The Endocrine Function of the Heart: Physiology and Involvements of Natriuretic Peptides and Cyclic Nucleotide Phosphodiesterases in Heart Failure



 ,
,
Abstract
1. Introduction
2. Physiology
2.1. The Family of Natriuretic Peptides
2.2. Structure, Synthesis, and Secretion
2.3. Receptors and Clearance
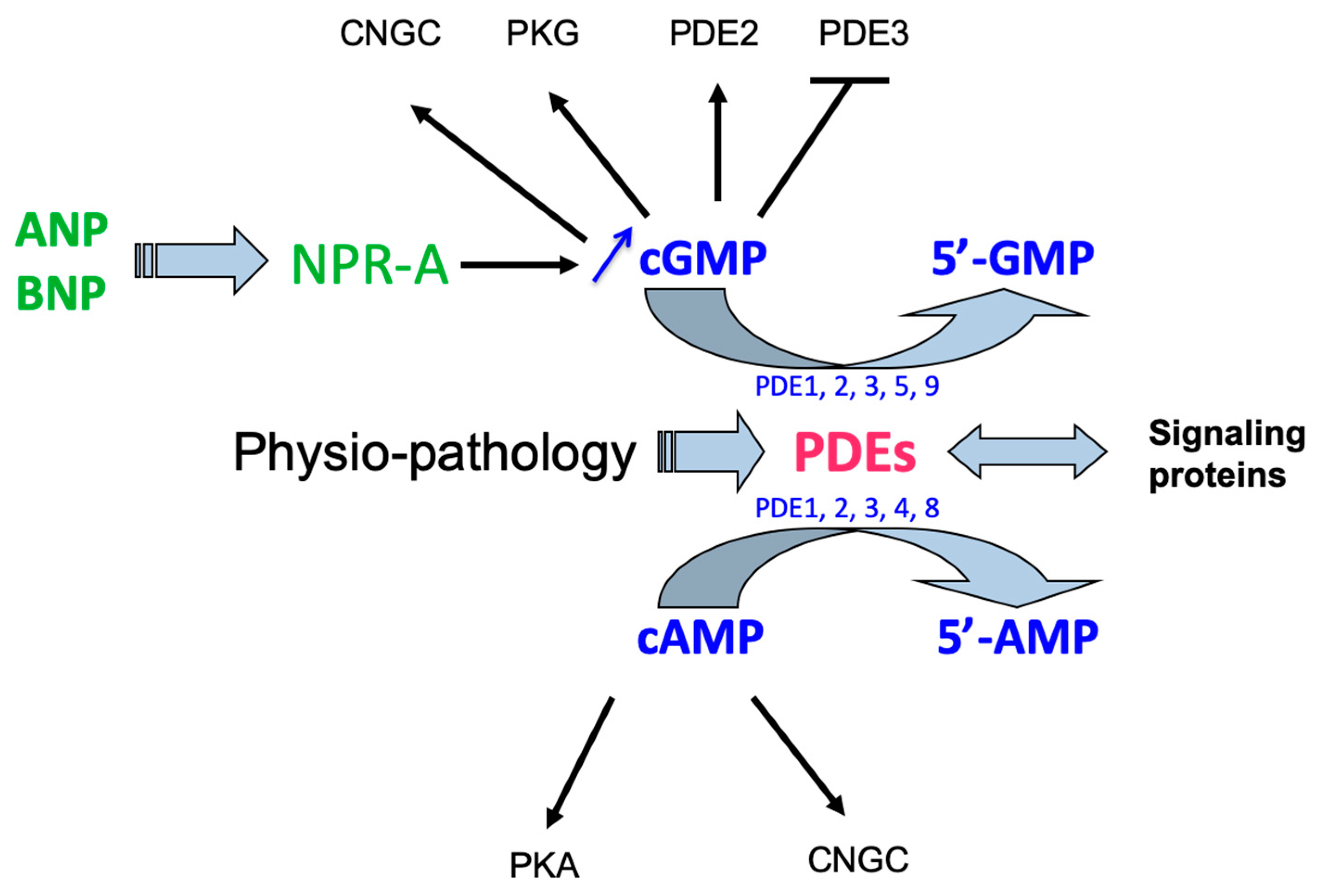
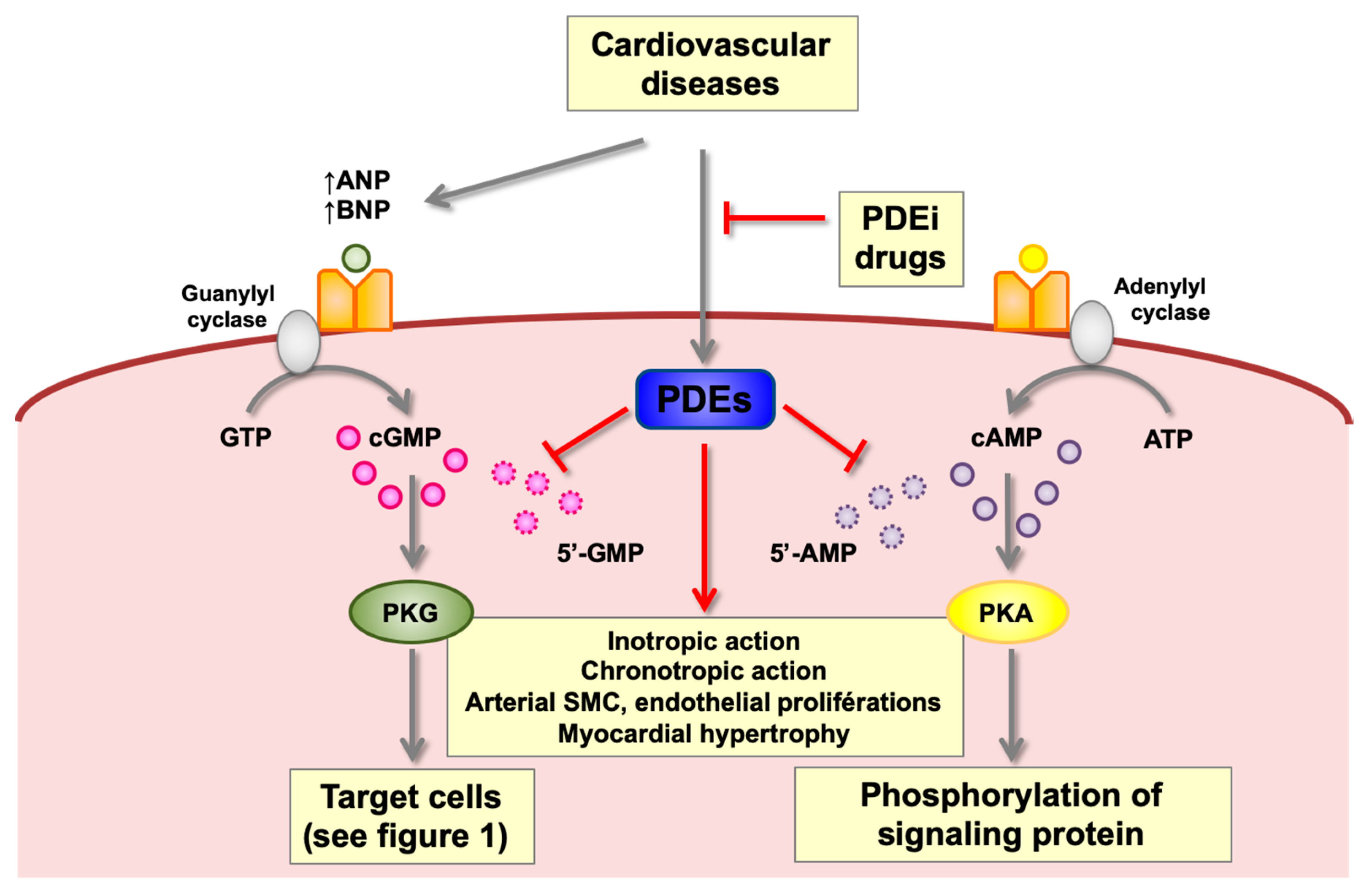
2.4. Signaling Pathways
2.4.1. Protein Kinase G of Types I and II
2.4.2. Cyclic Nucleotide Phosphodiesterases (PDEs)
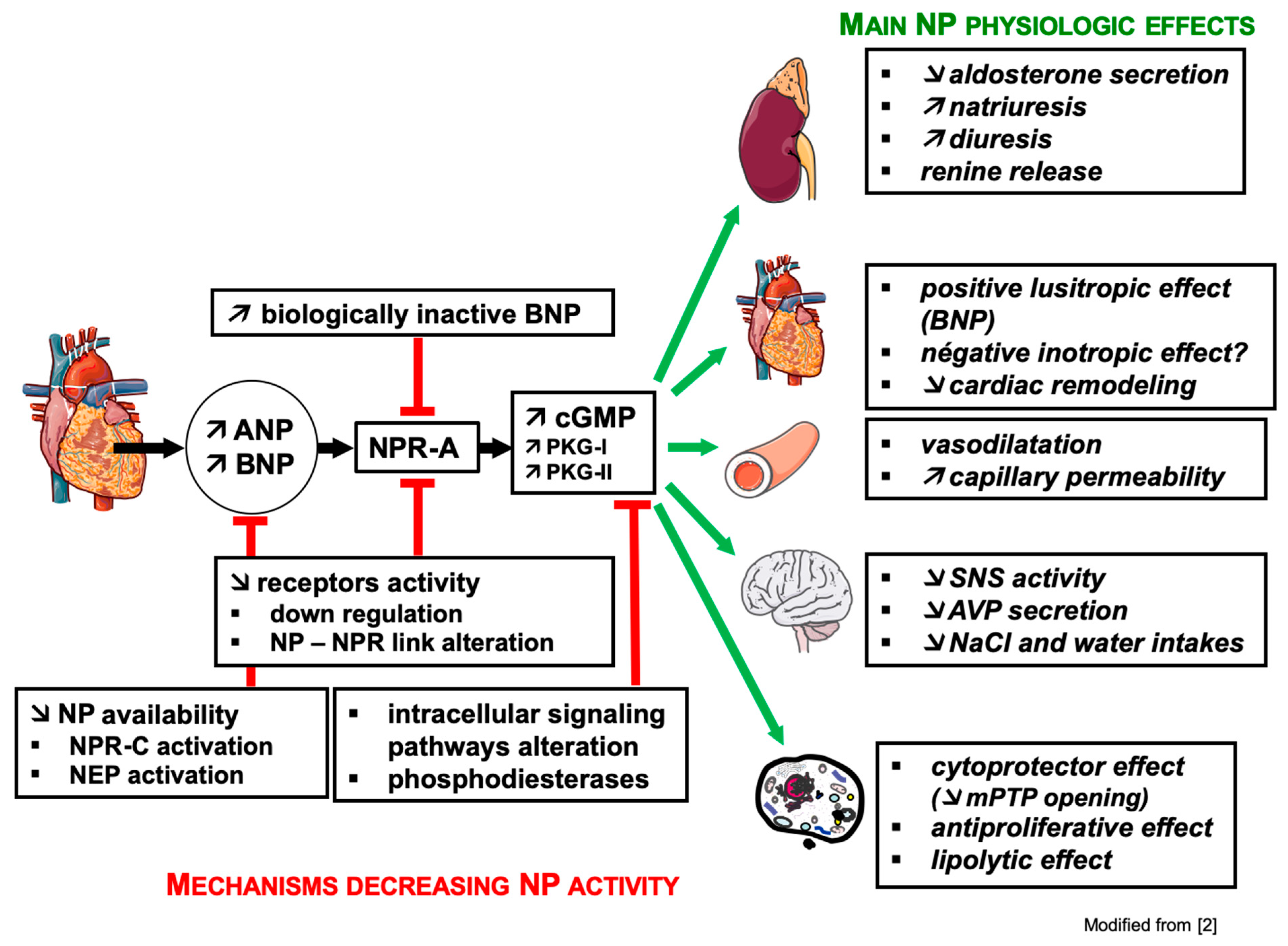
2.5. Physiological Effects of ANP and BNP
2.5.1. Renal Effects
2.5.2. Cardiovascular Effects
2.5.3. Effects on Neuro-Hormonal Systems
2.5.4. Cellular Effects
2.5.5. Effects on Adipose Tissue
3. Pathophysiology of Cardiac Natriuretic Peptides
3.1. Mechanisms Stimulating the Cardiac Natriuretic System in Pathological Situations
3.1.1. Hemodynamics
3.1.2. Heart Transplantation
Chronic Inflammation
Chronic Hypoxia
3.1.3. Neuroendocrine Factors
3.2. Mechanisms of Resistance to Natriuretic Peptides (Figure 2)
3.2.1. Renal Resistance
3.2.2. The BNP Paradox
3.3. Involvement of PDEs in Cardiac Pathologies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dussaule, J.-C.; Ledoux, S.; Pham, I. Système endocrinien cardiaque, coeur et vaisseaux. In Hormones, Coeur et Vaisseaux; Hittinger, L., Berthezène, F., Castaigne, A., Dubois-Randé, J.-L., Plouin, P.F., Eds.; Les Éditions INSERM: Paris, France, 1998; pp. 227–267. [Google Scholar]
- Talha, S.; Piquard, F.; Geny, B. Fonction endocrine du cœur. EMC Cardiol. 2015, 10, 1–8. [Google Scholar]
- Chiba, A.; Watanabe-Takano, H.; Miyazaki, T.; Mochizuki, N. Cardiomyokines from the heart. Cell. Mol. Life Sci. 2018, 75, 1349–1362. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, M.; Sorriento, D.; Coscioni, E.; Iaccarino, G.; Santulli, G. Adrenergic Receptors. In Endocrinology of the Heart in Health and Disease: Integrated, Cellular, and Molecular Endocrinology of the Heart (Chapter: 11); Schisler, J.C., Lang, C.H., Willis, M.S., Eds.; Academic Press, Elsevier Inc.: San Diego, CA, USA, 2017; pp. 285–315. [Google Scholar]
- de Bold, A.J.; Borenstein, H.B.; Veress, A.T.; Sonnenberg, H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 1981, 28, 89–94. [Google Scholar] [CrossRef]
- Sudoh, T.; Kangawa, K.; Minamino, N.; Matsuo, H. A new natriuretic peptide in porcine brain. Nature 1988, 332, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Moyes, A.J.; Hobbs, A.J. C-type Natriuretic Peptide: A Multifaceted Paracrine Regulator in the Heart and Vasculature. Int. J. Mol. Sci. 2019, 20, 2281. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Richter, R.; Forssmann, W.G. Urodilatin, a natriuretic peptide with clinical implications. Eur. J. Med. Res. 1998, 21, 103–110. [Google Scholar]
- Nakamura, S.; Naruse, M.; Naruse, K.; Kawana, M.; Nishikawa, T.; Hosoda, S.; Tanaka, I.; Yoshimi, T.; Yoshihara, I.; Inagami, T.; et al. Atrial natriuretic peptide and brain natriuretic peptide coexist in the secretory granules of human cardiac myocytes. Am. J. Hypertens. 1991, 4, 909–912. [Google Scholar] [CrossRef]
- Durocher, D.; Grepin, C.; Nemer, M. Regulation of gene expression in the endocrine heart. Recent. Prog. Horm. Res. 1998, 53, 7–23. [Google Scholar]
- Fu, S.; Ping, P.; Wang, F.; Luo, L. Synthesis, secretion, function, metabolism and application of natriuretic peptides in heart failure. J. Biol. Eng. 2018, 12, 2–21. [Google Scholar] [CrossRef]
- Edwards, B.S.; Zimmerman, R.S.; Schwab, T.R.; Heublein, D.M.; Burnett, J.C., Jr. Atrial stretch, not pressure, is the principal determinant controlling the acute release of atrial natriuretic factor. Circ. Res. 1988, 62, 191–195. [Google Scholar] [CrossRef]
- Stoupakis, G.; Klapholz, M. Natriuretic peptides: Biochemistry, physiology, and therapeutic role in heart failure. Heart Dis. 2003, 5, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R.; Abbey-Hosch, S.; Dickey, D.M. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr. Rev. 2006, 27, 47–72. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.; Koh, G.Y.; Nikonova, L.N.; Porter, J.G.; Maack, T. Molecular determinants of the clearance function of type C receptors of natriuretic peptides. J. Biol. Chem. 1996, 27, 9863–9869. [Google Scholar] [CrossRef] [PubMed]
