New Molecular Technologies for Minimal Residual Disease Evaluation in B-Cell Lymphoid Malignancies



Abstract
1. Introduction
1.1. The Early Steps and Prognostic Importance of Minimal Residual Disease
1.2. MRD Potential Molecular Targets in B-Cell Hematologic Malignancies
1.3. qPCR-MRD Approaches: Technical Considerations and Feasibility on Different Tissues
1.4. Limitations of Standard PCR-Based MRD Techniques
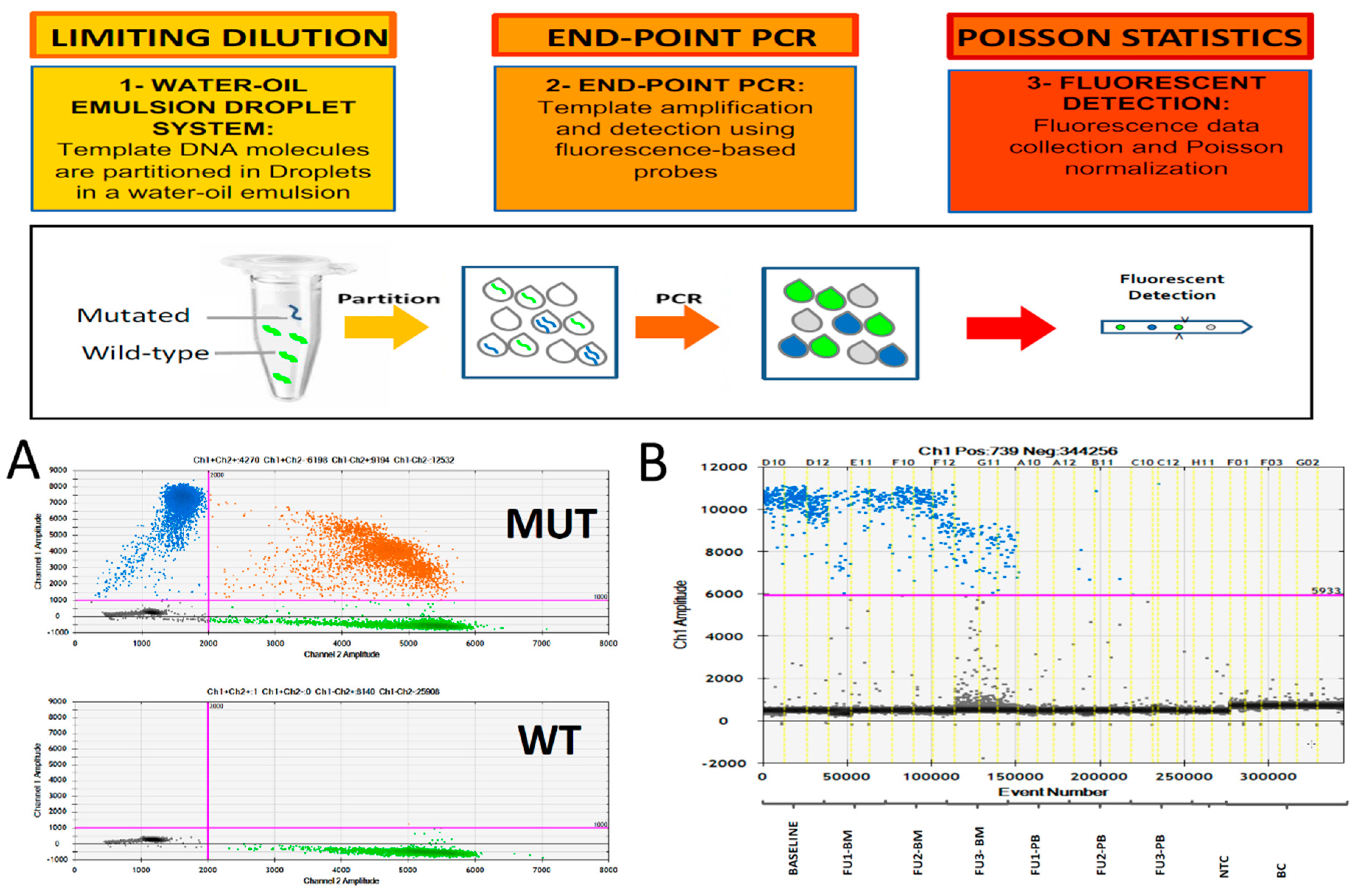
2. Digital PCR for MRD Monitoring
3. Next Generation Sequencing
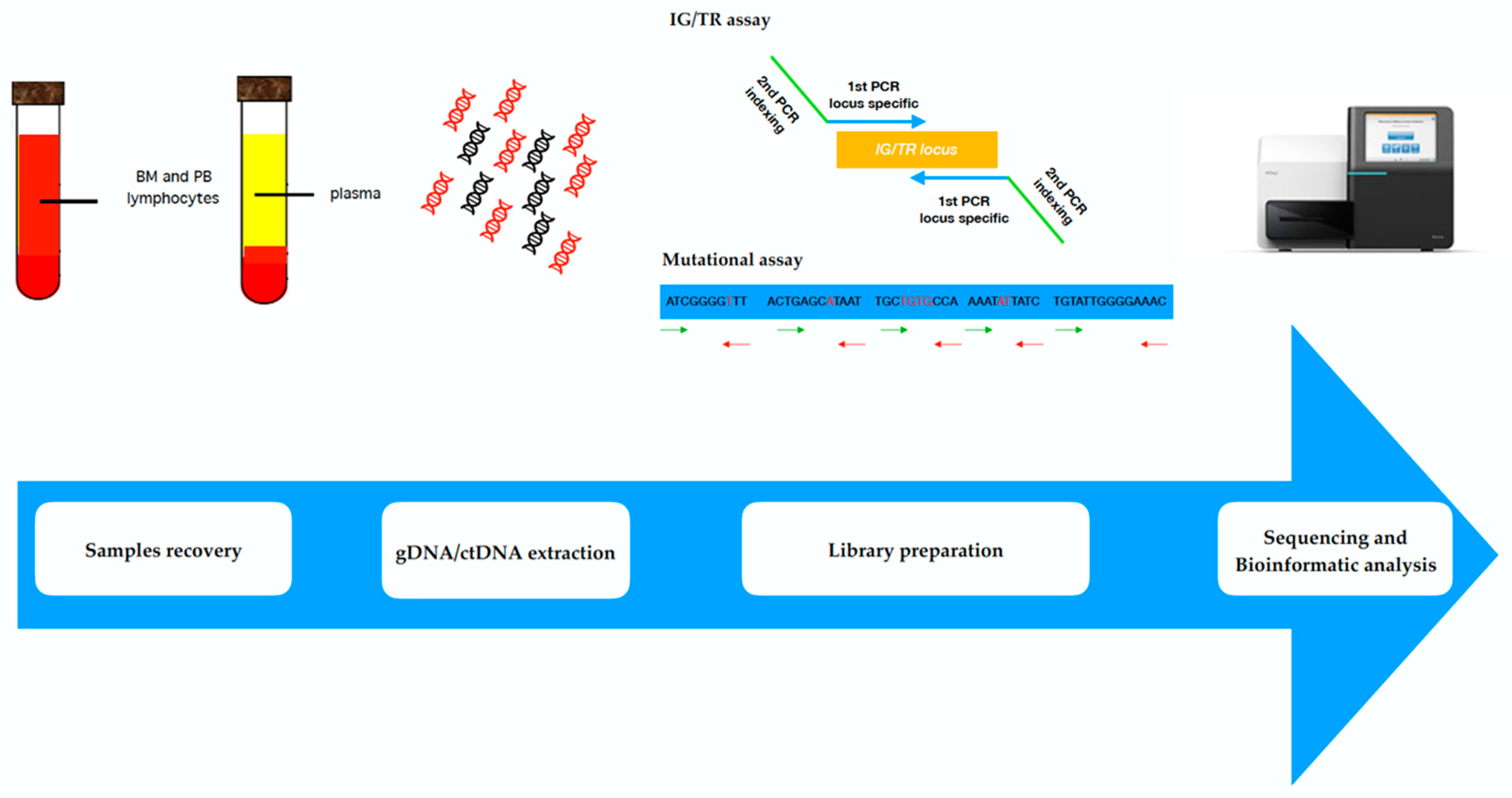
4. Liquid Biopsies: Novel Techniques for ctDNA Detection in Lymphoproliferative Malignancies
5. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Elrich, H. Specific enzymatic amplification of DNA in vitro: The polymerase chain reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Gribben, J.G.; Freedman, A.S.; Neuberg, D.; Roy, D.C.; Blake, K.W.; Woo, S.D.; Grossbard, M.L.; Rabinowe, S.N.; Coral, F.; Freeman, G.J. Immunologic purging of marrow assessed by PCR before autologous bone marrow transplantation for B-cell lymphoma. N. Engl. J. Med. 1991, 325, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Ladetto, M.; Donovan, J.W.; Harig, S.; Trojan, A.; Poor, C.; Schlossnan, R.; Anderson, K.C.; Gribben, J.G. Real-Time polymerase chain reaction of immunoglobulin rearrangements for quantitative evaluation of minimal residual disease in multiple myeloma. Biol. Blood Marrow Transplant. 2000, 6, 241–253. [Google Scholar] [CrossRef]
- Brüggemann, M.; Droese, J.; Bolz, I.; Lüth, P.; Pott, C.; Von Neuhoff, N.; Scheuering, U.; Kneba, M. Improved assessment of minimal residual disease in B cell malignancies using fluorogenic consensus probes for real-time quantitative PCR. Leukemia 2000, 14, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, V.H.; Cazzaniga, G.; Schrauder, A.; Hancock, J.; Bader, P.; Panzer-Grumayer, E.R.; Flohr, T.; Sutton, R.; Cave, H.; Madsen, H.O.; et al. Analysis of minimal residual disease by Ig/TCR gene rearrangements: Guidelines for interpretation of real-time quantitative PCR data. Leukemia 2007, 21, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Pott, C.; Bruggemann, M.; Ritgen, M.; Van der Velden, V.H.; Van Dongen, J.J.; Kneba, M. MRD detection in B-cell non-Hodgkin lymphomas using Ig gene rearrangements and chromosomal translocations as targets for real-time quantitative PCR. Methods Mole. Biol. 2013, 971, 175–200. [Google Scholar]
- Rawstron, A.C.; Child, J.A.; De tute, R.M.; Davies, F.E.; Gregory, W.M.; Bell, S.E.; Szubert, A.J.; Navarro-Coy, N.; Drayson, M.T.; Feyler, S.; et al. Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: Impact on outcome in the Medical Research Council Myeloma IX Study. J. Clin. Oncol. 2013, 31, 2540–2547. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, S.; Ritgen, M.; Fischer, K.; Stilgenbauer, S.; Busch, R.M.; Fingerle-Rowson, G.; Fink, A.M.; Bühler, A.; Zenz, T.; Wenger, M.K.; et al. Minimal residual disease quantification is an independent predictor of progression-free and overall survival in chronic lymphocytic leukemia: A multivariate analysis from the randomized GCLLSG CLL8 trial. J. Clin. Oncol. 2012, 30, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.J.; Van der Velden, V.H.J.; Brüggemann, M.; Orfao, A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: Need for sensitive, fast, and standardized technologies. Blood 2015, 125, 3996–4009. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.M.; Seriu, T.; Panzer-Grümayer, E.R.; Biondi, A.; Pongers-Willemse, M.J.; Corral, L.; Stolz, F.; Schrappe, M.; Masera, G.; Kamps, W.A.; et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 1998, 352, 1731–1738. [Google Scholar] [CrossRef]
- Dogliotti, I.; Ferrero, S. Personalized Medicine in Lymphoma: Tailoring Treatment According to Minimal Residual Disease. Med. Res. Arch. 2017, 5, 1–30. [Google Scholar] [CrossRef]
