Traumatic Brain Injury: At the Crossroads of Neuropathology and Common Metabolic Endocrinopathies


{kind=link}
Abstract
1. Introduction
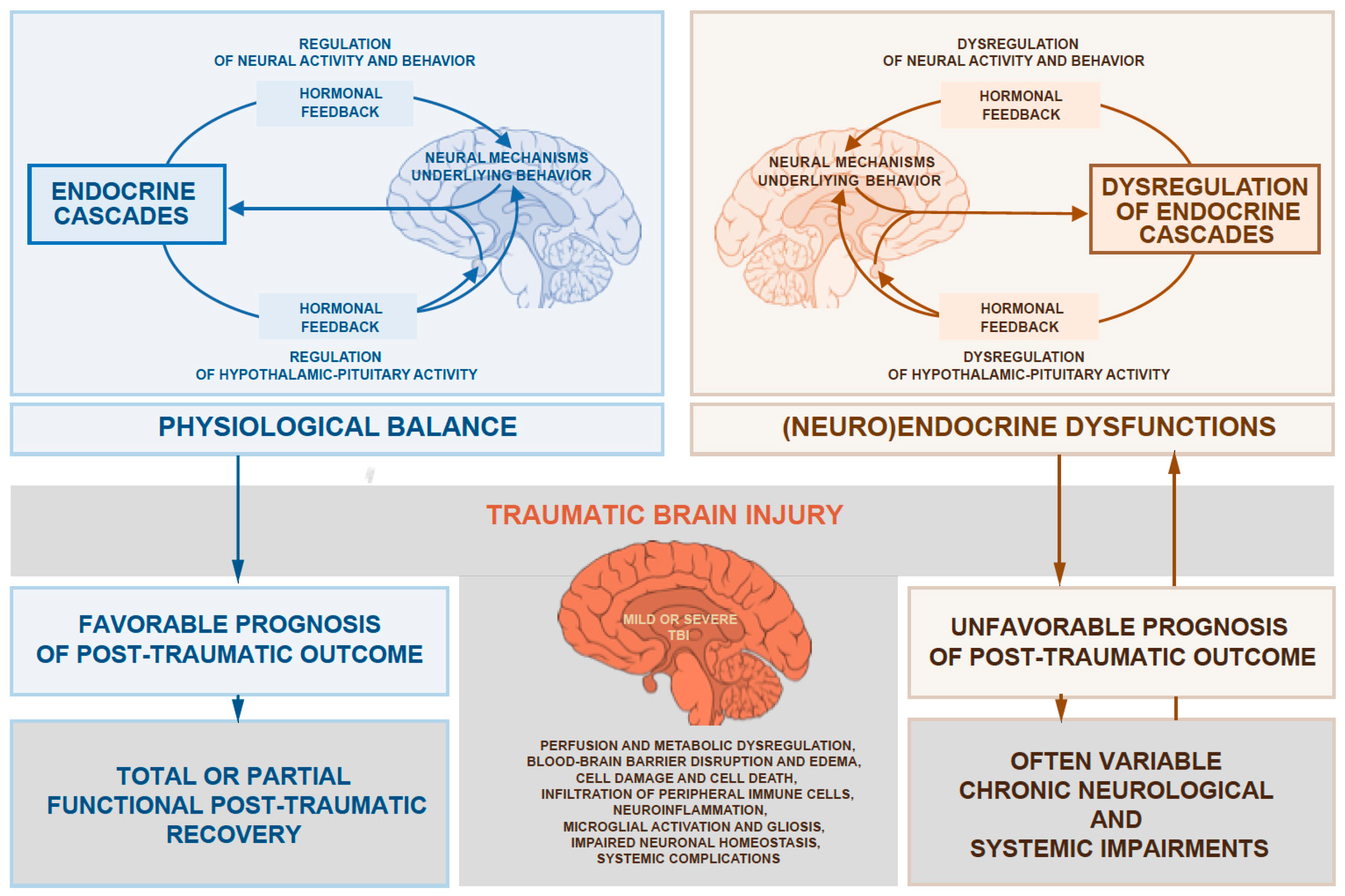
2. All Ranges of TBI Can Cause Significant Hormonal Dysfunction
3. Systemic Endocrine Disorders Can Impact the Outcome of TBI
3.1. Glucose Metabolism
3.1.1. How Insulin Affects the Brain
3.1.2. Hyperglycemia as a Predictor of Mortality Following TBI
3.1.3. Diabetes Mellitus as a Risk Factor for Higher TBI Mortality
3.2. Obesity
3.2.1. Obesity Causes Chronic Low-Grade Systemic Immunological Dysfunction
3.2.2. Obesity Can Cause Hypothalamic and Diffuse Brain Inflammation
3.2.3. Consequences of Obesity on TBI Outcomes
3.2.4. The Metabolic Syndrome
3.3. Thyroid Dysfunction
3.3.1. Neuroprotective Capacities of Thyroid Hormone in Post-Traumatic CNS
3.3.2. Low Thyroid Hormone Levels Correlate with Bad TBI Outcomes in the Critically Ill
4. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lechan, R.M.; Toni, R. Functional Anatomy of the Hypothalamus and Pituitary. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2016. [Google Scholar]
- Quiring, D.P. Transplantation of testes (by A. A. Berthold). Bull. Hist. Med. 1944, 16, 399–401. [Google Scholar]
- Harris, G.W. Effects of the nervous system on the pituitary-adrenal activity. Prog. Brain Res. 1970, 32, 86–88. [Google Scholar] [PubMed]
- Harris, G.W.; Naftolin, F. The hypothalamus and control of ovulation. Br. Med. Bull. 1970, 26, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Schunke, M.; Schulte, E.; Schumacher, U. Prometheus Lernatlas der Anatomie; Thieme: Stuttgart, Germany, 2015; Volume 2. [Google Scholar]
- Wijayatilake, D.S.; Sherren, P.B.; Jigajinni, S.V. Systemic complications of traumatic brain injury. Curr. Opin. Anaesthesiol. 2015, 28, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Bondanelli, M.; De Marinis, L.; Ambrosio, M.R.; Monesi, M.; Valle, D.; Zatelli, M.C.; Fusco, A.; Bianchi, A.; Farneti, M.; degli Uberti, E.C. Occurrence of pituitary dysfunction following traumatic brain injury. J. Neurotrauma 2004, 21, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, F.; Senyurek, H.; Unluhizarci, K.; Selcuklu, A.; Casanueva, F.F.; Kelestimur, F. High risk of hypopituitarism after traumatic brain injury: A prospective investigation of anterior pituitary function in the acute phase and 12 months after trauma. J. Clin. Endocrinol. Metab. 2006, 91, 2105–2111. [Google Scholar] [CrossRef] [PubMed]
- Peeters, W.; van den Brande, R.; Polinder, S.; Brazinova, A.; Steyerberg, E.W.; Lingsma, H.F.; Maas, A.I. Epidemiology of traumatic brain injury in Europe. Acta Neurochir. (Wien) 2015, 157, 1683–1696. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Lawrence, D.W. Top-cited articles in traumatic brain injury. Front. Hum. Neurosci. 2014, 8, 879. [Google Scholar] [CrossRef] [PubMed]