- Ralat, L.A.; Guo, Q.; Ren, M.; Funke, T.; Dickey, D.M.; Potter, L.R.; Tang, W.J. Insulin-degrading enzyme modulates the natriuretic peptide-mediated signaling response. J. Biol. Chem. 2011, 286, 4670–4679. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.K.; Wang, T.J. Natriuretic Peptides and Cardiometabolic Health. Circ. J. 2015, 79, 1647–1655. [Google Scholar] [CrossRef]
- De Bold, A.J.; Bruneau, B.G.; Kuroski de Bold, M.L. Mechanical and neuroendocrine regulation of the endocrine heart. Cardiovasc. Res. 1996, 31, 7–18. [Google Scholar] [CrossRef]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef]
- Lincoln, T.M.; Dey, N.; Sellak, H. Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: From the regulation of tone to gene expression. J. Appl. Physiol. 2001, 91, 1421–1430. [Google Scholar] [CrossRef]
- Vaandrager, A.B.; Hogema, B.M.; de Jonge, H.R. Molecular properties and biological functions of cGMP-dependent protein kinase II. Front. Biosci. 2005, 10, 2150–2164. [Google Scholar] [CrossRef]
- Rubattu, S.; Sciarretta, S.; Morriello, A.; Calvieri, C.; Battistoni, A.; Volpe, M. NPR-C: A component of the natriuretic peptide family with implications in human diseases. J. Mol. Med. (Berl.) 2010, 88, 889–897. [Google Scholar] [CrossRef]
- Azevedo, M.F.; Faucz, F.R.; Bimpaki, E.; Horvath, A.; Levy, I.; de Alexandre, R.B.; Ahmad, F.; Manganiello, V.; Stratakis, C.A. Clinical and molecular genetics of the phosphodiesterases (PDEs). Endocr. Rev. 2014, 35, 195–233. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug. Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Murata, T.; Shimizu, K.; Degerman, E.; Maurice, D.; Manganiello, V. Cyclic nucleotide phosphodiesterases: Important signaling modulators and therapeutic targets. Oral Dis. 2015, 21, e25–e50. [Google Scholar] [CrossRef] [PubMed]
- Komas, N.; Lugnier, C.; Le Bec, A.; Serradeil-Le Gal, C.; Barthélémy, G.; Stoclet, J.-C. Differential sensitivity to cardiotonic drugs of cyclic AMP phosphodiesterases isolated from canine ventricular and sinoatrial-enriched tissues. J. Cardiovasc. Pharmacol. 1989, 14, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C.; Schoeffter, P.; Le Bec, A.; Strouthou, E.; Stoclet, J.-C. Selective inhibition of cyclic nucleotide phosphodiesterases of human, bovine and rat aorta. Biochem. Pharmacol. 1986, 35, 1743–1751. [Google Scholar] [CrossRef]
- Stoclet, J.-C.; Boulanger-Saunier, C.; Lassegue, B.; Lugnier, C. Cyclic nucleotides and calcium regulation in heart and smooth muscle cells. Ann. N. Y. Acad. Sci. 1988, 522, 106–115. [Google Scholar] [CrossRef]
- Lugnier, C.; Schini, V.B. Characterization of cyclic nucleotide from cultured bovine aortic endothelial cells. Biochem. Pharmacol. 1990, 39, 75–84. [Google Scholar] [CrossRef]
- Komas, N.; Lugnier, C.; Stoclet, J.-C. Endothelium-dependent and independent relaxation of the rat aorta by cyclic nucleotide phosphodiesterase inhibitors. Br. J. Pharmacol. 1991, 104, 495–503. [Google Scholar] [CrossRef]
- Bristol, J.A.; Sircar, I.; Moos, W.H.; Evans, D.B.; Weishaar, R.E. Cardiotonic agents: 1. 4,5-Dihydro-6-[4-(1H-imidazol-1-yl)phenyl]-3 (2H)-pyridazinones: Novel positive inotropic agents for the treatment of congestive heart failure. J. Med. Chem. 1984, 27, 1099–1101. [Google Scholar] [CrossRef]
- Lugnier, C.; Stierlé, A.; Beretz, A.; Schoeffter, P.; Lebec, A.; Wermuth, C.G.; Cazenave, J.-P.; Stoclet, J.-C. Tissue and substrate specificity of inhibition by alkoxy-aryl-lactams of platelet and arterial smooth muscle cyclic nucleotide phosphodiesterases relationship to pharmacological activity. Biochem. Biophys. Res. Commun. 1983, 113, 954–959. [Google Scholar] [CrossRef]
- Boolell, M.; Allen, M.J.; Ballard, S.A.; Gepi-Attee, S.; Muirhead, G.J.; Naylor, A.M.; Osterloh, I.H.; Gingell, C. Sildenafil: An orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction. Int. J. Impot. Res. 1996, 8, 47–52. [Google Scholar] [PubMed]