- Van dongen, J.J.; Langerak, A.W.; Brüggemann, M.; Evans, P.A.; Hummel, M.; Lavender, F.L.; Delabesse, E.; Davi, F.; Schuuring, E.; García-Sanz, R.; et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003, 17, 2257–2317. [Google Scholar] [CrossRef] [PubMed]
- Donovan, J.W.; Ladetto, M.; Zou, G.; Neuberg, D.; Poor, C.; Bowers, D.; Gribben, J.G. Immunoglobulin heavy-chain consensus probes for real-time PCR quantification of residual disease in acute lymphoblastic leukemia. Blood 2000, 95, 2651–2658. [Google Scholar] [PubMed]
- Gonzalez, D.; Garcia-Sanz, R. Incomplete DJH rearrangements. Methods Mol. Med. 2005, 113, 165–173. [Google Scholar] [PubMed]
- González, D.; González, M.; Balanzategui, A.; Sarasquete, M.E.; López-Pérez, R.; Chillón, M.C.; García-Sanz, R.; San Miguel, J.F. Molecular characteristics and gene segment usage in IGH gene rearrangements in multiple myeloma. Haematologica 2005, 90, 906–913. [Google Scholar] [PubMed]
- Scherer, F.; Kurtz, D.M.; Diehn, M.; Alizadeh, A.A. High-throughput sequencing for noninvasive disease detection in hematologic malignancies. Blood 2017, 130, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Della Starza, I.; De Novi, L.A.; Cavalli, M.; Novelli, N.; Genuardi, E.; Mantoan, B.; Drandi, D.; Ferrante, M.; Monitillo, L.; Barbero, D.; et al. Immunoglobulin Kappa Deleting Element (IGK-Kde) Rearrangements as Possible Target for Minimal Residual Disease (MRD) Evaluation in Mantle Cell Lymphoma (MCL). Available online: https://learningcenter.ehaweb.org/eha/2018/stockholm/215093/irene.della.starza.immunoglobulin.kappa.deleting.element.28igk-kde29.html?f=ce_id=1346*ot_id=19042*media=2 (accessed on 20 August 2018).
- Ferrero, S.; Drandi, D.; Mantoan, B.; Ghione, P.; Omedè, P.; Ladetto, M. Minimal residual disease detection in lymphoma and multiple myeloma: Impact on therapeutic paradigms. Hematol. Oncol. 2011, 29, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hunter, Z.R.; Yang, G.; Cao, Y.; Liu, X.; Manning, R.; Tripsas, C.; Chen, J.; Patterson, C.J.; Kluk, M.; et al. Detection of MYD88 L265P in peripheral blood of patients with Waldenstrom’s Macroglobulinemia and IgM monoclonal gammopathy of undetermined significance. Leukemia 2014, 28, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Sebastian, E.; Chillon, M.C.; Giraldo, P.; Mariano Hernández, J.; Escalante, F.; González-López, T.J.; Aguilera, C.; De Coca, A.G.; Murillo, I.; et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenstrom’s macroglobulinemia. Leukemia 2013, 27, 1722–1728. [Google Scholar] [CrossRef] [PubMed]
- Hunter, Z.R.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Manning, R.J.; Tripsas, C.; Patterson, C.J.; Sheehy, P.; et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood 2014, 123, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Stamatoullas, A.; Mareschal, S.; Viailly, P.J.; Sarafan-Vasseur, N.; Bohers, E.; Dubois, S.; Picquenot, J.M.; Ruminy, P.; Maingonnat, C.; et al. Detection and prognostic value of recurrent exportin 1 mutations in tumor and cell-free circulating DNA of patients with classical Hodgkin lymphoma. Haematologica 2016, 101, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Penson, A.; Schiavoni, G.; Ladewig, E.; Fortini, E.; Wang, Y.; Spanhol-Rosseto, A.; Venanzi, A.; Gianni, A.M.; Viviani, S.; et al. New Recurrently Mutated Genes in Classical Hodgkin Lymphoma Revealed BY Whole-Exome Sequencing of Microdissected Tumor Cells. Blood 2016, 128, 1088. [Google Scholar]
- Rossi, D.; Diop, F.; Spaccarotella, E.; Monti, S.; Zanni, M.; Rasi, S.; Deambrogi, C.; Spina, V.; Bruscaggin, A.; Favini, C.; et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood 2017, 129, 1947–1957. [Google Scholar] [CrossRef] [PubMed]