- Margulies, S.; Hicks, R.; Combination Therapies for Traumatic Brain Injury Workshop Leaders. Combination therapies for traumatic brain injury: Prospective considerations. J. Neurotrauma 2009, 26, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Zygun, D.A.; Kortbeek, J.B.; Fick, G.H.; Laupland, K.B.; Doig, C.J. Non-neurologic organ dysfunction in severe traumatic brain injury. Crit. Care Med. 2005, 33, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Byrnes, K.R. Role of microglia in neurotrauma. Neurotherapeutics 2010, 7, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Mendelow, D.A.; Crawford, P.J. Primary and secondary brain injury. In Head Injury; Reilly, P., Bullock, R., Eds.; Chapman and Hall: London, UK, 1997; pp. 71–88. [Google Scholar]
- English, S.W.; Turgeon, A.F.; Owen, E.; Doucette, S.; Pagliarello, G.; McIntyre, L. Protocol management of severe traumatic brain injury in intensive care units: A systematic review. Neurocrit. Care 2013, 18, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Hackenberg, K.; Unterberg, A. Traumatic brain injury. Nervenarzt 2016, 87, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Teasdale, G.; Jennett, B. Assessment of coma and impaired consciousness. A practical scale. Lancet 1974, 2, 81–84. [Google Scholar] [CrossRef]
- Marshall, L.F.; Marshall, S.B.; Klauber, M.R.; Van Berkum Clark, M.; Eisenberg, H.; Jane, J.A.; Luerssen, T.G.; Marmarou, A.; Foulkes, M.A. The diagnosis of head injury requires a classification based on computed axial tomography. J. Neurotrauma 1992, 9 (Suppl. 1), S287–S292. [Google Scholar] [PubMed]
- Zetterberg, H.; Blennow, K. Fluid markers of traumatic brain injury. Mol. Cell. Neurosci. 2015, 66, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Nellgard, B. Amyloid beta 1-42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology 2004, 62, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Neselius, S.; Brisby, H.; Theodorsson, A.; Blennow, K.; Zetterberg, H.; Marcusson, J. CSF-biomarkers in olympic boxing: Diagnosis and effects of repetitive head trauma. PLoS ONE 2012, 7, e33606. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Zetterberg, H.; Hampel, H.; Blennow, K. Biomarker-based dissection of neurodegenerative diseases. Prog. Neurobiol. 2011, 95, 520–534. [Google Scholar] [CrossRef] [PubMed]
