1. Introduction
The immune system is comprised of many biological structures and processes that protect the host against disease via adaptive and innate mechanisms. Adaptive immunity is mediated by T cell receptors, such as CD4, CD8, and CD25 that recognize specific antigens and neutralize them. Innate immunity, on the other hand, involves an immune response against antigens that is mediated by dendritic cells, macrophages, mast cells, neutrophils, basophils, and eosinophils that produce the cytokines that activate the adaptive immune system [
1].
Regulatory T cells (Tregs) are immune-suppressive T cells that maintain tolerance to self-antigens and prevent autoimmune diseases. Tregs are characterized by the presence of the transcriptional regulator Forkhead box protein P3 (FOXP3) that plays an important role in immune suppression of effector T cell proliferation [
2]. T cells follow one of two developmental pathways to enter the FOXP3
+ Treg lineage. First, a developing thymocyte may recognize self-antigen presented within the thymus during T cell maturation and become a natural Treg (‘nTreg’). Alternatively, a conventional naïve CD4
+ T cell may encounter a tumor-associated (‘self’) or tumor-specific (‘neo’) antigen in the tumor environment, become activated, and under the influence of an immunosuppressive tumor microenvironment, differentiate into an induced FOXP3+ (‘iTreg’) [
3]. Within the tumor environment, however, Tregs also respond to context-dependent inflammatory signals (e.g., Th1, Th2, or Th17 inflammation). From these tumor environmental cues, Tregs are also capable of other functions, such as promotion of angiogenesis or metastasis, regulation of inflammation, and suppression of anti-tumor adaptive immune responses [
4]. Since Tregs are frequently enriched within the tumor microenvironment, many emerging cancer therapeutic strategies involve depletion or modulation of Tregs, with the aim of enhancing anti-tumor immune responses [
5,
6,
7,
8].
Exosomes are small extracellular microvesicles (30–100 nm) of endocytic membrane origin that carry a repertoire of functional biomolecules, including genomic DNA, RNA, microRNA, and protein [
9]. Upon secretion, exosomes, especially those derived from tumors (tumor-derived exosomes or TDEs), target other cells in the tumor microenvironment and deliver their payload, which can affect the targeted cell in multiple ways [
10,
11]. For example, H-RAS-mediated transformation of intestinal epithelial cells results in the emission of exosomes containing genomic DNA, HRAS oncoprotein, and transcript that can mediate transformation of normal cells [