- Rambaldi, A.; Lazzari, M.; Manzoni, C.; Carlotti, E.; Arcaini, L.; Baccarani, M.; Barbui, T.; Bernasconi, C.; Dastoli, G.; Fuga, G.; et al. Monitoring of minimal residual disease after CHOP and rituximab in previously untreated patients with follicular lymphoma. Blood 2002, 99, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Summers, K.E.; Davies, A.J.; Matthews, J.; Jenner, M.J.; Cornelius, V.; Amess, J.A.; Norton, A.J.; Rohatiner, A.Z.; Fitzgibbon, J.; Lister, T.A.; et al. The relative role of peripheral blood and bone marrow for monitoring molecular evidence of disease in follicular lymphoma by quantitative real-time polymerase chain reaction. Br. J. Haematol. 2002, 118, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Ladetto, M.; Lobetti-Bodoni, C.; Mantoan, B.; Ceccarelli, M.; Boccomini, C.; Genuardi, E.; Chiappella, A.; Baldini, L.; Rossi, G.; Pulsoni, A.; et al. Persistence of minimal residual disease in bone marrow predicts outcome in follicular lymphomas treated with a rituximab-intensive program. Blood 2013, 122, 3759–3766. [Google Scholar] [CrossRef] [PubMed]
- Gustine, J.M.K.; Xu, L.; Hunter, Z.R.; Hunter, Z.R.; Castillo, J.J.; Treon, S.P. To select or not to select? The role of B-cell selection in determining the MYD88 mutation status in Waldenström Macroglobulinaemia. Br. J. Haematol. 2017, 176, 822–824. [Google Scholar] [CrossRef] [PubMed]
- Ladetto, M.; Pagliano, G.; Ferrero, S.; Cavallo, F.; Drandi, D.; Santo, L.; Crippa, C.; De Rosa, L.; Pregno, P.; Grasso, M.; et al. Major tumor shrinking and persistent molecular remissions after consolidation with bortezomib, thalidomide, and dexamethasone in patients with autografted myeloma. J. Clin. Oncol. 2010, 28, 2077–2084. [Google Scholar] [CrossRef] [PubMed]
- Oliva, S.; Gambella, M.; Gilestro, M.; Muccio, V.E.; Gay, F.; Drandi, D.; Ferrero, S.; Passera, R.; Pautasso, C.; Bernardini, A.; et al. Minimal residual disease after transplantation or lenalidomide-based consolidation in myeloma patients: A prospective analysis. Oncotarget 2017, 8, 5924. [Google Scholar] [CrossRef] [PubMed]
- Corradini, P.; Ladetto, M.; Pileri, A.; Tarella, C. Clinical relevance of minimal residual disease monitoring in non-Hodgkin’s lymphomas: A critical reappraisal of molecular strategies. Leukemia 1999, 13, 1691–1695. [Google Scholar] [CrossRef] [PubMed]
- Pott, C.; Hoster, E.; Delfau-Larue, M.H.; Beldjord, K.; Böttcher, S.; Asnafi, V.; Plonquet, A.; Siebert, R.; Callet-Bauchu, E.; Andersen, N.; et al. Molecular remission is an independent predictor of clinical outcome in patients with mantle cell lymphoma after combined immunochemotherapy: A European MCL intergroup study. Blood 2010, 115, 3215–3223. [Google Scholar] [CrossRef] [PubMed]
- Geisler, C.H.; Kolstad, A.; Laurell, A.; Andersen, N.S.; Pedersen, L.B.; Jerkeman, M.; Eriksson, M.; Nordström, M.; Kimby, E.; Boesen, A.M.; et al. Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem cell rescue: A nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 2008, 112, 2687–2693. [Google Scholar] [CrossRef] [PubMed]
- Pott, C.; Tiemann, M.; Linke, B.; Ott, M.M.; Von Hofen, M.; Bolz, I.; Hiddemann, W.; Parwaresch, R.; Kneba, M. Structure of Bcl-1 and IgH-CDR3 rearrangements as clonal markers in mantle cell lymphomas. Leukemia 1998, 12, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Kotrova, M.; Van der Velden, V.H.J.; Van Dongen, J.J.M.; Formankova, R.; Sedlacek, P.; Brüggemann, M.; Zuna, J.; Stary, J.; Trka, J.; Fronkova, E. Next-generation sequencing indicates false-positive MRD results and better predicts prognosis after SCT in patients with childhood ALL. Bone Marrow Transplant. 2017, 52, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Brüggemann, M.; Knecht, H.; Bartram, J. International multi-laboratory next-generation sequencing for MRD analysis in ALL. A pilot study by the Euroclonality-NGS Consortium. Haematologica 2015, 100, 31. [Google Scholar]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [PubMed]
- Morley, A.A. Digital PCR: A. brief history. Biomol. Detect. Quantif. 2014, 1, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.; Garson, J.A.; Grant, P.; Nastouli, E.; Foy, C.A.; Huggett, J.F. Comparative study of sensitivity, linearity, and resistance to inhibition of digital and nondigital polymerase chain reaction and loop mediated isothermal amplification assays for quantification of human cytomegalovirus. Anal. Chem. 2014, 86, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- Dingle, T.C.; Sedlak, R.H.; Cook, L.; Jerome, K.R. Tolerance of droplet-digital PCR vs. real-time quantitative PCR to inhibitory substances. Clin. Chem. 2013, 59, 1670–1672. [Google Scholar] [CrossRef] [PubMed]