- Yafi, F.A.; Sharlip, I.D.; Becher, E.F. Update on the safety of phosphodiesterase type 5 inhibitors for the treatment of erectile dysfunction. Sex. Med. Rev. 2018, 6, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Champion, H.C.; Belardi, D.; Moslehi, J.; Mongillo, M.; Mergia, E.; Montrose, D.C.; Isoda, T.; Aufiero, K.; Zaccolo, M.; et al. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ. Res. 2005, 96, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Ghazi, M.; Vicenzi, M.; Arena, R.; Guazzi, M.D. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: Results of a 1-year, prospective, randomized, placebo-controlled study. Circ. Heart Fail. 2011, 4, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Klinger, J.R.; Thaker, S.; Houtchens, J.; Preston, I.R.; Hill, N.S.; Farber, H.W. Pulmonary hemodynamic responses to brain natriuretic peptide and sildenafil in patients with pulmonary arterial hypertension. Chest 2006, 129, 417–425. [Google Scholar] [CrossRef]
- Patrucco, E.; Albergine, M.S.; Santana, L.F.; Beavo, J.A. Phosphodiesterase 8A (PDE8A) regulates excitation-contraction coupling in ventricular myocytes. J. Mol. Cell. Cardiol. 2010, 49, 330–333. [Google Scholar] [CrossRef]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9A controls nitric-oxide independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef]
- Theilig, F.; Wu, Q. ANP-induced signaling cascade and its implications in renal pathophysiology. Am. J. Physiol. Renal. Physiol. 2015, 308, F1047–F1055. [Google Scholar] [CrossRef]
- Maack, T. Role of atrial natriuretic factor in volume control. Kidney Int. 1996, 49, 1732–1737. [Google Scholar] [CrossRef][Green Version]
- Bae, E.H.; Ma, S.K.; Lee, J.; Kim, S.W. Altered regulation of renal nitric oxide and atrial natriuretic peptide systems in angiotensin II-induced hypertension. Regul. Pept. 2011, 10, 31–37. [Google Scholar] [CrossRef]
- Chen, H.H.; Burnett, J.C. Natriuretic peptides in the pathophysiology of congestive heart failure. Curr. Cardiol. Rep. 2000, 2, 198–205. [Google Scholar] [CrossRef] [PubMed]
- McGrath, M.F.; de Bold, M.L.; de Bold, A.J. The endocrine function of the heart. Trends Endocrinol. Metab. 2005, 16, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Tarjan, E.; Denton, D.A.; Weisinger, R.S. Atrial natriuretic peptide inhibits water and sodium intake in rabbits. Regul. Pept. 1988, 23, 63–75. [Google Scholar] [CrossRef]
- Matsukawa, T.; Miyamoto, T. Angiotensin II-stimulated secretion of arginine vasopressin is inhibited by atrial natriuretic peptide in humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R624–R629. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Pratt, R.E.; Dzau, V.J. Interaction of atrial natriuretic polypeptide and angiotensin II on protooncogene expression and vascular cell growth. Biochem. Biophys. Res. Commun. 1991, 176, 1601–1609. [Google Scholar] [CrossRef]
- Tamura, N.; Ogawa, Y.; Chusho, H.; Nakamura, K.; Nakao, K.; Suda, M.; Kasahara, M.; Hashimoto, R.; Katsuura, G.; Mukoyama, M.; et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc. Natl. Acad. Sci. USA 2000, 97, 4239–4244. [Google Scholar] [CrossRef]
- Das, B.B.; Raj, S.; Solinger, R. Natriuretic peptides in cardiovascular diseases of fetus, infants and children. Cardiovasc. Hematol. Agents Med. Chem. 2009, 7, 43–51. [Google Scholar] [CrossRef]
- Schwachtgen, L.; Herrmann, M.; Georg, T.; Schwarz, P.; Marx, N.; Lindinger, A. Reference values of NT-proBNP serum concentrations in the umbilical cord blood and in healthy neonates and children. Z. Kardiol. 2005, 94, 399–404. [Google Scholar] [CrossRef]
- Becker, J.R.; Chatterjee, S.; Robinson, T.Y.; Bennett, J.S.; Panáková, D.; Galindo, C.L.; Zhong, L.; Shin, J.T.; Coy, S.M.; Kelly, A.E.; et al. Differential activation of natriuretic peptide receptors modulates cardiomyocyte proliferation during development. Development 2014, 141, 335–345. [Google Scholar] [CrossRef]
- D’Souza, S.P.; Yellon, D.M.; Martin, C.; Schulz, R.; Heusch, G.; Onody, A.; Ferdinandy, P.; Baxter, G.F. B-type natriuretic peptide limits infarct size in rat isolated hearts via KATP channel opening. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1592–H1600. [Google Scholar] [CrossRef]