12]. TDEs are key players in epithelial to mesenchymal transition (EMT) and are notorious for orchestrating the invasion-metastasis cascade [
9]. Additionally, TDEs from breast tumor cells that are resistant to antiestrogen drugs and metformin can mediate the transfer of the resistance phenotype to cells that are initially sensitive to these drugs [
13]. In one of the most striking examples involving TDEs, transfer of mitochondrial DNA was shown to act as an oncogenic signal, promoting an exit from dormancy of therapy-induced cancer stem-like cells and leading to endocrine therapy resistance in OXPHOS-dependent breast cancer [
14].
Taken together, these observations suggest that TDEs can mediate specific cell-to-cell communication to horizontally transfer information. By activating signaling pathways in the cells they fuse or interact with, TDEs can effect significant changes in recipient cells. Indeed, the concept of horizontal information transfer, previously thought to be confined to the prokaryotic world, is now gaining acceptance in biology, in general, and cancer biology, in particular [
10,
15].
In addition to their role in horizontal information transfer between tumor cells, emerging evidence has also uncovered a similar role of exosomes concerning immune regulation through direct interaction or by activating specific receptors present on immune regulatory cells. It is, therefore, not surprising that TDEs have been the focus of intense investigation, especially in cancer [
16]. For example, when murine bone marrow precursor cells were incubated with TDEs, they caused expression of interleukin-6 (IL-6), which inhibited the differentiation of dendritic cells. This led to suppression of maturation, thereby causing a decrease in immune response [
17]. Similarly, TDEs can activate macrophage-induced tumor invasion and metastasis function by inhibiting interferon gamma (IFNγ) and IL-16 and increasing secretion of IL-8 and C-C motif ligand 2 (CCL2) in target cells [
18]. Finally, TDEs isolated from KRAS mutant (MT) human colon cancer cells can induce conventional CD4
+ CD25
− T cells to convert to Tregs (CD4
+ CD25
+ FOXP3
+) in a cell-extrinsic manner [
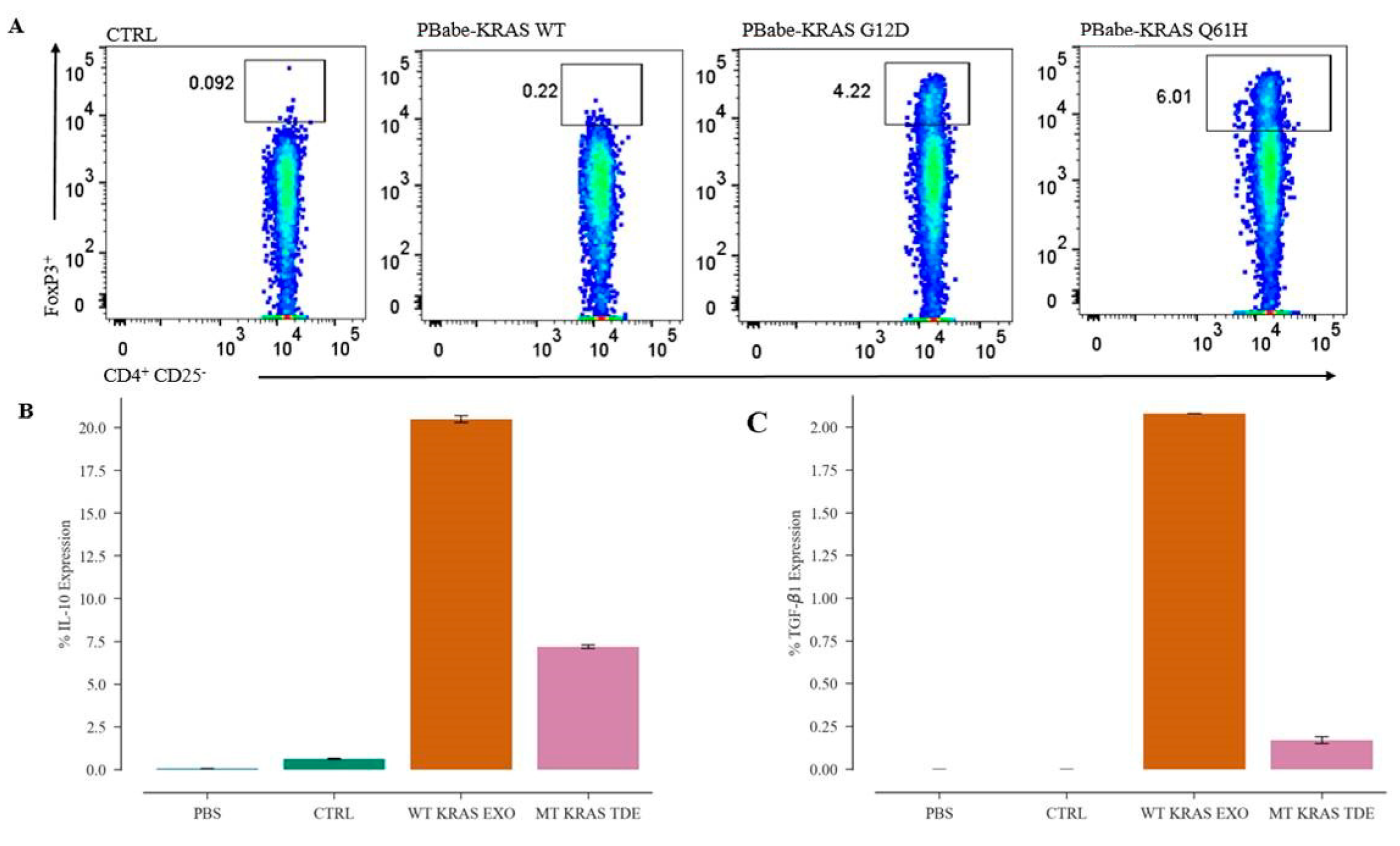
19]. However, the phenomenon was found to be dependent on the secretion of IL-10 and TGF-β1 via the activation of the MEK-ERK-AP1 pathway. Indeed, disrupting KRAS signaling by knocking down MT KRAS using siRNA led to ~50% attenuation in the expression of the mRNAs expressing IL-10 and TGF-β1. Conversely, overexpression of MT KRAS in tumor cells harboring wild type (WT) KRAS resulted in an increase of mRNAs for the two cytokines [
19].