- Drandi, D.; Ferrero, S.; Ladetto, M. Droplet Digital PCR for Minimal Residual Disease Detection in Mature Lymphoproliferative Disorders. Methods Mol. Biol. 2018, 1768, 229–256. [Google Scholar] [PubMed]
- Drandi, D.; Kubiczkova-Besse, L.; Ferrero, S.; Dani, N.; Passera, R.; Mantoan, B.; Gambella, M.; Monitillo, L.; Saraci, E.; Ghione, P.; et al. Minimal Residual Disease Detection by Droplet Digital PCR in Multiple Myeloma, Mantle Cell Lymphoma, and Follicular Lymphoma: A Comparison with Real-Time PCR. J. Mol. Diagn. 2015, 17, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Della Starza, I.; Nunes, V.; Cavalli, M.; De Novi, L.A.; Ilari, C.; Apicella, V.; Vitale, A.; Testi, A.M.; Del Giudice, I.; Chiaretti, S.; et al. Comparative analysis between RQ-PCR and digital-droplet-PCR of immunoglobulin/T-cell receptor gene rearrangements to monitor minimal residual disease in acute lymphoblastic leukaemia. Br. J. Haematol. 2016, 174, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, M.; De Novi, L.A.; Della Starza, I.; Cappelli, L.V.; Nunes, V.; Pulsoni, A.; Del Giudice, I.; Guarini, A.; Foà, R. Comparative analysis between RQ-PCR and digital droplet PCR of BCL2/IGH gene rearrangement in the peripheral blood and bone marrow of early stage follicular lymphoma. Br. J. Haematol. 2017, 177, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, F.; Paolicchi, M.; Ghio, F.; Ciabatti, E.; Grassi, S.; Salehzadeh, S.; Ercolano, G.; Metelli, M.R.; Del Re, M.; Iovino, L.; et al. The Droplet Digital PCR: A New Valid Molecular Approach for the Assessment of B-RAF V600E Mutation in Hairy Cell Leukemia. Front. Pharmacol. 2016, 7, 363. [Google Scholar] [CrossRef] [PubMed]
- Camus, V.; Sarafan-vasseur, N.; Bohers, E.; Dubois, S.; Mareschal, S.; Bertrand, P.; Viailly, P.J.; Ruminy, P.; Maingonnat, C.; Lemasle, E.; et al. Digital PCR for quantification of recurrent and potentially actionable somatic mutations in circulating free DNA from patients with diffuse large B.-cell lymphoma. Leuk. Lymphoma 2016, 57, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Alikian, M.; Whale, A.S.; Akiki, S.; Piechocki, K.; Torrado, C.; Myint, T.; Cowen, S.; Griffiths, M.; Reid, A.G.; Apperley, J.; et al. RT-qPCR and RT-Digital PCR: A Comparison of Different Platforms for the Evaluation of Residual Disease in Chronic Myeloid Leukemia. Clin. Chem. 2017, 63, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Fava, C.; Gottardi, E.; Berchialla, P.; Rege-Cambrin, G.; Varotto, M.; Daraio, F.; Crasto, F.; Lorenzatti, R.; Volpengo, A.; Fantino, C.; et al. Comparison of Droplet Digital PCR and Standard PCR in Chronic Myeloid Leukemia Patients in MR4. In Proceedings of the 45th Congress of the Italian-Society-of-Hematology, Florence, Italy, 4–7 October 2015; pp. 128–129. [Google Scholar]
- Wang, W.J.; Zheng, C.F.; Liu, Z.; Tan, Y.H.; Chen, X.H.; Zhao, B.L.; Li, G.X.; Xu, Z.F.; Ren, F.G.; Zhang, Y.F.; et al. Droplet Digital PCR for BCR/ABL(P210) Detecting of CML: A High Sensitive Method of the Minimal Residual Disease& Disease Progression. Eur. J. Haematol. 2018, 101, 291–296. [Google Scholar] [PubMed]
- Jennings, L.J.; George, D.; Czech, J.; Yu, M.; Joseph, L. Detection and quantification of BCR-ABL1 fusion transcripts by droplet digital PCR. J. Mol. Diagn. 2014, 16, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Goh, H.G.; Lin, M.; Fukushima, T.; Saglio, G.; Kim, D.; Choi, S.Y.; Kim, S.H.; Lee, J.; Lee, Y.S.; Oh, S.M.; et al. Sensitive quantitation of minimal residual disease in chronic myeloid leukemia using nanofluidic digital polymerase chain reaction assay. Leuk. Lymphoma 2011, 52, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Drandi, D.; Genuardi, E.; Dogliotti, I.; Ferrante, M.; Jiménez, C.; Guerrini, F.; Schirico, M.L.; Mantoan, B.; Muccio, V.; Lia, G.; et al. Highly sensitive MYD88L252P (L265P) mutation detection by droplet digital PCR in Waldenström Macroglobulinemia. Haematologica 2018, 103, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Bea, S.; Valdes-Mas, R.; Navarro, A.; Salaverria, I.; Martín-Garcia, D.; Jares, P.; Giné, E.; Pinyol, M.; Royo, C.; Nadeu, F.; et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18250–18255. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed]
