; Groot, A.P.; Rosendaal, F.R.; Büller, H.R. A C1173T dimorphism in the VKORC1 gene determines coumarin sensitivity and bleeding risk. PLoS Med. 2005, 2, e312. [Google Scholar] [CrossRef]
- Schwarz, U.I.; Ritchie, M.D.; Bradford, Y.; Li, C.; Dudek, S.M.; Frye-Anderson, A.; Kim, R.B.; Roden, D.M.; Stein, C.M. Genetic determinants of response to warfarin during initial anticoagulation. N. Engl. J. Med. 2008, 358, 999–1008. [Google Scholar] [CrossRef]
- Ferder, N.S.; Eby, C.S.; Deych, E.; Li, C.; Dudek, S.M.; Frye-Anderson, A.; Kim, R.B.; Roden, M.D.; Michael Stein, C. Ability of VKORC1 and CYP2C9 to predict therapeutic warfarin dose during the initial weeks of therapy. J. Thromb. Haemost. 2010, 8, 95–100. [Google Scholar] [CrossRef]
- Meckley, L.M.; Wittkowsky, A.K.; Rieder, M.J.; Rettie, A.E.; Veenstra, D.L. An analysis of the relative effects of VKORC1 and CYP2C9 variants on anticoagulation related outcomes in warfarin-treated patients. Thromb. Haemost. 2008, 100, 229–239. [Google Scholar] [CrossRef]
- Marusic, S.; Gojo-Tomic, N.; Franic, M.; Bozina, N. Therapeutic efficacy of acenocoumarol in a warfarin-resistant patient with deep venous thrombosis: A case report. Eur. J. Clin. Pharmacol. 2009, 65, 1265–1266. [Google Scholar] [CrossRef] [PubMed]
- Loebstein, R.; Dvoskin, I.; Halkin, H.; Vecsler, M.; Lubetsky, A.; Rechavi, G.; Amariglio, N.; Cohen, Y.; Ken-Dror, G.; Almog, S.; et al. A coding VKORC1 Asp36Tyr polymorphism predisposes to warfarin resistance. Blood 2007, 109, 2477–2480. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.A.; Edelmann, L.; Kornreich, R.; Desnick, R.J. Warfarin pharmacogenetics: CYP2C9 and VKORC1 genotypes predict different sensitivity and resistance frequencies in the Ashkenazi and Sephardi Jewish populations. Am. J. Hum. Genet. 2008, 82, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Anton, A.I.; Cerezo-Manchado, J.J.; Padilla, J.; Perez-Andreu, V.; Corral, J.; Vicente, V.; Roldan, V.; Gonzalez-conejero, R. Novel associations of VKORC1 variants with higher acenocoumarol requirements. PLoS ONE 2013, 8, 1–7. [Google Scholar] [CrossRef]
- Dean, L. Warfarin therapy and VKORC1 and CYP genotype. In Medical Genetics Summaries; Pratt, V., McLeod, H., Rubinstein, W., Dean, L., Kattman, B., Malheiro, A., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Gage, B.F.; Eby, C.; Johnson, J.; Deych, E.; Rieder, M.; Ridker, P.; Milligan, P.E.; Grice, G.; Lenzini, P.; Rettie, A.E.; et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of Warfarin. Clin. Pharmacol. Ther. 2008, 84, 326–331. [Google Scholar] [CrossRef]
- Ruff, C.T.; Giugliano, R.P.; Braunwald, E.; Hoffman, E.B.; Deenadayalu, N.; Ezekowitz, M.D.; Camm, A.J.; Weitz, J.I.; Lewis, B.S.; Parkhomenkoe, A.; et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: A meta-analysis of randomised trials. Lancet 2014, 383, 955–962. [Google Scholar] [CrossRef]
- Ntaios, G.; Papavasileiou, V.; Makaritsis, K.; Vemmos, K.; Michel, P.; Lip, G.Y.H. Real-world setting comparison of nonvitamin-K antagonist oral anticoagulants versus vitamin-K antagonists for stroke prevention in atrial fibrillation. Stroke 2017, 48, 2494–2503. [Google Scholar] [CrossRef]
- Hohnloser, S.; Basic, E.; Hohmann, C.; Nabauer, M. Effectiveness and safety of non–vitamin K oral anticoagulants in comparison to Phenprocoumon: Data from 61,000 patients with atrial fibrillation. Thromb. Haemost. 2018, 118, 526–538. [Google Scholar] [CrossRef]
- Joppa, S.; Salciccioli, J.; Adamski, J.; Patel, S.; Wysokinski, W.; McBane, R.; Al-Saffar, F.; Esser, H.; Shamoun, F. A practical review of the emerging direct anticoagulants, laboratory monitoring, and reversal agents. J. Clin. Med. 2018, 7, 29. [Google Scholar] [CrossRef]
- Ghanny, S.; Crowther, M. Treatment with novel oral anticoagulants. Curr. Opin. Hematol. 2013, 20, 430–436. [Google Scholar] [CrossRef]