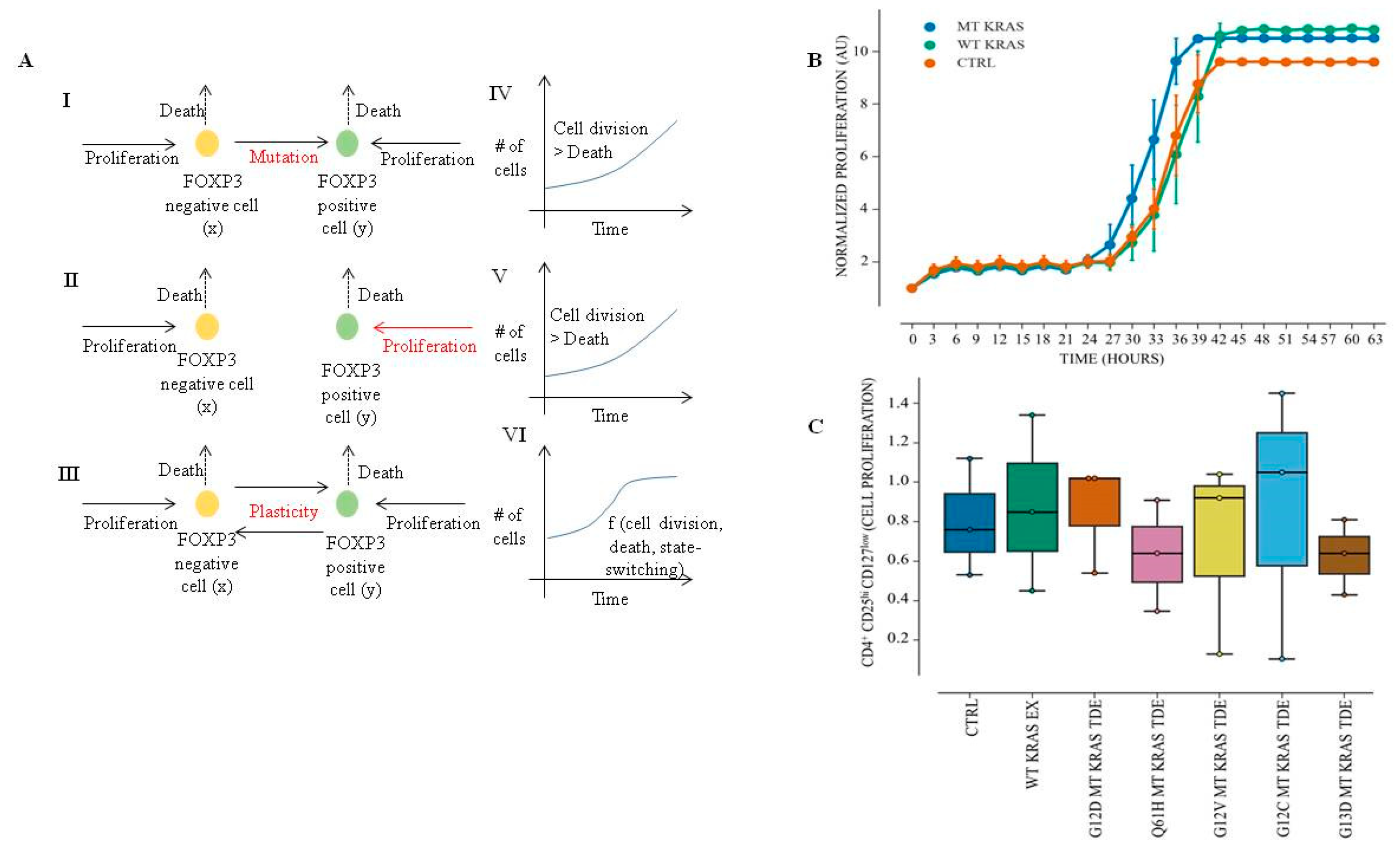
In this manuscript, we interrogated the role of TDEs from non-small-cell lung cancer (NSCLC) in contributing to immune-suppression by horizontally transferring information to immune regulatory cells and inducing them to switch their phenotype. We show that TDEs isolated from MT KRAS NSCLC cells can convert naïve T cells to FOXP3+ Treg-like cells. Furthermore, we show that this phenomenon is independent of cytokine signaling, given that transfection of MT KRAS cDNA alone is sufficient to actuate the switch of naïve CD4+ T cells to a FOXP3+ phenotype with immune suppressive function, even in the absence of other biomolecules, such as cytokines. We present a mathematical model supporting these conclusions. Although the converted T cells express FOXP3 and are functional, they appear to be distinct from bona fide Tregs at the molecular level. Finally, we identified the interferon (IFN) pathway as the mechanism underlying the immune-protective to immune-suppressive switch.
2. Experimental Section
2.1. Study Design
The experiments described in this study used cell lines and live cells isolated from serum samples. For reproducibility and statistical analyses, experiments were each performed at least three times and in triplicate. For the in vivo studies, 4–5 mice were used per group. The details of each experiment are described below. Cut-off dates for data collection, data inclusion/exclusion criteria, outliers, randomization, blinding, etc., were not applicable to this study. All subjects gave their informed consent for inclusion before they participated in the study. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of the Institutional Review Board (IRB) at City of Hope under IRB 17281.
2.2. Isolation of Tumor-Derived Exosomes (TDEs) from Patient Serum Samples
Serum from lung cancer patients with MT KRAS or WT KRAS were collected at the City of Hope National Medical Center under Institutional Review Board approval. TDEs were isolated using the Total Exosome Isolation Reagent (Invitrogen™ Waltham, MA, USA) following the manufacturer’s protocol.
2.3. Isolation and Transmission Electron Microscopy (TEM) of TDEs from Lung Cancer Cell Lines
TDEs were isolated from MT KRAS or WT KRAS cells as follows. Because cell culture media contains bovine pituitary extract enriched in microvesicles, all the experiments in which exosomes were collected or counted were performed using culture medium without fetal bovine serum (FBS). The culture medium was first centrifuged at 300× g for 10 min to remove cells and debris and subsequently centrifuged at 10,000× g for 45 min to remove large particles. Finally, the medium was ultracentrifuged twice at 110,000× g at 4 °C for 2 h in a Beckman Coulter Optima L-100XP ultracentrifuge to pellet the TDEs. TDEs were then suspended in a small volume of PBS and the samples were stored at −80 °C until used.