- Ladetto, M.; Bruggemann, M.; Monitillo, L.; Ferrero, S.; Pepin, F.; Drandi, D.; Barbero, D.; Palumbo, A.; Passera, R.; Boccadoro, M.; et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B.-cell disorders. Leukemia 2014, 28, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lopez, J.; Lahuerta, J.J.; Pepin, F.; González, M.; Barrio, S.; Ayala, R.; Puig, N.; Montalban, M.A.; Paiva, B.; Weng, L.; et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood 2014, 123, 3073–3079. [Google Scholar] [CrossRef] [PubMed]
- Gargis, A.S.; Kalman, L.; Berry, M.W.; Bick, D.P.; Dimmock, D.P.; Hambuch, T.; Lu, F.; Lyon, E.; Voelkerding, K.V.; Zehnbauer, B.A.; et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol. 2012, 30, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Pulsipher, M.A.; Carlson, C.; Langholz, B.; Wall, D.A.; Schultz, K.R.; Bunin, N.; Kirsch, I.; Gastier-Foster, J.M.; Borowitz, M.; Desmarais, C.; et al. IgH-V(D)J NGS-MRD measurement pre- and early post-allotransplant defines very low- and very high-risk ALL patients. Blood 2015, 125, 3501–3508. [Google Scholar] [CrossRef] [PubMed]
- Logan, A.C.; Zhang, B.; Narasimhan, B.; Carlton, V.; Zheng, J.; Moorhead, M.; Krampf, M.R.; Jones, C.D.; Waqar, A.N.; Faham, M.; et al. Minimal residual disease quantification using consensus primers and high-throughput IGH sequencing predicts post-transplant relapse in chronic lymphocytic leukemia. Leukemia 2013, 27, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Faham, M.; Zheng, J.; Moorhead, M.; Carlton, V.; Stow, P.; Coustan-Smith, E.; Pui, C.H.; Campana, D. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood 2012, 120, 5173–5180. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Pepin, F.; Carlton, V.E.; Klinger, M.; Logan, A.C.; Miklos, D.B.; Faham, M.; Dahl, G.; Lacayo, N. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood 2012, 120, 4407–4417. [Google Scholar] [CrossRef] [PubMed]
- Logan, A.C.; Vashi, N.; Faham, M.; Carlton, V.; Kong, K.; Buño, I.; Zheng, J.; Moorhead, M.; Klinger, M.; Zhang, B.; et al. Immunoglobulin and T cell receptor gene high-throughput sequencing quantifies minimal residual disease in acute lymphoblastic leukemia and predicts post-transplantation relapse and survival. Biol. Blood Marrow Transplant. 2014, 20, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Mannis, G.N.; Martin, T.G.; Damon, L.E.; Andreadis, C.; Olin, R.L.; Kong, K.A.; Faham, M.; Hwang, J.; Ai, W.Z.; Gaensler, K.M.L.; et al. Quantification of Acute Lymphoblastic Leukemia Clonotypes in Leukapheresed Peripheral Blood Progenitor Cells Predicts Relapse Risk after Autologous Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2016, 22, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, Y.; Xu, Y.; Muramatsu, H.; Okuno, Y.; Narita, A.; Suzuki, K.; Wang, X.; Kawashima, N.; Sakaguchi, H.; Yoshida, N.; et al. Clinical utility of next-generation sequencing-based minimal residual disease in paediatric B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2017, 176, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Sala torra, O.; Othus, M.; Williamson, D.W.; Wood, B.; Kirsch, I.; Robins, H.; Beppu, L.; O’Donnell, M.R.; Forman, S.J.; Appelbaum, F.R.; et al. Next-Generation Sequencing in Adult B Cell Acute Lymphoblastic Leukemia Patients. Biol. Blood Marrow Transplant. 2017, 23, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.F.; Kim, H.T.; Kong, K.A.; Faham, M.; Sun, H.; Sohani, A.R.; Alyea, E.P.; Carlton, V.E.; Chen, Y.B.; Cutler, C.S.; et al. Next-generation sequencing-based detection of circulating tumour DNA After allogeneic stem cell transplantation for lymphoma. Br. J. Haematol. 2016, 175, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Logan, A.C.; Gao, H.; Wang, C.; Sahaf, B.; Jones, C.D.; Marshall, E.L.; Buño, I.; Armstrong, R.; Fire, A.Z.; Weinberg, K.I.; et al. High-throughput VDJ sequencing for quantification of minimal residual disease in chronic lymphocytic leukemia and immune reconstitution assessment. Proc. Natl. Acad. Sci. USA 2011, 108, 21194–21199. [Google Scholar] [CrossRef] [PubMed]