- Fiscus, R.R.; Tu, A.W.; Chew, S.B. Natriuretic peptides inhibit apoptosis and prolong the survival of serum-deprived PC12 cells. Neuroreport 2001, 12, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, Y.; Yan, M.; Wu, Y.; Zheng, X. B-type natriuretic peptide-induced cardioprotection against reperfusion is associated with attenuation of mitochondrial permeability transition. Biol. Pharm. Bull. 2009, 32, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.N.; Ge, Y.K.; Li, J.Y.; Zeng, X.H.; Zheng, X.X. B-type natriuretic peptide enhances mild hypoxia-induced apoptotic cell death in cardiomyocytes. Biol. Pharm. Bull. 2007, 30, 1084–1090. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Vito, P.; Incerpi, S.; Pedersen, J.Z.; Luly, P. Atrial natriuretic peptide and oxidative stress. Peptides 2010, 31, 1412–1419. [Google Scholar] [CrossRef]
- Talha, S.; Bouitbir, J.; Charles, A.-L.; Zoll, J.; Goette-Di Marco, P.; Meziani, F.; Piquard, F.; Geny, B. Pretreatment with brain natriuretic peptide reduces skeletal muscle mitochondrial dysfunction and oxidative stress after ischemia-reperfusion. J. Appl. Physiol. 2013, 114, 172–179. [Google Scholar] [CrossRef]
- Schlueter, N.; de Sterke, A.; Willmes, D.M.; Spranger, J.; Jordan, J.; Birkenfeld, A.L. Metabolic actions of natriuretic peptides and therapeutic potential in the metabolic syndrome. Pharmacol. Ther. 2014, 144, 12–27. [Google Scholar] [CrossRef]
- Sengenès, C.; Berlan, M.; De Glisezinski, I.; Lafontan, M.; Galitzky, J. Natriuretic peptides: A new lipolytic pathway in human adipocytes. FASEB J. 2000, 14, 1345–1351. [Google Scholar] [CrossRef]
- Goharian, T.S.; Goetze, J.P.; Faber, J.; Andersen, L.B.; Grøntved, A.; Jeppesen, J.L. Associations of proatrial natriuretic peptide with components of the metabolic syndrome in adolescents and young adults from the general population. Am. J. Hypertens. 2017, 30, 561–568. [Google Scholar] [CrossRef]
- Magri, P.; Rao, M.A.; Cangianiello, S.; Bellizzi, V.; Russo, R.; Mele, A.F.; Andreucci, M.; Memoli, B.; De Nicola, L.; Volpe, M. Early impairment of renal hemodynamic reserve in patients with asymptomatic heart failure is restored by angiotensin II antagonism. Circulation 1988, 98, 2849–2854. [Google Scholar] [CrossRef]
- Mastromarino, V.; Volpe, M.; Musumeci, M.B.; Autore, C.; Conti, E. Erythropoietin and the heart: Facts and perspectives. Clin. Sci. 2011, 120, 51–63. [Google Scholar] [CrossRef]
- Liang, F.; Gardner, D.G. Mechanical strain activates BNP gene transcription through a p38/NF-κB—dependent mechanism. J. Clin. Investig. 1999, 104, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Sadoshima, J.; Jahn, L.; Takahashi, T.; Kulik, T.J.; Izumo, S. Molecular characterization of the stretch-induced adaptation of cultured cardiac cells. An in vitro model of load-induced cardiac hypertrophy. J. Biol. Chem. 1992, 267, 10551–10560. [Google Scholar] [PubMed]
- Ogawa, T.; de Bold, A.J. The heart as an endocrine organ. Endocr. Connect. 2014, 3, R31–R44. [Google Scholar] [CrossRef] [PubMed]
- Kuroski de Bold, M.L.; de Bold, A.J. Stretch-secretion coupling in atrial cardiocytes. Dissociation between atrial natriuretic factor release and mechanical activity. Hypertension 1991, 18, III169–III178. [Google Scholar] [CrossRef] [PubMed]
- Yokota, N.; Bruneau, B.G.; Fernandez, B.E.; de Bold, M.L.; Piazza, L.A.; Eid, H.; de Bold, A.J. Dissociation of cardiac hypertrophy, myosin heavy chain isoform expression, and natriuretic peptide production in DOCA-salt rats. Am. J. Hypertens. 1995, 8, 301–310. [Google Scholar] [CrossRef]
- Gulati, G.; Heck, S.L.; Røsjø, H.; Ree, A.H.; Hoffmann, P.; Hagve, T.A.; Norseth, J.; Gravdehaug, B.; Steine, K.; Geisler, J.; et al. Neurohormonal blockade and circulating cardiovascular biomarkers during anthracycline therapy in breast cancer patients: Results from the PRADA (Prevention of Cardiac Dysfunction During Adjuvant Breast Cancer Therapy) study. J. Am. Heart Assoc. 2017, 11, e006513. [Google Scholar] [CrossRef] [PubMed]
- Geny, B.; Charloux, A.; Lampert, E.; Lonsdorfer, J.; Haberey, P.; Piquard, F. Enhanced brain natriuretic peptide response to peak exercise in heart transplant recipients. J. Appl. Physiol. 1998, 85, 2270–2276. [Google Scholar] [CrossRef]