2.4. Nanosight Analysis and Concentration Determination
Nanoparticle tracking analysis was used to determine TDE concentration. TDE samples were diluted 1:10 in PBS and visualized with the NanoSight NS300 nanoparticles detector (Malvern, Westborough, MA, USA). The preparations were introduced into the sample chamber of the instrument equipped with a 635 nm laser. All samples were diluted to give counts in the linear range of the instrument (up to 7 × 108 per mL). The particles in the laser beam undergo Brownian motion and videos of these particle movements are recorded. The Nanosight Tracking Analysis (NTA) 2.3 software (Malvern Analytical, Malver, PA 19355, USA) then analyzes the video and determines the particle concentration and the size distribution of the particles. Three videos of 30 s duration were recorded for each sample at appropriate dilutions with a shutter speed setting of 1500 (exposure time 30 ms) and camera gain of 560. The detection threshold was set at 6 and at least 1000 tracks were analyzed for each video.
2.5. Genomic and TDE DNA Isolation
Total DNA from cells was isolated using the DNeasy Blood and Tissue Kit (Qiagen, Germantown, MD 20874, USA; Qiagen, hilden, Germany). TDE DNA was isolated from the serum-depleted cell culture supernatants treated with proteinase K, lysis buffer, and precipitated with ethanol (100%) followed by heat inactivation at 56 °C.
2.6. Isolation of CD4+ T and Naïve CD4+ CD25− T Cells from Donor PBMCs
PBMCs from healthy donors were processed for isolation of CD4+, and naïve CD4+ T cells (CD4+ CD25− T) cells using Histopaque (Sigma Aldrich, Munich, Germany). Briefly, 5 mL of donor blood was diluted with PBS and upon centrifugation over Histopaque solution, PBMCs were isolated. Approximately, 1 × 107 mL of PBMCs were used for isolation of CD4+ T cells using the MojoSort™ Human CD4+ T Cell Isolation Kit (catalog; 480009). For isolation of naïve CD4+ CD25− T cells, the MojoSortTM Human CD4 naïve T cell isolation kit (catalog; 480041) (BioLegend, San Diego, CA, USA) was used.
2.7. Isolation of Human CD4+ CD127low CD25+ Regulatory T Cells from Donor PBMCs
PBMCs from healthy donors were processed for isolation of CD4+ CD127low CD25+ Regulatory T cells using Histopaque (Sigma Aldrich, Munich, Germany). Briefly, 5 mL of donor blood was diluted with PBS and upon centrifugation over Histopaque solution, PBMCs were isolated. Approximately, 1 × 107 mL of PBMCs were used for isolation of CD4+ CD127low CD25+ Regulatory T cells using the EasySep™ CD4+ CD127low CD25+ Human Regulatory T Cell Isolation Kit (STEMCELL Technologies, Cambridge, MA, USA) following the manufacturer’s protocol.
2.8. Cell Culture and Transfection
The human NSCLC cell lines A549, H358, H460, and H1299 were maintained in complete growth medium containing RPMI from (Life Technologies, Camarillo, CA, USA) with 10% FBS and antibiotics penicillin and streptomycin. The CRISPR/Cas9 plasmid encoding the target wild type sgKRAS sequence was purchased from Addgene. CRISPR/Cas9 plasmid at 2 µg concentration was transfected by Turbofectin 8.0 following the protocol from OriGene (Rockville, MD, USA).
2.9. Site-Directed Mutagenesis and TOPO® TA Cloning
Plasmid pBabe-KRas WT KRAS (Plasmid# 75282) and pBabe-KRAS G12D (Plasmid # 58902) were purchased from Addgene. The pBabe-KRAS point mutation Q61H was created by using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies) following the recommended protocol. For TOPO® TA Cloning, pCR™4-TOPO™ Vector kit was purchased from (Thermo Fisher Scientific, Waltham, MA, USA) and the PCR product was cloned into the TOPO vector following the manufacturer’s protocol. The pCMV-AC- KRAS GFP fusion plasmid was purchased from OriGene (Rockville, MD, USA) and used as a template for construction of Q61H KRAS mutation by the site-directed mutagenesis.
2.10. Western Blotting
The TDE pellet was dissolved in RIPA buffer and quantified using the BCA Protein Assay method (ThermoFisher Scientific, Waltham, MA, USA). Reducing buffer was added to the samples and the proteins were analyzed by SDS-Mini Protean TGX gels (Bio-Rad, Irvine, CA, USA) and transferred to Immobilon-P PVDF Membrane (Millipore Sigma, Darmstadt, Germany). The membrane was then incubated with target antibodies at 4 °C overnight. After washing the membrane with tris-buffered saline (TBS) and Tween20 (TBS-T) three times for 30 min, the membrane was incubated with secondary antibodies for 1 h at room temperature. After washing five times, 10 min each time, a chemiluminescent detection system (Bio-Rad Clarity western ECL substrate) was used to detect the secondary antibody. Finally, the membranes were exposed to X-ray films to detect the proteins of interest. Antibodies used were a mouse monoclonal KRAS antibody (Santa Cruz Biotechnology, Dallas,TX, USA) and for detection of exosomes, anti-CD9, anti-CD63, and anti-CD81 antibodies (System Biosciences, Palo Alto, CA, USA).
