Immune Checkpoints as Promising Targets for the Treatment of Idiopathic Pulmonary Fibrosis?


Abstract
1. Introduction
2. IPF a Cancer-Like Disease
3. Immune Checkpoints in Cancer
3.1. PD-1/PD-L1
3.2. CTLA-4 and Other Checkpoints
4. What Can We Learn?
5. Immune Checkpoints in IPF
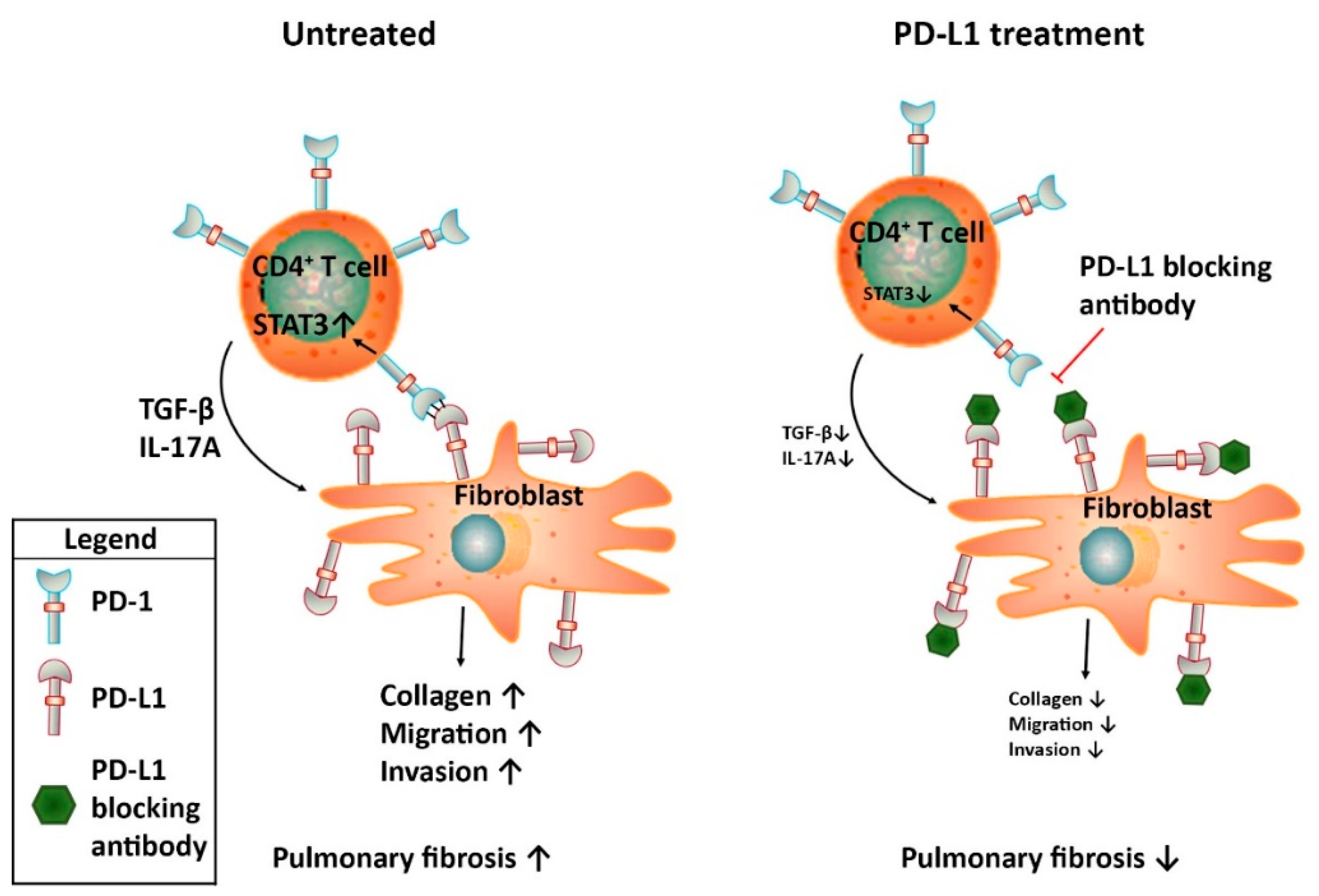
5.1. PD-1/PD-L1 Axis
5.2. CTLA-4 Axis
5.3. TIM-3
6. Immune Checkpoints; a New Horizon or a False Flag?
7. To Bear in Mind
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic Pulmonary Fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Strongman, H.; Kausar, I.; Maher, T.M. Incidence, Prevalence, and Survival of Patients with Idiopathic Pulmonary Fibrosis in the UK. Adv. Ther. 2018, 35, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, R.B.; Burke, N.; Fell, C.; Dion, G.; Kolb, M. Epidemiology and survival of idiopathic pulmonary fibrosis from national data in Canada. Eur. Respir. J. 2016, 48, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Navaratnam, V.; Fleming, K.M.; West, J.; Smith, C.J.; Jenkins, R.G.; Fogarty, A.; Hubbard, R.B. The rising incidence of idiopathic pulmonary fibrosis in the UK. Thorax 2011, 66, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Crestani, B.; Huggins, J.T.; Kaye, M.; Costabel, U.; Glaspole, I.; Ogura, T.; Song, J.W.; Stansen, W.; Quaresma, M.; Stowasser, S.; et al. Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: Results from the open-label extension study, INPULSIS-ON. Lancet Respir. Med. 2019, 7, 60–68. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. Ascend Study Group. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; du Bois, R.M.; Fagan, E.A.; Fishman, R.S.; Glaspole, I.; Glassberg, M.K.; Lancaster, L.; et al. Pirfenidone for Idiopathic Pulmonary Fibrosis: Analysis of Pooled Data from Three Multinational Phase 3 Trials. Eur. Respir. J. 2016, 47, 243–253. [Google Scholar] [CrossRef]
- Wuyts, W.A.; Antoniou, K.M.; Borensztajn, K.; Costabel, U.; Cottin, V.; Crestani, B.; Grutters, J.C.; Maher, T.M.; Poletti, V.; Richeldi, L.; et al. Combination Therapy: The Future of Management for Idiopathic Pulmonary Fibrosis? Lancet Respir. Med. 2014, 2, 933–942. [Google Scholar] [CrossRef]
- Vancheri, C. Idiopathic Pulmonary Fibrosis and Cancer: Do They Really Look Similar? BMC Med. 2015, 13, 220. [Google Scholar] [CrossRef] [PubMed]
- Hokland, P.; Hokland, M.; Cotter, F. The Nobel Prize for Medicine Awarded for Cancer Therapy by Inhibition of Negative Immune Regulation. Br. J. Haematol. 2018, 183, 698–700. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal. Similarities between Tumor Stroma Generation and Wound Healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Vancheri, C.; Failla, M.; Crimi, N.; Raghu, G. Idiopathic Pulmonary Fibrosis: A Disease with Similarities and Links to Cancer Biology. Eur. Respir. J. 2010, 35, 496–504. [Google Scholar] [CrossRef] [PubMed]