- Torre-Amione, G.; MacLellan, W.; Kapadia, S.; Weilbaecher, D.; Farmer, J.; Young, J.; Mann, D. Tumor necrosis factor-alpha is persistently expressed in cardiac allografts in the absence of histological or clinical evidence of rejection. Transplant. Proc. 1998, 30, 875–877. [Google Scholar] [CrossRef]
- Ma, K.K.; Ogawa, T.; De Bold, A.J. Selective upregulation of cardiac brain natriuretic peptide at the transcriptional and translational levels by pro-inflammatory cytokines and by conditioned medium derived from mixed lymphocyte reactions via p38 MAP kinase. J. Mol. Cell. Cardiol. 2004, 36, 505–513. [Google Scholar] [CrossRef]
- Vesely, D.L.; de Bold, A.J. Cardiac natriuretic peptides gene expression and secretion in inflammation. J. Investig. Med. 2009, 57, 29–32. [Google Scholar] [CrossRef]
- Talha, S.; Charloux, A.; Enache, I.; Piquard, F.; Geny, B. Mechanisms involved in increased plasma brain natriuretic peptide after heart transplantation. Cardiovasc. Res. 2011, 89, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Gramley, F.; Lorenzen, J.; Pezzella, F.; Kettering, K.; Himmrich, E.; Plumhans, C.; Koellensperger, E.; Munzel, T. Hypoxia and myocardial remodeling in human cardiac allografts: A time-course study. J. Heart Lung Transplant. 2009, 28, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.S.; Hyun, J.Y.; Kwak, Y.G.; Kim, I.S.; Kim, C.H.; Choi, E.; Kim, M.S.; Park, J.W. Hypoxic activation of the atrial natriuretic peptide gene promoter through direct and indirect actions of hypoxia-inducible factor-1. Biochem. J. 2003, 370, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, A.; Klanke, B.; Wagner, M.; Volk, T.; Willam, C.; Wiesener, M.S.; Eckardt, K.U.; Warnecke, C. Hypoxia, via stabilization of the hypoxia-inducible factor HIF-1alpha, is a direct and sufficient stimulus for brain-type natriuretic peptide induction. Biochem. J. 2008, 409, 233–242. [Google Scholar] [CrossRef]
- Stockmann, P.T.; Will, D.H.; Sides, S.D.; Brunnert, S.R.; Wilner, G.D.; Leahy, K.M.; Wiegand, R.C.; Needleman, P. Reversible induction of right ventricular atriopeptin synthesis in hypertrophy due to hypoxia. Circ. Res. 1988, 63, 207–213. [Google Scholar] [CrossRef]
- Arjamaa, O. Physiology of natriuretic peptides: The volume overload hypothesis revisited. World. J. Cardiol. 2014, 6, 4–7. [Google Scholar] [CrossRef]
- Anttila, K.; Streng, T.; Pispa, J.; Vainio, M.; Nikinmaa, M. Hypoxia exposure and B-type natriuretic peptide release from Langendorff heart of rats. Acta Physiol. (Oxf.) 2017, 220, 28–35. [Google Scholar] [CrossRef]
- Kim, M.; Platt, M.J.; Shibasaki, T.; Quaggin, S.E.; Backx, P.H.; Seino, S.; Simpson, J.A.; Drucker, D.J. GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat. Med. 2013, 19, 567–575. [Google Scholar] [CrossRef]
- Abassi, Z.; Burnett, J.C., Jr.; Grushka, E.; Hoffman, A.; Haramati, A.; Winaver, J. Atrial natriuretic peptide and renal cGMP in rats with experimental heart failure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1991, 261, R858–R864. [Google Scholar] [CrossRef]
- Chen, H.H.; Schirger, J.A.; Chau, W.L.; Jougasaki, M.; Lisy, O.; Redfield, M.M.; Barclay, P.T.; Burnett, J.C., Jr. Renal response to acute neutral endopeptidase inhibition in mild and severe experimental heart failure. Circulation 1999, 100, 2443–2448. [Google Scholar] [CrossRef]
- Margulies, K.B.; Burnett, J.C., Jr. Inhibition of cyclic GMP phosphodiesterases augments renal responses to atrial natriuretic factor in congestive heart failure. J. Card. Fail. 1994, 1, 71–80. [Google Scholar] [CrossRef]
- Supaporn, T.; Sandberg, S.M.; Borgeson, D.D.; Heublein, D.M.; Luchner, A.; Wei, C.M.; Dousa, T.P.; Burnett, J.C., Jr. Blunted cGMP response to agonists and enhanced glomerular cyclic 3′, 5′-nucleotide phosphodiesterase activities in experimental congestive heart failure. Kidney Int. 1996, 50, 1718–1725. [Google Scholar] [CrossRef] [PubMed]
- Bryan, P.M.; Xu, X.; Dickey, D.M.; Chen, Y.; Potter, L.R. Renal hyporesponsiveness to atrial natriuretic peptide in congestive heart failure results from reduced atrial natriuretic peptide receptor concentrations. Am. J. Physiol. Renal Physiol. 2007, 292, F1636–F1644. [Google Scholar] [CrossRef] [PubMed]