2.11. Incubation of Exosomes with CD4+ T and Naïve CD4+ CD25− T Cells
CD4 T cells at a density of 2 × 106 cells/well in 24-well plates were seeded in RPMI medium without FBS. Naïve CD4+ CD25− T cells at a density of 1 × 106 cells/well in 24-well plates were seeded in RPMI medium without FBS. After 3–4 h, TDEs isolated from control, WT KRAS TDEs and Q61H mutant KRAS lung cancer cells were incubated with CD4 and naïve CD4 T cells for 48 h and analyzed by flow cytometry.
2.12. Transfecting CD4+ and Naïve CD4+ CD25− T Cells with KRAS cDNAs
CD4+ T cells at density of 3 × 106 mL and naïve CD4+ CD25− T cells at a density of 1.5 × 106 mL were seeded in 24-well plates, and 3–4 h later were transfected with 2 µg of pBabe KRAS WT and pBabe KRAS G12D or pBabe KRAS Q61H plasmid DNA by electroporation using the Amaxa® Human T Cell Nucleofector® Kit (Lonza, Basel, Switzerland) following the manufacturer’s protocol. Twenty-four hours post transfection, 1 µg/mL of puromycin was added, and after 48 h, the cells were analyzed by flow cytometry.
2.13. Cell Surface and Nuclear Staining
The cells were stained with anti-human monoclonal antibody (mAb) CD4–FITC clone RPA-T4 (BioLegend, San Diego, CA, USA). For nuclear staining, the cells were fixed, and permeabilized using the True Nuclear Transcription Factor Buffer (BioLegend, San Diego, CA, USA). The anti-human mAb FOXP3-PE antibody (BioLegend, San Diego, CA, USA) was used for staining intranuclear FOXP3. Flow cytometry data were analyzed using FlowJo.
2.14. TDE Flow Cytometry Analysis
TDEs isolated from the NSCLC cells and patient serum samples were directly analyzed for intracellular cytokine IL-10 and TGF-β1 expression by Attune NxT flow cytometer. For this purpose, TDEs in 100 μL final volume of PBS were stained using the PE anti-human CD9 antibody (Clone HI9a) (BioLegend, San Diego, CA, USA) and incubated for 1 h at 4 °C. The TDEs were then washed and stained for intracellular cytokines using the PE/Cy7 anti-human IL-10 antibody (clone JES3-9D7) and Brilliant Violet 421™ anti-human LAP (TGF-β1) antibody (TW4-2F8) (BioLegend, San Diego, CA, USA). TDEs isolated from mice serum were stained with APC anti-human CD63 Antibody (Clone H5C6) (BioLegend, San Diego, CA, USA) and FITC anti-human KRAS antibody (Catalog#: OACA01930, Aviva Systems Bio, CA, USA).
2.15. Immune Treg Phenotype Cell Metabolism Assay
To assay for the Treg phenotype cell metabolism assay, CD4+ CD127low CD25+ regulatory T cells or naïve CD4+ CD25− T cells alone or after incubation with MT KRAS TDEs were seeded at cell density of 20,000 cells in 50 µL/well in a PM-M1 Microplate™ (Biolog, Hayward, CA, USA) for analysis of carbon metabolism. Briefly, the regulatory T cells, naïve CD4+ CD25− T cells alone or after incubation with MT KRAS TDEs for 24–48 h were mixed with 10 µL of Dye MB and the plates were incubated in the OmniLog® incubator. Cell metabolism of the immune Tregs was calculated based on the redox dye intensity in units. Mutant and WT KRAS NSCLC were also assessed for carbon metabolism using the PM-M1 Microplate™ (Biolog, Hayward, CA, USA).
2.16. IncuCyte® S3 Cell Count Proliferation Assay
Immune cell proliferation assay was performed using the IncuCyte® Cell Count Live Cell Imaging System S3 (IncuCyte, Ann Arbor, MI, USA). Briefly, naïve CD4+ CD25− T cells at density of 1 × 104 cells/well were incubated with MT KRAS or WT KRAS TDEs in 96-well plates for 24 h and cell proliferation was analyzed by counting the cells in real time. For CD4+ CD127low CD25+ Treg proliferation assay, 1.5 × 104 were incubated with MT KRAS TDEs or WT KRAS TDEs in a 96-well plate for 24 h, and cell proliferation was analyzed by counting the cells using in real time IncuCyte ® S3 Live Cell Imaging System.