- Paré, G.; Eriksson, N.; Lehr, T.; Connolly, S.; Eikelboom, J.; Ezekowitz, M.D.; Axelsson, T.; Haertter, S.; Oldgren, J.; Reilly, P.; et al. Genetic determinants of Dabigatran plasma levels and their relation to bleeding. Circulation 2013, 127, 1404–1412. [Google Scholar]
- Merali, Z.; Ross, S.; Paré, G. The pharmacogenetics of carboxylesterases: CES1 and CES2 genetic variants and their clinical effect. Drug Metabol. Drug Interact. 2014, 29, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Chin, P. Which patients may benefit from dose adjustment of non-vitamin K antagonist oral anticoagulants? Semin. Thromb. Hemost. 2015, 41, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Gouin-Thibault, I.; Delavenne, X.; Blanchard, A.; Siguret, V.; Salem, J.E.; Narjoz, C.; Gaussem, P.; Beaune, P.; Funck-Brentano, C.; Azizi, M.; et al. Interindividual variability in dabigatran and rivaroxaban exposure: contribution of ABCB1 genetic polymorphisms and interaction with clarithromycin. J. Thromb. Haemost. 2017, 15, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Wessler, J.D.; Grip, L.T.; Mendell, J.; Giugliano, R.P. The P-glycoprotein transport system and cardiovascular drugs. J. Am. Coll. Cardiol. 2013, 61, 2495–2502. [Google Scholar] [CrossRef] [PubMed]
- Dimatteo, C.; D’Andrea, G.; Vecchione, G.; Paoletti, O.; Cappucci, F.; Tiscia, G.L.; Buono, M.; Grandone, E.; Testa, S.; Margaglion, M. Pharmacogenetics of dabigatran etexilate interindividual variability. Thromb. Res. 2016, 144, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mueck, W.; Stampfuss, J.; Kubitza, D.; Becka, M. Clinical pharmacokinetic and pharmacodynamic profile of rivaroxaban. Clin. Pharmacokinet. 2014, 53, 1–16. [Google Scholar] [CrossRef]
- Amin, H.; Nowak, R.J.; Schindler, J.L. Cardioembolic stroke: Practical considerations for patient risk management and secondary prevention. Postgrad. Med. 2014, 126, 55–65. [Google Scholar] [CrossRef]
- Gnoth, M.J.; Buetehorn, U.; Muenster, U.; Schwarz, T.; Sandmann, S. In vitro and in vivo P-Glycoprotein transport characteristics of rivaroxaban. J. Pharmacol. Exp. Ther. 2011, 338, 372–380. [Google Scholar] [CrossRef]
- Stöllberger, C.; Finsterer, J. Relevanz von P-Glykoprotein in der schlaganfallprävention mit Dabigatran, rivaroxaban und Apixaban. Herz 2015, 40, 140–145. [Google Scholar] [CrossRef]
- Gong, I.Y.; Kim, R.B. Importance of pharmacokinetic profile and variability as determinants of dose and response to dabigatran, rivaroxaban, and apixaban. Can. J. Cardiol. 2013, 29, S24–S33. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Joseph, J.; Young, L.; McRae, S.; Curnow, J.; Nandurkar, H.; Wood, P.; McLintock, C. New oral anticoagulants: A practical guide on prescription, laboratory testing and peri-procedural/bleeding management. Intern. Med. J. 2014, 44, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; He, K.; Herbst, J.J.; Kolb, J.; Shou, W.; Wang, L.; Balimane, P.V.; Han, Y.-E.; Gan, J.; Frost, C.E.; et al. Characterization of efflux transporters involved in distribution and disposition of Apixaban. Drug Metab. Dispos. 2013, 41, 827–835. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cîmpan, P.L.; Chira, R.I.; Mocan, M.; Anton, F.P.; Farcaş, A.D. Oral Anticoagulant Therapy—When Art Meets Science. J. Clin. Med. 2019, 8, 1747. https://doi.org/10.3390/jcm8101747
Cîmpan PL, Chira RI, Mocan M, Anton FP, Farcaş AD. Oral Anticoagulant Therapy—When Art Meets Science. Journal of Clinical Medicine. 2019; 8(10):1747. https://doi.org/10.3390/jcm8101747
Chicago/Turabian StyleCîmpan, Patricia Lorena, Romeo Ioan Chira, Mihaela Mocan, Florin Petru Anton, and Anca Daniela Farcaş. 2019. "Oral Anticoagulant Therapy—When Art Meets Science" Journal of Clinical Medicine 8, no. 10: 1747. https://doi.org/10.3390/jcm8101747
APA StyleCîmpan, P. L., Chira, R. I., Mocan, M., Anton, F. P., & Farcaş, A. D. (2019). Oral Anticoagulant Therapy—When Art Meets Science. Journal of Clinical Medicine, 8(10), 1747. https://doi.org/10.3390/jcm8101747