- Charloux, A.; Piquard, F.; Doutreleau, S.; Brandenberger, G.; Geny, B. Mechanisms of renal hyporesponsiveness to ANP in heart failure. Eur. J. Clin. Investig. 2003, 33, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Díez, J. Chronic heart failure as a state of reduced effectiveness of the natriuretic peptide system: Implications for therapy. Eur. J. Heart Fail. 2017, 19, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Heublein, D.M.; Huntley, B.K.; Boerrigter, G.; Cataliotti, A.; Sandberg, S.M.; Redfield, M.M.; Burnett, J.C., Jr. Immunoreactivity and guanosine 3′,5′-cyclic monophosphate activating actions of various molecular forms of human B-type natriuretic peptide. Hypertension 2007, 49, 1114–1119. [Google Scholar] [CrossRef]
- Lewis, L.K.; Raudsepp, S.D.; Yandle, T.G.; Prickett, T.C.; Richards, A.M. Development of a BNP1-32 immunoassay that does not cross-react with proBNP. Clin. Chem. 2017, 63, 1110–1117. [Google Scholar] [CrossRef]
- Yan, W.; Sheng, N.; Seto, M.; Morser, J.; Wu, Q. Corin, a mosaic transmembrane serine protease encoded by a novel cDNA from human heart. J. Biol. Chem. 1999, 274, 14926–14935. [Google Scholar] [CrossRef]
- Semenov, A.G.; Tamm, N.N.; Seferian, K.R.; Postnikov, A.B.; Karpova, N.S.; Serebryanaya, D.V.; Koshkina, E.V.; Krasnoselsky, M.I.; Katrukha, A.G. Processing of pro-B-type natriuretic peptide: Furin and corin as candidate convertases. Clin. Chem. 2010, 56, 1166–1176. [Google Scholar] [CrossRef]
- Huntley, B.K.; Sandberg, S.M.; Heublein, D.M.; Sangaralingham, S.J.; Burnett, J.C., Jr.; Ichiki, T. Pro-B-type natriuretic peptide-1-108 processing and degradation in human heart failure. Circ. Heart Fail. 2015, 8, 89–97. [Google Scholar] [CrossRef]
- Ichiki, T.; Huntley, B.K.; Burnett, J.C., Jr. BNP molecular forms and processing by the cardiac serine protease corin. Adv. Clin. Chem. 2013, 61, 1–31. [Google Scholar] [PubMed]
- Brandt, I.; Lambeir, A.M.; Ketelslegers, J.M.; Vanderheyden, M.; Scharpé, S.; De Meester, I. Dipeptidyl-peptidase IV converts intact B-type natriuretic peptide into its des-SerPro form. Clin. Chem. 2006, 52, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Volpe, M.; Carnovali, M.; Mastromarino, V. The natriuretic peptides system in the pathophysiology of heart failure: From molecular basis to treatment. Clin. Sci. (Lond.) 2016, 130, 57–77. [Google Scholar] [CrossRef] [PubMed]
- Pankow, K.; Wang, Y.; Gembardt, F.; Krause, E.; Sun, X.; Krause, G.; Schultheiss, H.P.; Siems, W.E.; Walther, T. Successive action of meprin A and neprilysin catabolizes B-type natriuretic peptide. Circ. Res. 2007, 101, 875–882. [Google Scholar] [CrossRef]
- Ala-Kopsala, M.; Magga, J.; Peuhkurinen, K.; Leipälä, J.; Ruskoaho, H.; Leppäluoto, J.; Vuolteenaho, O. Molecular heterogeneity has a major impact on the measurement of circulating N-terminal fragments of A- and B-type natriuretic peptides. Clin. Chem. 2004, 50, 1576–1588. [Google Scholar] [CrossRef]
- Suzuki, T.; Israr, M.Z.; Heaney, L.M.; Takaoka, M.; Squire, I.B.; Ng, L.L. Prognostic role of molecular forms of B-Type natriuretic peptide in acute heart failure. Clin. Chem. 2017, 63, 880–886. [Google Scholar] [CrossRef]
- Niederkofler, E.E.; Kiernan, U.A.; O’Rear, J.; Menon, S.; Saghir, S.; Protter, A.A.; Nelson, R.W.; Schellenberger, U. Detection of endogenous B-type natriuretic peptide at very low concentrations in patients with heart failure. Circ. Heart Fail. 2008, 1, 258–264. [Google Scholar] [CrossRef]
- Hawkridge, A.M.; Heublein, D.M.; Bergen, H.R., 3rd; Cataliotti, A.; Burnett, J.C., Jr.; Muddiman, D.C. Quantitative mass spectral evidence for the absence of circulating brain natriuretic peptide (BNP-32) in severe human heart failure. Proc. Natl. Acad. Sci. USA 2005, 102, 17442–17447. [Google Scholar] [CrossRef]
- Seferian, K.R.; Tamm, N.N.; Semenov, A.G.; Mukharyamova, K.S.; Tolstaya, A.A.; Koshkina, E.V.; Kara, A.N.; Krasnoselsky, M.I.; Apple, F.S.; Esakova, T.V.; et al. The brain natriuretic peptide (BNP) precursor is the major immunoreactive form of BNP in patients with heart failure. Clin. Chem. 2007, 53, 866–873. [Google Scholar] [CrossRef]