- De Groot, P.; Munden, R.F. Lung Cancer Epidemiology, Risk Factors, and Prevention. Radiol. Clin. N. Am. 2012, 50, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Ballester, B.; Milara, J.; Cortijo, J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, A.; Gomatou, G.; Bouros, E.; Trigidou, R.; Tzilas, V.; Bouros, D. Common Pathogenic Mechanisms between Idiopathic Pulmonary Fibrosis and Lung Cancer. Chest 2019, 156, 383–391. [Google Scholar] [CrossRef]
- Kinoshita, T.; Goto, T. Molecular Mechanisms of Pulmonary Fibrogenesis and Its Progression to Lung Cancer: A Review. Int. J. Mol. Sci. 2019, 20, 1461. [Google Scholar] [CrossRef] [PubMed]
- Reyfman, P.A.; Gottardi, C.J. IPF and Lung Cancer: Finding Similarities within Differences. Am. J. Respir. Cell Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Ulke, H.M.; Mutze, K.; Lehmann, M.; Wagner, D.E.; Heinzelmann, K.; Gunther, A.; Eickelberg, O.; Konigshoff, M. The Oncogene Ect2 Contributes to a Hyperplastic, Proliferative Lung Epithelial Cell Phenotype in IPF. Am. J. Respir. Cell Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Spek, C.A.; Duitman, J. Is Idiopathic Pulmonary Fibrosis a Cancer-Like Disease? Transcriptome Analysis to Fuel the Debate. ERJ Open Res. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Mercer, P.F.; Woodcock, H.V.; Eley, J.D.; Plate, M.; Sulikowski, M.G.; Durrenberger, P.F.; Franklin, L.; Nanthakumar, C.B.; Man, Y.; Genovese, F.; et al. Exploration of a Potent PI3 Kinase/Mtor Inhibitor as a Novel Anti-Fibrotic Agent in IPF. Thorax 2016, 71, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Al-Tamari, H.M.; Dabral, S.; Schmall, A.; Sarvari, P.; Ruppert, C.; Paik, J.; DePinho, R.A.; Grimminger, F.; Eickelberg, O.; Guenther, A.; et al. Foxo3 an Important Player in Fibrogenesis and Therapeutic Target for Idiopathic Pulmonary Fibrosis. EMBO Mol. Med. 2018, 10, 276–293. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Ramaiya, N.H.; Hatabu, H.; Hodi, F.S. Monitoring Immune-Checkpoint Blockade: Response Evaluation and Biomarker Development. Nat. Rev. Clin. Oncol. 2017, 14, 655–668. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 Antigen on the Surface of Stimulated Mouse T and B Lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed Cell Death 1 Forms Negative Costimulatory Microclusters That Directly Inhibit T Cell Receptor Signaling by Recruiting Phosphatase Shp2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The Evolving Landscape of Biomarkers for Checkpoint Inhibitor Immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Lanier, L.L.; O’Fallon, S.; Somoza, C.; Phillips, J.H.; Linsley, P.S.; Okumura, K.; Ito, D.; Azuma, M. CD80 (B7) and Cd86 (B70) Provide Similar Costimulatory Signals for T Cell Proliferation, Cytokine Production, and Generation of Ctl. J. Immunol. 1995, 154, 97–105. [Google Scholar] [PubMed]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. Pd-L2 Is a Second Ligand for Pd-1 and Inhibits T Cell Activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.F.; Huang, C.Y.; Ke, T.W.; Chen, T.W.; Lan, Y.C.; You, Y.S.; Chen, W.T.; Chao, K.S.C. Upregulation of Tumor Pd-L1 by Neoadjuvant Chemoradiotherapy (Neocrt) Confers Improved Survival in Patients with Lymph Node Metastasis of Locally Advanced Rectal Cancers. Cancer Immunol. Immunother. 