2.17. In vitro Immune Suppression Assay
Naïve CD4+ CD25− T cells at density of 1 × 106 /well were seeded in a 24-well plate with MT KRAS or WT KRAS TDEs for 24 h. CD4+ CD127low CD25+ Tregs at a density of 3–5 × 105 well were seeded in a 24-well plate. In a separate well, naïve CD4+ CD25− T cells at density of 1 × 106 well were pre-stained with CellTrace™ CFSE Proliferation Kit (ThermoFisher Scientific, MA). After 24 h, the naïve CD4 T carboxyfluorescein succinimidyl ester (CFSE) pre-stained cells were then added to the naïve CD4+ CD25− T cells that were pre-incubated with WT or MT KRAS TDEs. After 24 h, cells were fixed and permeabilized and analyzed by flow cytometry. For flow cytometry analysis, cells were analyzed by fixing and staining with intranuclear antihuman mAb FOXP3-PE antibody (BioLegend, San Diego, CA, USA) and counted for cells that are positive for CFSE dye intensity. CD4+ CD127low CD25+ Tregs or naïve CD4+ CD25− T cells alone were used as control.
2.18. Salmonella typhimurium Host Strains
YS1646 was purchased from ATCC
® (202165, Manassas, VA, USA). The bioluminescence expression vector for bacterial plasmid pAKlux2 was purchased from Addgene (Watertown, MA, USA) [
20]. Bacteria were grown to the OD = 0.7, washed three times with equal volumes of sterile PBS and diluted in PBS to the final concentration of 1 × 10
5 CFU/μL. To inactivate bacteria, the aliquoted preparation was incubated at 70 °C for 1 h.
2.19. NSCLC Xenograft Mouse Model
Human NSCLC cells A549 (5 × 106) and H358 (5 × 106 cells) were injected, in left and right flanks of NSG mice (n = 12), respectively, to create a two-sided xenograft model. After injection, tumor size was monitored three times/week and tumor volume ((length × width)2 × 0.5) was calculated. When tumor volumes reached 100 mm3 mice were randomly assigned in 3 groups (n = 4) to receive three consecutive daily retro-orbital injections (50 μL) of sterile PBS (Group 1), or 5 × 106 CFU preparation of heat-inactivated (Group 2) and live bioluminescent Salmonella typhimurium YS1646 (Group 3). After injection, the tumor growth was monitored for 21 days. Animals were then euthanized and blood was collected. Mice PBMC fractions were isolated using Histopaque (Sigma Aldrich, Munich, Germany) by collecting the PBMC supernatant after centrifugation at 2000 rpm for 20 min. Exosomes from mice serum were isolated using the total Exosome Isolation Reagent (Invitrogen™, Waltham, MA, USA) following the manufacturer’s protocol.
2.20. Ex vivo Human MT KRAS Exosomes and Immune Phenotype Conversion Assay
NSCLC xenograft serum exosomes containing MT KRAS from the control PBS (Group 1), or heat-inactivated Salmonella typhimurium YS1646 (Group 2)- or live bioluminescent Salmonella typhimurium YS1646 (Group 3)-treated mice were incubated with the naïve CD4+ CD25− T cells isolated from healthy donor PBMCs at a density of 1 × 106 well in a 24-well plate for 48 h. The cells were stained with antihuman mAb CD4–FITC clone RPA-T4 (BioLegend, San Diego, CA, USA). For nuclear staining, the cells were fixed, permeabilized using the True Nuclear Transcription Factor Buffer (BioLegend, San Diego, CA, USA). The anti-human mAb FOXP3-PE antibody (BioLegend, San Diego, CA, USA) was used.
2.21. Infection with Salmonella typhimurium
Naïve CD4+ CD25− T cells at a density of 5 × 105 well were incubated with WT KRAS or MT KRAS TDEs for 24 h. The live “T37” or heat-inactivated “T70” bioluminescent Salmonella typhimurium preparations (1 × 105 CFU/µL) were diluted in PBS (1:10) and co-cultured with the naïve CD4 T cells that were pre-incubated with TDEs and after 24 h, the cells were fixed and stained for CD4+ and FOXP3+ and analyzed by flow cytometry.
2.22. In vitro Salmonella typhimurium Human MT KRAS TDEs and Immune Phenotype Conversion Assay
Naïve CD4+ CD25− T cells isolated from healthy donor PBMCs were seeded in 24-well plate at a density of 5 × 105 cells/well. WT KRAS or MT KRAS TDEs were incubated with the naïve CD4+ CD25− T cells. Next, we diluted the preparation (1 × 105 CFU/µL) of heat-inactivated “T70” or live “T37” bioluminescent Salmonella typhimurium in 1:10 PBS and then incubated with the naïve CD4+ CD25− T cells pre-incubated with the WT KRAS or MT KRAS TDEs. PBS was used as control in the experiments. After 24 h, the cells were fixed and stained by anti-human mAb FOXP3-PE antibody (BioLegend, CA, USA) and anti-human mAb CD4–FITC clone RPA-T4 (BioLegend, San Diego, CA, USA). The flow cytometry data analysis was carried out using FlowJo® (v10.6.1, Ashland, OR, USA).