- Semenov, A.G.; Postnikov, A.B.; Tamm, N.N.; Seferian, K.R.; Karpova, N.S.; Bloshchitsyna, M.N.; Koshkina, E.V.; Krasnoselsky, M.I.; Serebryanaya, D.V.; Katrukha, A.G. Processing of pro-brain natriuretic peptide is suppressed by O-glycosylation in the region close to the cleavage site. Clin. Chem. 2009, 55, 489–498. [Google Scholar] [CrossRef]
- Leduc, R. Characterization of a secreted form of human furin endoprotease. Biochem. Biophys. Res. Commun. 1993, 15, 1011–1018. [Google Scholar]
- Dong, N.; Chen, S.; Yang, J.; He, L.; Liu, P.; Zheng, D.; Li, L.; Zhou, Y.; Ruan, C.; Plow, E.; et al. Plasma soluble corin in patients with heart failure. Circ. Heart Fail. 2010, 3, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Ping, P.; Zhu, Q.; Ye, P.; Luo, L. Brain natriuretic peptide and its biochemical, analytical, and clinical issues in heart failure: A narrative review. Front. Phys. 2018, 9, 692. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, L.; Salles, T.A.; Arruda-Junior, D.F.; Campos, L.C.; Pereira, A.C.; Barreto, A.L.; Antonio, E.L.; Mansur, A.J.; Tucci, P.J.; Krieger, J.E.; et al. Circulating dipeptidyl peptidase IV activity correlates with cardiac dysfunction in human and experimental heart failure. Circ. Heart Fail. 2013, 6, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Gomez, N.; Touihri, K.; Matheeussen, V.; Mendes Da Costa, A.; Mahmoudabady, M.; Mathieu, M.; Baerts, L.; Peace, A.; Lybaert, P.; Scharpé, S.; et al. Dipeptidyl peptidase IV inhibition improves cardiorenal function in overpacing-induced heart failure. Eur. J. Heart Fail. 2012, 14, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Talha, S.; Charloux, A.; Piquard, F.; Geny, B. Brain natriuretic peptide and right heart dysfunction after heart transplantation. Clin. Transplant. 2017, 31. [Google Scholar] [CrossRef]
- Van der Meer, P.; Gaggin, H.K.; Dec, G.W. ACC/AHA Versus ESC Guidelines on Heart Failure: JACC Guideline Comparison. J. Am. Coll. Cardiol. 2019, 21, 2756–2768. [Google Scholar] [CrossRef]
- Wang, T.; Liu, J.; McDonald, C.; Lupino, K.; Zhai, X.; Wilkins, B.J.; Hakonarson, H.; Pei, L. GDF15 is a heart-derived hormone that regulates body growth. EMBO Mol. Med. 2017, 8, 1150–1164. [Google Scholar] [CrossRef]
- Dalos, D.; Spinka, G.; Schneider, M.; Wernly, B.; Paar, V.; Hoppe, U.; Litschauer, B.; Strametz-Juranek, J.; Sponder, M. New cardiovascular biomarkers in ischemic heart disease-GDF-15, a probable predictor for ejection fraction. J. Clin. Med. 2019, 8, 924. [Google Scholar] [CrossRef]
- Moghtadaei, M.; Polina, I.; Rose, R.A. Electrophysiological effects of natriuretic peptides in the heart are mediated by multiple receptor subtypes. Prog. Biophys. Mol. Biol. 2016, 120, 37e49. [Google Scholar] [CrossRef]
- Kim, G.E.; Kass, D.A. Cardiac phosphodiesterases and their modulation for treating heart disease. Handb. Exp. Pharmacol. 2017, 243, 249–269. [Google Scholar] [PubMed]
- Cruickshank, J.M. Phosphodiesterase III inhibitors: Long-term risks and short-term benefits. Cardiovasc. Drugs Ther. 1993, 7, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Muller, B.; Lugnier, C.; Stoclet, J.-C. Involvement of rolipram-sensitive cyclic AMP phosphodiesterase in the regulation of cardiac contraction. J. Cardiovasc. Pharmacol. 1990, 16, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Abi-Gerges, A.; Richter, W.; Lefebvre, F.; Mateo, P.; Varin, A.; Heymes, C.; Samuel, J.L.; Lugnier, C.; Conti, M.; Fischmeister, R.; et al. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ. Res. 2009, 105, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Mokni, W.; Keravis, T.; Etienne-Selloum, N.; Walter, A.; Kane, M.O.; Schini-Kerth, V.B.; Lugnier, C. Concerted regulation of cGMP and cAMP phosphodiesterases in early cardiac hypertrophy induced by angiotensin II. PLoS ONE 2010, 5, e14227. [Google Scholar] [CrossRef]
- Molina, C.E. Cyclic Adenosine monophosphate phosphodiesterase type 4 protects against atrial arrhythmias. J. Am. Coll. Cardiol. 2012, 59, 2182–2190. [Google Scholar] [CrossRef]
- Nagendran, J.; Archer, S.L.; Soliman, D.; Gurtu, V.; Moudgil, R.; Haromy, A.; St Aubin, C.; Webster, L.; Rebeyka, I.M.; Ross, D.B. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 2007, 116, 238–248. [Google Scholar] [CrossRef]