- Rawstron, A.C.; Fazi, C.; Agathangelidis, A.; Villamor, N.; Letestu, R.; Nomdedeu, J.; Palacio, C.; Stehlikova, O.; Kreuzer, K.A.; Liptrot, S.; et al. A complementary role of multiparameter flow cytometry and high-throughput sequencing for minimal residual disease detection in chronic lymphocytic leukemia: An European Research Initiative on CLL study. Leukemia 2016, 30, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Vij, R.; Mazumder, A.; Klinger, M.; O’Dea, D.; Paasch, J.; Martin, T.; Weng, L.; Park, J.; Fiala, M.; Faham, M.; et al. Deep sequencing reveals myeloma cells in peripheral blood in majority of multiple myeloma patients. Clin. Lymphoma Myeloma Leuk. 2014, 14, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Korde, N.; Roschewski, M.; Zingone, A.; Kwok, M.; Manasanch, E.E.; Bhutani, M.; Tageja, N.; Kazandjian, D.; Mailankody, S.; Wu, P.; et al. Treatment with Carfilzomib-Lenalidomide-Dexamethasone with Lenalidomide Extension in Patients with Smoldering or Newly Diagnosed Multiple Myeloma. JAMA Oncol. 2015, 1, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Suvannasankha, A.; Fay, J.W.; Arnulf, B.; Kaufman, J.L.; Ifthikharuddin, J.J.; Weiss, B.M.; Krishnan, A.; Lentzsch, S.; Comenzo, R.; et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood 2017, 130, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Orfao, A.; Chim, C.S. Molecular detection of minimal residual disease in multiple myeloma. Br. J. Haematol. 2018, 181, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Bartram, J.; Mountjoy, E.; Brooks, T.; Hancock, J.; Williamson, H.; Wright, G.; Moppett, J.; Goulden, N.; Hubank, M. Accurate Sample Assignment in a Multiplexed, Ultrasensitive, High-Throughput Sequencing Assay for Minimal Residual Disease. J. Mol. Diagn. 2016, 18, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Langerak, A.W.; Brüggemann, M.; Davi, F.; Darzentas, N.; Van Dongen, J.J.M.; Gonzalez, D.; Cazzaniga, G.; Giudicelli, V.; Lefranc, M.P.; Giraud, M.; et al. High-Throughput Immunogenetics for Clinical and Research Applications in Immunohematology: Potential and Challenges. J. Immunol. 2017, 198, 3765–3774. [Google Scholar] [CrossRef] [PubMed]
- Bystry, V.; Reigl, T.; Krejci, A.; Demko, M.; Hanakova, B.; Grioni, A.; Knecht, H.; Schlitt, M.; Dreger, P.; Sellner, L.; et al. ARResT/Interrogate: An interactive immunoprofiler for IG/TR NGS data. Bioinformatics 2017, 33, 435–437. [Google Scholar] [CrossRef] [PubMed]
- Giraud, M.; Salson, M.; Duez, M.; Villenet, C.; Quief, S.; Caillault, A.; Grardel, N.; Roumier, C.; Preudhomme, C.; Figeac, M. Fast multiclonal clusterization of V(D)J recombinations from high-throughput sequencing. BMC Genom. 2014, 15, 409. [Google Scholar] [CrossRef] [PubMed]
- Kotrova, M.; Muzikova, K.; Mejstrikova, E.; Novakova, M.; Bakardjieva-Mihaylova, V.; Fiser, K.; Stuchly, J.; Giraud, M.; Salson, M.; Pott, C.; et al. The predictive strength of next-generation sequencing MRD detection for relapse compared with current methods in childhood ALL. Blood 2015, 126, 1045–1047. [Google Scholar] [CrossRef] [PubMed]
- Pott, C.; Knecht, H.; Herzog, A.; Genuardi, E.; Unterhalt, M.; Mantoan, B.; Dogliotti, I.; Hiddemann, W.; Boccadoro, M.; Herrmann, D.; et al. Standardized IGH-Based Next-Generation Sequencing for MRD Detection in Follicular Lymphoma. Blood 2017, 130, 1491. [Google Scholar]