2.23. Single-Cell Analysis
Sequences from 10× genomics platform were de-multiplexed and aligned to genome build hg19 using CellRanger 3.0.1. (
https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger). To balance the size differences of the Treg group to KRAS MT and WT groups, 1000 cells were randomly selected from MT and WT groups for the clustering analysis over all three groups. Data were then imported into Seurat package version 2.3.4 for further filtering, variable gene selection, dimensional reduction and clustering analysis. We filtered out cells that have unique gene counts less than 200 or over 2000 and high mitochondrial expression (>10% mitochondria counts). After normalization, 1619 genes were identified as variable genes for PCA. Using embedded jackstraw analysis in the Seurat package, we identify the first 10 statistically significant principal components from a previous PCA step to be used in the non-linear dimensional reduction clustering analysis (tSNE). Marker genes for each group were identified by performing differential expression analysis in Seurat, with the exclusion of genes that had less than 25% detection percentage in either of the two comparison groups.
2.24. Fractal Dimension (FD) and Lacunarity (LC) Analysis
Immunohistochemistry (IHC) staining images with three markers (DAPI, CD4 and FOXP3) were scanned for the presence of both tumor cells and surrounding stroma and immune cells in the tissue microenvironment. The DAPI-stained nuclei were used to identify the overall cells in the tissue. CD4 and FOXP3
+ staining images were used to identify T-helper cell and Treg cell distribution. IHC staining images were segmented to identify the cells on the tissue slides using Fiji/ImageJ ver 1.52 [
21]. In this study, we measured the Fractal Dimension (FD) and Lacunarity (LC) of the merged binary IHC images using box counting scan method implemented in the FracLac plugin ver2016Apr120248a502 [
22] from Fiji/ImageJ version 1.52 [
21]. A total of 558 IHC staining images with 95% tissue coverage were collected from 15 lung cancer patients. A total of 264 slides were from KRAS mutant tumor samples, and 294 slides were from wild type tumor samples. The difference of FD and lacunarity between the KRAS mutant and wild type samples were tested using Mann–Whitney
U-test. The
p-value indicates the level of distance between the two data sets. All tests were performed using R (version 3.5.1, R Foundation, Vienna, Austria) [
22]. The test results show that the mean of the KRAS mutant FD is significantly higher than the mean of wild type KRAS FD (
p-value = 7.152 × 10
5). The mean of the KRAS mutant lacunarity is significantly less than the mean of the wild type KRAS lacunarity (
p-value = 3.297 × 10
6).
2.25. Statistical Analyses
A two-tailed Wilcoxon rank-sum test was used to test the null hypothesis that two sets of measurements were drawn from the same distribution. This was used instead of the paired Student’s t-test as the populations could not be assumed to be normally distributed.
4. Discussion
A major hurdle for effective cancer immunotherapy is the immunosuppressive tumor microenvironment. Although large numbers of tumor-specific T cells can be generated in patients by active immunization or adoptive transfer, these T cells do not readily translate to tumor cell killing in situ. In fact, the Treg subpopulation of T cells plays an important role in suppressing tumor-specific immunity [
31,
32]. Thus, our observations (
Figure S8) employing FD/LC analyses of clinical samples from patients with MT KRAS NSCLC had a higher percentage of FOXP3
+ Treg-like cells underscore the significance of the present study.
We used a variety of experimental approaches to demonstrate that mutant KRAS TDEs from lung cancer cell lines, patient serum, and xenograft mice can actuate phenotypic switching in T cells independent of cytokines produced by the tumor cells. Furthermore, we demonstrated that in addition to the molecular markers that characterize Tregs, converted cells have a metabolic profile that parallels bona fide Tregs and also function like authentic Tregs by suppressing proliferation of naïve T cells. However, despite these similarities, single-cell sequencing data revealed that the conversion results in a new class of immune regulatory cells that share many similarities with bona fide Tregs. Our data are consistent with previous observations demonstrating phenotypic and functional diversity in human Treg cells [
33]. In fact, such heterogeneity may allow subsets of Tregs with unique specificities and immunomodulatory functions to be targeted to define immune environments during different types of inflammatory responses [
33]. Additional studies aimed at discerning the function(s) of this new class of Tregs are needed to fully appreciate their role in tumorigenesis.
Nonetheless, the present study raises some important questions. How does mutant KRAS cause phenotypic switching in T cells? One potential mechanism by which mutant KRAS could modulate FOXP3 expression is by acting at the transcriptional level. Consistent with this possibility, it has been reported that H-RAS is localized to the nucleus [
34,
35]. However, it should be noted that another study that detected KRAS in the nucleus pointed to potential limitations imposed by the non-specificity of the antibodies or contaminations in cellular preparations [
36]. However, our attempts employing immunofluorescence microscopy of green fluorescence protein (GFP) WT KRAS fusion protein or MT KRAS GFP fusion protein did not show nuclear localization of either protein.
An alternate possibility is that KRAS could regulate FOXP3 expression without entering the nucleus via aberrant signaling. For example, oncogenic N-RAS was shown to act as the most potent regulator of SRF-, NF-
κB-, and AP-1-dependent transcription. The N-RAS and RGL2 (Ral guanine nucleotide exchange factor) axis is a distinct signaling pathway for SRF-targeted gene expression such as Egr1 and JunB as the RGL2 RAS binding domain significantly impairs oncogenic N-RAS-induced SRE activation. Indeed, oncogenic N-RAS elevated acetylated histone H3K9 and H3K23 levels globally in the chromatin, and chromatin immunoprecipitation assays revealed that acetylated H3K9 is significantly enriched at the promoter and coding regions of Egr1 and JunB [
37].