2019, 68, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Dahan, R.; Sega, E.; Engelhardt, J.; Selby, M.; Korman, A.J.; Ravetch, J.V. FcγRs Modulate the Anti-Tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell 2015, 28, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed Death-1 Ligand 1 Interacts Specifically with the B7-1 Costimulatory Molecule to Inhibit T Cell Responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 Can Function as a Negative Regulator of T Cell Activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Brunner, M.C.; Chambers, C.A.; Chan, F.K.; Hanke, J.; Winoto, A.; Allison, J.P. CTLA-4-Mediated Inhibition of Early Events of T Cell Proliferation. J. Immunol. 1999, 162, 5813–5820. [Google Scholar]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) Bind with Similar Avidities but Distinct Kinetics to CD28 and CTLA-4 Receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Van der Merwe, P.A.; Bodian, D.L.; Daenke, S.; Linsley, P.; Davis, S.J. CD80 (B7-1) Binds Both CD28 and CTLA-4 with a Low Affinity and Very Fast Kinetics. J. Exp. Med. 1997, 185, 393–403. [Google Scholar] [CrossRef]
- Jain, N.; Nguyen, H.; Chambers, C.; Kang, J. Dual Function of Ctla-4 in Regulatory T Cells and Conventional T Cells to Prevent Multiorgan Autoimmunity. Proc. Natl. Acad. Sci. USA 2010, 107, 1524–1528. [Google Scholar] [CrossRef]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 Control over Foxp3+ Regulatory T Cell Function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Fehlings, M.; Simoni, Y.; Penny, H.L.; Becht, E.; Loh, C.Y.; Gubin, M.M.; Ward, J.P.; Wong, S.C.; Schreiber, R.D.; Newell, E.W. Checkpoint Blockade Immunotherapy Reshapes the High-Dimensional Phenotypic Heterogeneity of Murine Intratumoural Neoantigen-Specific CD8+ T Cells. Nat. Commun. 2017, 8, 562. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. FC-Dependent Depletion of Tumor-Infiltrating Regulatory T Cells Co-Defines the Efficacy of Anti-CTLA-4 Therapy against Melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, J.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 Antibodies of IGG2a Isotype Enhance Antitumor Activity through Reduction of Intratumoral Regulatory T Cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Fares, C.M.; van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.C.; Lopez-Verges, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. TIM-3 Marks Human Natural Killer Cell Maturation and Suppresses Cell-Mediated Cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef]
- Sul, J.; Blumenthal, G.M.; Jiang, X.; He, K.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Patients with Metastatic Non-Small Cell Lung Cancer Whose Tumors Express Programmed Death-Ligand 1. Oncologist 2016, 21, 643–650. [Google Scholar] [CrossRef]
- American Association for Cancer Research. Nivolumab Gets FDA Nod for Bladder Cancer. Cancer Discov. 2017, 7, OF7. [Google Scholar] [CrossRef]