- Bolotin, D.A.; Shugay, M.; Mamedov, I.Z.; Putintseva, E.V.; Turchaninova, M.A.; Zvyagin, I.V.; Britanova, O.V.; Chudakov, D.M. MiTCR: Software for T-cell receptor sequencing data analysis. Nat. Methods 2013, 10, 813–814. [Google Scholar] [CrossRef] [PubMed]
- Aouinti, S.; Giudicelli, V.; Duroux, P.; Malouche, D.; Kossida, S.; Lefranc, M.P. IMGT/StatClonotype for Pairwise Evaluation and Visualization of NGS IG and TR IMGT Clonotype (AA) Diversity or Expression from IMGT/HighV-QUEST. Front. Immunol. 2016, 7, 339. [Google Scholar] [CrossRef] [PubMed]
- Duez, M.; Giraud, M.; Herbert, R.; Rocher, T.; Salson, M.; Thonier, F. Vidjil: A Web Platform for Analysis of High-Throughput Repertoire Sequencing. PLoS ONE 2016, 11, e0166126. [Google Scholar] [CrossRef] [PubMed]
- Bystry, V.; Agathangelidis, A.; Bikos, V.; Sutton, L.A.; Baliakas, P.; Hadzidimitriou, A.; Stamatopoulos, K.; Darzentas, N. ARResT/AssignSubsets: A novel application for robust subclassification of chronic lymphocytic leukemia based on B cell receptor IG stereotypy. Bioinformatics 2015, 31, 3844–3846. [Google Scholar] [CrossRef] [PubMed]
- Beccuti, M.; Genuardi, E.; Romano, G.; Monitillo, L.; Barbero, D.; Boccadoro, M.; Ladetto, M.; Calogero, R.; Ferrero, S.; Cordero, F. HashClone: A new tool to quantify the minimal residual disease in B-cell lymphoma from deep sequencing data. BMC Bioinform. 2017, 18, 516. [Google Scholar] [CrossRef] [PubMed]
- Benichou, J.; Ben-hamo, R.; Louzoun, Y.; Efroni, S. Rep-Seq: Uncovering the immunological repertoire through next-generation sequencing. Immunology 2012, 135, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Rustad, E.H.; Coward, E.; Skytoen, E.R.; Misund, K.; Holien, T.; Standal, T.; Børset, M.; Beisvag, V.; Myklebost, O.; Meza-Zepeda, L.; et al. Monitoring multiple myeloma by quantification of recurrent mutations in serum. Haematologica 2017, 102, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Dynamic monitoring of circulating tumor DNA in non-Hodgkin lymphoma. Blood 2016, 127, 3127–3132. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, D.M.; Green, M.R.; Bratman, S.V.; Scherer, F.; Liu, C.L.; Kunder, C.A.; Takahashi, K.; Glover, C.; Keane, C.; Kihira, S.; et al. Noninvasive monitoring of diffuse large B-cell lymphoma by immunoglobulin high-throughput sequencing. Blood 2015, 125, 3679–3687. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Oki, Y.; Neuberg, D.S.; Faham, M.; Cummings, C.; Klinger, M.; Weng, L.; Bhattar, S.; Lacasce, A.S.; Jacobsen, E.D.; et al. Detection of circulating tumour DNA in patients with aggressive B-cell non-Hodgkin lymphoma. Br. J. Haematol. 2013, 163, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Oberle, A.; Brandt, A.; Voigtlaender, M.; Thiele, B.; Radloff, J.; Schulenkorf, A.; Alawi, M.; Akyüz, N.; März, M.; Ford, C.T.; et al. Monitoring multiple myeloma by next-generation sequencing of V(D)J rearrangements from circulating myeloma cells and cell-free myeloma DNA. Haematologica 2017, 102, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Roschewski, M.; Dunleavy, K.; Pittaluga, S.; Moorhead, M.; Pepin, F.; Kong, K.; Shovlin, M.; Jaffe, E.S.; Staudt, L.M.; Lai, C.; et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: A correlative biomarker study. Lancet Oncol. 2015, 16, 541–549. [Google Scholar] [CrossRef]
- Oki, Y.; Neelapu, S.S.; Fanale, M.; Kwak, L.W.; Fayad, L.; Rodriguez, M.A.; Wallace, M.; Klinger, M.; Carlton, V.; Kong, K.; et al. Detection of classical Hodgkin lymphoma specific sequence in peripheral blood using a next-generation sequencing approach. Br. J. Haematol. 2015, 169, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, C.; Huet, S.; Carlton, V.E.; Fabiani, B.; Delmer, A.; Jardin, F.; Delfau-Larue, M.H.; Hacini, M.; Ribrag, V.; Guidez, S.; et al. The prognostic value of clonal heterogeneity and quantitative assessment of plasma circulating clonal IG-VDJ sequences at diagnosis in patients with follicular lymphoma. Oncotarget 2017, 8, 8765–8774. [Google Scholar] [CrossRef] [PubMed]