Our single-cell sequencing results suggest that MT KRAS may also modulate phenotypic switching without actually entering the nucleus (
Figure S8). To this end, we identified an IFN gene expression signature induced by MT KRAS in CD4
+ naïve T cells. Since WT KRAS did not induce the expression of the same genes, we conclude that MT KRAS is responsible for upregulation of type I IFN expression. This conclusion is further corroborated by the fact that the transcription factor IRF7, a key transcriptional regulator of type I IFN-dependent immune responses, was upregulated in the converted cells but not in the naïve T cell control Group. Other groups have also found a link between different types of IFNs (including Type I) and FOXP3 expression in mice and humans [
28,
29,
30]. Although more work will be needed to fully understand the exact mechanism linking mutation of KRAS and cell signaling, leading to IFN expression and phenotypic conversion of CD4
+ naïve T cells to Tregs, we propose a working model (
Figure S8). TDEs shed from NSCLC cells carrying MT KRAS are incorporated into CD4
+ naïve T cells and induce the IRF7 signaling pathway. This leads to increased gene expression and induction of key proteins involved in expression and secretion of type I IFN. Type I IFN, in turn, induces the conversion of CD4
+ naïve T to Treg-like cells by upregulating genes that characterize Tregs, including FOXP3. However, we note that the switch is not a binary event but a gradual process that appears to involve a series of bifurcations. Consequently, MT KRAS-mediated conversion results in a heterogeneous population with varying Treg-like phenotypes with cells that have a close resemblance to bona fide Tregs, expressing the full complement of Treg-restricted genes including FOXP3.
Another possibility is that the KRAS protein could upregulate FOXP3 protein post-transcriptionally, as has been reported with the zinc finger protein ZNF304 [
38]. A majority of KRAS-positive colorectal cancers (CRCs) have a CpG island methylator phenotype (CIMP). CIMP is characterized by aberrant DNA hypermethylation and transcriptional silencing of many genes that are encoded by the INK4-ARF locus. An RNA interference screen identified the zinc-finger DNA-binding protein ZNF304 as the pivotal factor required for INK4-ARF silencing and CIMP. In these cells, ZNF304 is bound at the promoters of INK4-ARF and other CIMP genes and recruits a corepressor complex that includes the DNA methyltransferase DNMT1, resulting in DNA hypermethylation and transcriptional silencing. Thus, KRAS promotes silencing through upregulation of ZNF304. Therefore, it is conceivable that KRAS may promote upregulation of the transcription factor(s) (e.g., NFAT) [
39] that, in turn, binds to the FOXP3 promoter and drives its expression in T cells and thereby, activates the cascade of events, culminating in their conversion to Treg-like cells. Additional studies that are currently underway in our laboratory could help uncover the identity of this new class of converted T cells in NSCLC.
The present study also begs the question as to why only MT KRAS, but not WT KRAS, can actuate phenotypic switching in T cells? Indeed, this is related to the more general question of why only mutant KRAS is oncogenic, and, to date, this remains one of the most challenging questions in cancer biology. Perhaps some recent biophysical and computational studies may help shed new light. A study investigating the effect of point mutations on the structure of KRAS employing UV photo disassociation mass spectrometry [
40] revealed that different downstream effects occur due to differences in the long-range conformational dynamics specific to each of the commonly occurring KRAS point mutations. Similarly, a molecular dynamics study based on conditional time-delayed correlations, identified driver–follower relationships in correlated motions of KRAS residue pairs, revealing the direction of information flow during allosteric modulation of its nucleotide-dependent intrinsic activity. Thus, the more C-terminal active Switch-II region motions drive the more N-terminal Switch-I region motions in the KRAS molecule [
41]. Interestingly, the short region of the C-terminal region of KRAS (residues 167–188/189, also called the hypervariable region) is intrinsically disordered [
42]. Thus, in MT KRAS, intrinsic disorder appears to facilitate intramolecular interactions spanning long distances that appear to be crucial for activation mechanisms and intensified oncogenic signaling compared to WT KRAS [
40].
Lastly, it is unknown whether the phenotypic switch is transient or permanent, i.e., can be inherited. It is important to note that biological barriers may likely curtail a permanent horizontal transformation of normal cells through such a TDE-mediated mechanism [
12]. Although this issue is outside the scope of the present study, it is possible that the mutant KRAS-actuated phenotypic switch we observe with lung cancer-derived exosomes is transient and the number of T cells converted would depend on the tumor mass. Consistent with this argument, our in vivo studies showed that TDEs from control mice that harbored large tumors were able to convert a larger population of T cells compared to TDEs from
Salmonella-infected mice with attenuated tumors. As MT KRAS is present in 41% of NSCLC patients [
43], this newly discovered MT KRAS-induced phenotypic switching could have important therapeutic implications for lung cancer. However, given that KRAS is the most frequently mutated oncogene in a wide variety of cancers, the present work is likely to have a much wider impact.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