- Hazarika, M.; Chuk, M.K.; Theoret, M.R.; Mushti, S.; He, K.; Weis, S.L.; Putman, A.H.; Helms, W.S.; Cao, X.; Li, H.; et al. FDA Approval Summary: Nivolumab for Treatment of Unresectable or Metastatic Melanoma Following Progression on Ipilimumab. Clin. Cancer Res. 2017, 23, 3484–3488. [Google Scholar] [CrossRef]
- Pai-Scherf, L.; Blumenthal, G.M.; Li, H.; Subramaniam, S.; Mishra-Kalyani, P.S.; He, K.; Zhao, H.; Yu, J.; Paciga, M.; Goldberg, K.B.; et al. FDA Approval Summary: Pembrolizumab for Treatment of Metastatic Non-Small Cell Lung Cancer: First-Line Therapy and Beyond. Oncologist 2017, 22, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab Versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lin, Z.; Zhang, X.; Chen, C.; Zhao, H.; Hong, S.; Zhang, L. First-Line Treatment for Patients with Advanced Non-Small Cell Lung Carcinoma and High Pd-L1 Expression: Pembrolizumab or Pembrolizumab Plus Chemotherapy. J. Immunother. Cancer 2019, 7, 120. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Stein, J.E.; Rimm, D.L.; Wang, D.W.; Bell, J.M.; Johnson, D.B.; Sosman, J.A.; Schalper, K.A.; Anders, R.A.; Wang, H.; et al. Comparison of Biomarker Modalities for Predicting Response to PD-1/PD-L1 Checkpoint Blockade: A Systematic Review and Meta-Analysis. JAMA Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ready, N.; Hellmann, M.D.; Awad, M.M.; Otterson, A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M.; et al. First-Line Nivolumab Plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer (Checkmate 568): Outcomes by Programmed Death Ligand 1 and Tumor Mutational Burden as Biomarkers. J. Clin. Oncol. 2019, 37, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. PD-L1 Positivity Predicts Response. Nat. Rev. Clin. Oncol. 2019, 16, 337. [Google Scholar] [CrossRef] [PubMed]
- McQuade, J.L.; Daniel, C.R.; Helmink, B.A.; Wargo, J.A. Modulating the Microbiome to Improve Therapeutic Response in Cancer. Lancet Oncol. 2019, 20, e77–e91. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.A.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut Microbiome Modulates Response to Anti-PD-1 Immunotherapy in Melanoma Patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Guyard, A.; Danel, C.; Theou-Anton, N.; Debray, M.P.; Gibault, L.; Mordant, P.; Castier, Y.; Crestani, B.; Zalcman, G.; Blons, H.; et al. Morphologic and Molecular Study of Lung Cancers Associated with Idiopathic Pulmonary Fibrosis and Other Pulmonary Fibroses. Respir. Res. 2017, 18, 120. [Google Scholar] [CrossRef]
- Jovanovic, D.; Roksandic, M.M.; Kotu, S.J.; Markovic, J.; Ceriman, V.; Kontic, M.; Skodric, T.V. Membrane PD-L1 Expression and Soluble PD-L1 Plasma Levels in Idiopathic Pulmonary Fibrosis-a Pilot Study. J Thorac. Dis. 2018, 10, 6660–6669. [Google Scholar] [CrossRef]