- Fontanilles, M.; Marguet, F.; Bohers, E.; Viailly, P.J.; Dubois, S.; Bertrand, P.; Camus, V.; Mareschal, S.; Ruminy, P.; Maingonnat, C.; et al. Non-invasive detection of somatic mutations using next-generation sequencing in primary central nervous system lymphoma. Oncotarget 2017, 8, 48157–48168. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Scherer, F.; Kurtz, D.M.; Newman, A.M.; Stehr, H.; Craig, A.F.; Esfahani, M.S.; Lovejoy, A.F.; Chabon, J.J.; Klass, D.M.; Liu, C.L.; et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci. Transl. Med. 2016, 8, 364ra155. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, D.M.; Jin, M.; Soo, J. Circulating Tumor DNA Is a Reliable Measure of Tumor Burden at Diagnosis of Diffuse Large B Cell Lymphoma: An International Reproducibility Study. Blood 2017, 130, 310. [Google Scholar]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Di Trani, M.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood 2018, 131, 2413–2425. [Google Scholar] [CrossRef] [PubMed]
- Van Ginkel, J.H.; Van den Broek, D.A.; Van Kuik, J.; Linders, D.; De Weger, R.; Willems, S.M.; Huibers, M.M.H. Preanalytical blood sample workup for cell-free DNA analysis using Droplet Digital PCR for future molecular cancer diagnostics. Cancer Med. 2017, 6, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- El Messaoudi, S.; Rolet, F.; Mouliere, F.; Thierry, A.R. Circulating cell free DNA: Preanalytical considerations. Clin. Chim. Acta 2013, 424, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating Tumor DNA as a Liquid Biopsy for Cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Henry, N.L.; Paoletti, C.; Jiang, H.; Vats, P.; Chinnaiyan, A.M.; Hayes, D.F.; Merajver, S.D.; Rae, J.M.; Tewari, M. Comparative analysis of circulating tumor DNA stability In K3EDTA, Streck, and CellSave blood collection tubes. Clin. Biochem. 2016, 49, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Van Dongen, J.J.M.; Orfao, A. New criteria for response assessment: Role of minimal residual disease in multiple myeloma. Blood 2015, 125, 3059–3068. [Google Scholar] [CrossRef] [PubMed]
- Van dongen, J.J.; Orfao, A. EuroFlow: Resetting leukemia and lymphoma immunophenotyping. Basis for companion diagnostics and personalized medicine. Leukemia 2012, 26, 1899–1907. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Technique | Approach | Sensitivity | Advantages | Disadvantages |
|---|---|---|---|---|
| Droplet digital PCR | Targeted analysis | Up to 5.00 × 10−5 [23,47,53] | Absolute quantification of target (no need of standard curve) | Discovery of unknown mutations not possible |
| High precision measure even at low concentration targets | Not able to overcome the limitation of allele-specific design | |||
| NGS | (a) Multiple targets; (b) Whole genome; (c) Whole exome | Up to 1.00 × 10−4 [25,101] | Potentially highly sensitive | Long turn-around time, not fully standardized |
| Allows discovery approach | Bionformatic tools and expert personnel needed | |||
| No need for patient-specific reagents | Expensive |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dogliotti, I.; Drandi, D.; Genuardi, E.; Ferrero, S. New Molecular Technologies for Minimal Residual Disease Evaluation in B-Cell Lymphoid Malignancies. J. Clin. Med. 2018, 7, 288. https://doi.org/10.3390/jcm7090288
Dogliotti I, Drandi D, Genuardi E, Ferrero S. New Molecular Technologies for Minimal Residual Disease Evaluation in B-Cell Lymphoid Malignancies. Journal of Clinical Medicine. 2018; 7(9):288. https://doi.org/10.3390/jcm7090288
Chicago/Turabian StyleDogliotti, Irene, Daniela Drandi, Elisa Genuardi, and Simone Ferrero. 2018. "New Molecular Technologies for Minimal Residual Disease Evaluation in B-Cell Lymphoid Malignancies" Journal of Clinical Medicine 7, no. 9: 288. https://doi.org/10.3390/jcm7090288
APA StyleDogliotti, I., Drandi, D., Genuardi, E., & Ferrero, S. (2018). New Molecular Technologies for Minimal Residual Disease Evaluation in B-Cell Lymphoid Malignancies. Journal of Clinical Medicine, 7(9), 288. https://doi.org/10.3390/jcm7090288