- Yanaka, N.; Kurosawa, Y.; Minami, K.; Kawai, E.; Omori, K. cGMP-phosphodiesterase activity is up-regulated in response to pressure overload of rat ventricles. Biosci. Biochem. 2003, 67, 973–979. [Google Scholar] [CrossRef]
- Mehel, H.; Emons, J.; Vettel, C.; Wittköpper, K.; Seppelt, D.; Dewenter, M.; Lutz, S.; Sossalla, S.; Maier, L.S.; Lechêne, P.; et al. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts β-adrenergic responses in cardiomyocytes. J. Am. Coll. Cardiol. 2013, 62, 1596–1606. [Google Scholar] [CrossRef]
- Zoccarato, A.; Surdo, N.C.; Aronsen, J.M.; Fields, L.A.; Mancuso, L.; Dodoni, G.; Stangherlin, A.; Livie, C.; Jiang, H.; Sin, Y.Y.; et al. Cardiac hypertrophy is inhibited by a local pool of cAMP regulated by phosphodiesterase 2. Circ. Res. 2015, 117, 707–719. [Google Scholar] [CrossRef]
- Wagner, M.; Mehel, H.; Fischmeister, R.; El-Armouche, A. Phosphodiesterase 2: Anti-adrenergic friend or hypertrophic foe in heart disease? Naunyn Schmiedebergs Arch. Pharmacol. 2016, 389, 1139–1141. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, D.; Rybalkin, S.D.; Pi, X.; Wang, Y.; Zhang, C.; Munzel, T.; Beavo, J.A.; Berk, B.C.; Yan, C. Upregulation of phosphodiesterase 1A1 expression is associated with the development of nitrate tolerance. Circulation 2001, 104, 2338–2343. [Google Scholar] [CrossRef] [PubMed]
- Rybalkin, S.D.; Rybalkina, I.; Beavo, J.A.; Bornfeldt, K.E. Cyclic nucleotide phosphodiesterase 1C promotes human arterial smooth muscle cell proliferation. Circ. Res. 2002, 90, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Oikawa, M.; Cai, Y.; Wojtovich, A.P.; Nagel, D.J.; Xu, X.; Xu, H.; Florio, V.; Rybalkin, S.D.; Beavo, J.A.; et al. Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ. Res. 2009, 105, 956–964. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cardiac PDEs Family | Localization | Substrate | Action | Inhibition | References |
|---|---|---|---|---|---|
| PDE1 | Heart, vascular SMC | 1A and B: cGMP 1C: cAMP/cGMP | Regulation of cyclic nucleotides and calcium | [26,27,28] | |
| PDE2 | Heart, endothelial cells | cAMP/cGMP | Feedback of basal cGMP and cAMP in response to an increase in cGMP (production of NO, ANP and BNP) | [26,27,29] | |
| PDE3 | Heart, endothelial cells, vascular SMC | cGMP > cAMP | Interaction between the regulatory pathways of cAMP and cGMP | Increases cardiac strength while inducing vascular relaxation | [26,30,31] |
| PDE4 | Heart, vascular SMC, endothelial cells and immunocytes | cAMP | Insensitivity to cGMP differentiates from PDE3 | [32] | |
| PDE5 | Heart, vascular SMC and endothelium | cGMP | Treatment of erectile dysfunction, beneficial effect in HF, treatment of PAH | [33,34,35,36,37] | |
| PDE8 | Cardiomyocytes in the ventricle | cAMP | Control of cardiac function at the excitation-contraction coupling | [38] | |
| PDE9 | Dilated cardiomyopathies | cGMP | [39] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lugnier, C.; Meyer, A.; Charloux, A.; Andrès, E.; Gény, B.; Talha, S. The Endocrine Function of the Heart: Physiology and Involvements of Natriuretic Peptides and Cyclic Nucleotide Phosphodiesterases in Heart Failure. J. Clin. Med. 2019, 8, 1746. https://doi.org/10.3390/jcm8101746
Lugnier C, Meyer A, Charloux A, Andrès E, Gény B, Talha S. The Endocrine Function of the Heart: Physiology and Involvements of Natriuretic Peptides and Cyclic Nucleotide Phosphodiesterases in Heart Failure. Journal of Clinical Medicine. 2019; 8(10):1746. https://doi.org/10.3390/jcm8101746
Chicago/Turabian StyleLugnier, Claire, Alain Meyer, Anne Charloux, Emmanuel Andrès, Bernard Gény, and Samy Talha. 2019. "The Endocrine Function of the Heart: Physiology and Involvements of Natriuretic Peptides and Cyclic Nucleotide Phosphodiesterases in Heart Failure" Journal of Clinical Medicine 8, no. 10: 1746. https://doi.org/10.3390/jcm8101746
APA StyleLugnier, C., Meyer, A., Charloux, A., Andrès, E., Gény, B., & Talha, S. (2019). The Endocrine Function of the Heart: Physiology and Involvements of Natriuretic Peptides and Cyclic Nucleotide Phosphodiesterases in Heart Failure. Journal of Clinical Medicine, 8(10), 1746. https://doi.org/10.3390/jcm8101746