- Ni, K.; Liu, M.; Zheng, J.; Wen, L.; Chen, Q.; Xiang, Z.; Lam, K.T.; Liu, Y.; Chan, G.C.; Lau, Y.L.; et al. PD-1/PD-L1 Pathway Mediates the Alleviation of Pulmonary Fibrosis by Human Mesenchymal Stem Cells in Humanized Mice. Am. J. Respir. Cell Mol. Biol. 2018, 58, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Celada, L.J.; Kropski, J.A.; Herazo-Maya, J.D.; Luo, W.; Creecy, A.; Abad, A.T.; Chioma, O.S.; Lee, G.; Hassell, N.E.; Shaginurova, G.I.; et al. PD-1 up-Regulation on CD4+ T Cells Promotes Pulmonary Fibrosis through STAT3-Mediated IL-17a and TGF-beta1 Production. Sci. Transl. Med. 2018, 10, 8356. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Liu, X.; Liang, J.; Habiel, D.M.; Kulur, V.; Coelho, A.L.; Deng, N.; Xie, T.; Wang, Y.; Liu, N.; et al. PD-L1 on Invasive Fibroblasts Drives Fibrosis in a Humanized Model of Idiopathic Pulmonary Fibrosis. JCI Insight 2019, 4, 125326. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, K.M.; Karagiannis, K.; Tsitoura, E.; Tzanakis, N. Mesenchymal Stem Cell Treatment for IPF—Time for Phase 2 Trials? Lancet Respir. Med. 2017, 5, 472–473. [Google Scholar] [CrossRef]
- Habiel, D.M.; Espindola, M.S.; Kitson, C.; Azzara, A.V.; Coelho, A.L.; Stripp, B.; Hogaboam, C.M. Characterization of CD28(Null) T Cells in Idiopathic Pulmonary Fibrosis. Mucosal Immunol. 2019, 12, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.A.; Raez, L.E.; Oliveres, H.; Rolfo, C.C. Current Knowledge of Ipilimumab and Its Use in Treating Non-Small Cell Lung Cancer. Expert Opin. Biol. Ther. 2019, 19, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A.; Barrera, L.; Estrada, A.; Watson, S.R.; Wilson, K.; Aziz, N.; Kaminski, N.; Zlotnik, A. Gene Expression Profiles Distinguish Idiopathic Pulmonary Fibrosis from Hypersensitivity Pneumonitis. Am. J. Respir. Crit. Care Med. 2006, 173, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Kuwano, K.; Kunitake, R.; Kawasaki, M.; Hagimoto, N.; Hara, N. B7-1, B7-2 and Class II MHC Molecules in Idiopathic Pulmonary Fibrosis and Bronchiolitis Obliterans-Organizing Pneumonia. Eur. Respir. J. 2000, 15, 49–55. [Google Scholar]
- Isshiki, T.; Akiba, H.; Nakayama, M.; Harada, N.; Okumura, K.; Homma, S.; Miyake, S. Cutting Edge: Anti-TIM-3 Treatment Exacerbates Pulmonary Inflammation and Fibrosis in Mice. J. Immunol. 2017, 199, 3733–3737. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kuai, Q.; Gao, F.; Wang, Y.; He, M.; Zhou, H.; Han, G.; Jiang, X.; Ren, S.; Yu, Q. Overexpression of TIM-3 in Macrophages Aggravates Pathogenesis of Pulmonary Fibrosis in Mice. Am. J. Respir. Cell Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Johnson, D.B.; Chandra, S.; Sosman, J.A. Immune Checkpoint Inhibitor Toxicity in 2018. JAMA 2018, 320, 1702–1703. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Wang, X.; Woo, K.M.; Iyriboz, T.; Halpenny, D.; Cunningham, J.; Chaft, J.E.; Segal, N.H.; Callahan, M.K.; Lesokhin, A.M.; et al. Pneumonitis in Patients Treated with Anti-Programmed Death-1/Programmed Death Ligand 1 Therapy. J. Clin. Oncol. 2017, 35, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crino, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab Versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab Versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Perez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab Versus Docetaxel for Previously Treated, PD-L1-Positive, Advanced Non-Small-Cell Lung Cancer (Keynote-010): A Randomised Controlled Trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Shimizu, J.; Hasegawa, T.; Horio, Y.; Inaba, Y.; Yatabe, Y.; Hida, T. Pre-Existing Pulmonary Fibrosis Is a Risk Factor for Anti-PD-1-Related Pneumonitis in Patients with Non-Small Cell Lung Cancer: A Retrospective Analysis. Lung Cancer 2018, 125, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.B.A.G.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K. Esmo Guidelines Committee. Management of Toxicities from Immunotherapy: Esmo Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2017, 28, iv119–iv142. [Google Scholar] [CrossRef]
- Ide, M.; Tanaka, K.; Sunami, S.; Asoh, T.; Maeyama, T.; Tsuruta, N.; Nakanishi, Y.; Okamoto, I. Durable Response to Nivolumab in a Lung Adenocarcinoma Patient with Idiopathic Pulmonary Fibrosis. Thorac. Cancer 2018, 9, 1519–1521. [Google Scholar] [CrossRef]
- Kashiwada, T.; Minegishi, Y.; Saito, Y.; Kato, T.; Atsumi, K.; Seike, M.; Kubota, K.; Terasaki, Y.; Gemma, A. Organizing Pneumonia after Nivolumab Treatment in a Patient with Pathologically Proven Idiopathic Pulmonary Fibrosis. J. Nippon. Med. Sch. 2019, 86, 43–47. [Google Scholar] [CrossRef]
- Khunger, M.; Velcheti, V. A Case of a Patient with Idiopathic Pulmonary Fibrosis with Lung Squamous Cell Carcinoma Treated with Nivolumab. J. Thorac. Oncol. 2017, 12, e96–e97. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, D.; Morimoto, T.; Ito, J.; Sato, Y.; Ito, M.; Teraoka, S.; Otsuka, K.; Nagata, K.; Nakagawa, A.; Tomii, K. A Pilot Trial of Nivolumab Treatment for Advanced Non-Small Cell Lung Cancer Patients with Mild Idiopathic Interstitial Pneumonia. Lung Cancer 2017, 111, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Duchemann, B.; Didier, M.; Pailler, M.C.; Brillet, P.Y.; Kambouchner, M.; Uzunhan, Y.; Freynet, O.; Chouahnia, K.; Zelek, L.; Nunes, H. Can Nivolumab Be Used Safely in Idiopathic Pulmonary Fibrosis? Rev. Mal. Respir. 2019, 36, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Bellosillo, B.; Caux, C.; de Langen, A.; Mazieres, J.; Normanno, N.; Preusser, M.; Provencio, M.; Rojo, F.; Wolf, J.; et al. Immune checkpoint inhibitor treatment in patients with oncogene-addicted non-small cell lung cancer (NSCLC): Summary of a multidisciplinary round-table discussion. ESMO Open 2019, 4, e000498. [Google Scholar] [CrossRef]
- Gettinger, S.; Rizvi, N.A.; Chow, L.Q.; Borghaei, H.; Brahmer, J.; Ready, N.; Gerber, D.E.; Shepherd, F.A.; Antonia, S.; Goldman, J.W.; et al. Nivolumab Monotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 2980–2987. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-Year Overall Survival for Patients with Advanced Non‒Small-Cell Lung Cancer Treated with Pembrolizumab: Results from the Phase I KEYNOTE-001 Study. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Leighl, N.B.; Hellmann, M.D.; Hui, R.; Carcereny, E.; Felip, E.; Ahn, M.J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Pembrolizumab in patients with advanced non-small-cell lung cancer (KEYNOTE-001): 3-year results from an open-label.; phase 1 study. Lancet Respir. Med. 2019, 7, 347–357. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Ntolios, P.; Karameris, A.; Vilaras, G.; Boglou, P.; Koulelidis, A.; Archontogeorgis, K.; Kaltsas, K.; Zacharis, G.; Sarikloglou, E.; et al. Increased expression of epidermal growth factor receptor (EGF-R) in patients with different forms of lung fibrosis. Biomed. Res. Int. 2013, 2013, 654354. [Google Scholar] [CrossRef]
- Epstein Shochet, G.; Brook, E.; Eyal, O.; Edelstein, E.; Shitrit, D. Epidermal growth factor receptor paracrine upregulation in idiopathic pulmonary fibrosis fibroblasts is blocked by nintedanib. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L1025–L1034. [Google Scholar] [CrossRef]
- Derosa, L.; Routy, B.; Kroemer, G.; Zitvogel, L. The intestinal microbiota determines the clinical efficacy of immune checkpoint blockers targeting PD-1/PD-L1. Oncoimmunology 2018, 7, e1434468. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, D.N.; Ashley, S.L.; Gurczynski, S.J.; Xia, M.; Wilke, C.; Falkowski, N.R.; Norman, K.C.; Arnold, K.B.; Huffnagle, G.B.; Salisbury, M.L.; et al. Lung Microbiota Contribute to Pulmonary Inflammation and Disease Progression in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, P.L.; Cox, M.J.; Willis-Owen, S.A.; Mallia, P.; Russell, K.E.; Russell, A.M.; Murphy, E.; Johnston, S.L.; Schwartz, D.A.; Wells, A.U.; et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Cedars-Sinai. Available online: https://www.cedars-sinai.org/newsroom/study-protein-linked-to-cancer-growth-drives-deadly-lung-disease/ (accessed on 25 September 2019).
- Pulmonary Fibrosis News. Available online: https://pulmonaryfibrosisnews.com/2019/03/25/protein-that-drives-cancer-is-potential-new-therapeutic-target-for-idiopathic-pulmonary-fibrosis-study-suggests/ (accessed on 25 September 2019).


{kind=link}
| Query in PubMed | # of Publications |
|---|---|
| PD-1 AND IPF | 3 |
| PD-L1 AND IPF | 5 |
| PD-1 AND pulmonary fibrosis | 13 |
| PD-L1 AND pulmonary fibrosis | 11 |
| CTLA-4 AND IPF | 1 |
| CTLA4 AND IPF | 2 |
| CTLA-4 AND pulmonary fibrosis | 11 |
| CTLA4 AND pulmonary fibrosis | 7 |
| Immune checkpoint AND IPF | 4 |
| Immune checkpoint AND pulmonary fibrosis | 13 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duitman, J.; van den Ende, T.; Spek, C.A. Immune Checkpoints as Promising Targets for the Treatment of Idiopathic Pulmonary Fibrosis? J. Clin. Med. 2019, 8, 1547. https://doi.org/10.3390/jcm8101547
Duitman J, van den Ende T, Spek CA. Immune Checkpoints as Promising Targets for the Treatment of Idiopathic Pulmonary Fibrosis? Journal of Clinical Medicine. 2019; 8(10):1547. https://doi.org/10.3390/jcm8101547
Chicago/Turabian StyleDuitman, JanWillem, Tom van den Ende, and C. Arnold Spek. 2019. "Immune Checkpoints as Promising Targets for the Treatment of Idiopathic Pulmonary Fibrosis?" Journal of Clinical Medicine 8, no. 10: 1547. https://doi.org/10.3390/jcm8101547
APA StyleDuitman, J., van den Ende, T., & Spek, C. A. (2019). Immune Checkpoints as Promising Targets for the Treatment of Idiopathic Pulmonary Fibrosis? Journal of Clinical Medicine, 8(10), 1547. https://doi.org/10.3390/jcm8101547