- Nylen, K.; Ost, M.; Csajbok, L.Z.; Nilsson, I.; Hall, C.; Blennow, K.; Nellgard, B.; Rosengren, L. Serum levels of S100B, S100A1B and S100BB are all related to outcome after severe traumatic brain injury. Acta Neurochir. (Wien) 2008, 150, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Czeiter, E.; Mondello, S.; Kovacs, N.; Sandor, J.; Gabrielli, A.; Schmid, K.; Tortella, F.; Wang, K.K.; Hayes, R.L.; Barzo, P.; et al. Brain injury biomarkers may improve the predictive power of the impact outcome calculator. J. Neurotrauma 2012, 29, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Mondello, S.; Muller, U.; Jeromin, A.; Streeter, J.; Hayes, R.L.; Wang, K.K. Blood-based diagnostics of traumatic brain injuries. Expert Rev. Mol. Diagn. 2011, 11, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Mondello, S.; Robicsek, S.A.; Gabrielli, A.; Brophy, G.M.; Papa, L.; Tepas, J.; Robertson, C.; Buki, A.; Scharf, D.; Jixiang, M.; et al. Alphaii-spectrin breakdown products (SBDPs): Diagnosis and outcome in severe traumatic brain injury patients. J. Neurotrauma 2010, 27, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Smith, D.H.; Blennow, K. Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nat. Rev. Neurol. 2013, 9, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Gaddam, S.S.; Buell, T.; Robertson, C.S. Systemic manifestations of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 205–218. [Google Scholar] [PubMed]
- Schneider, H.J.; Schneider, M.; Saller, B.; Petersenn, S.; Uhr, M.; Husemann, B.; von Rosen, F.; Stalla, G.K. Prevalence of anterior pituitary insufficiency 3 and 12 months after traumatic brain injury. Eur. J. Endocrinol. 2006, 154, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Estes, S.M.; Urban, R.J. Hormonal replacement in patients with brain injury-induced hypopituitarism: Who, when and how to treat? Pituitary 2005, 8, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, E.; Masel, B.; Aimaretti, G.; Leon-Carrion, J.; Casanueva, F.F.; Dominguez-Morales, M.R.; Elovic, E.; Perrone, K.; Stalla, G.; Thompson, C.; et al. Consensus guidelines on screening for hypopituitarism following traumatic brain injury. Brain Inj. 2005, 19, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Menon, D.K.; Ercole, A. Critical care management of traumatic brain injury. Handb. Clin. Neurol. 2017, 140, 239–274. [Google Scholar] [PubMed]
- Prabhakar, V.K.; Shalet, S.M. Aetiology, diagnosis, and management of hypopituitarism in adult life. Postgrad. Med. J. 2006, 82, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Higham, C.E.; Johannsson, G.; Shalet, S.M. Hypopituitarism. Lancet 2016, 388, 2403–2415. [Google Scholar] [CrossRef]
- Pappachan, J.M.; Raskauskiene, D.; Kutty, V.R.; Clayton, R.N. Excess mortality associated with hypopituitarism in adults: A meta-analysis of observational studies. J. Clin. Endocrinol. Metab. 2015, 100, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Chemaitilly, W.; Li, Z.; Huang, S.; Ness, K.K.; Clark, K.L.; Green, D.M.; Barnes, N.; Armstrong, G.T.; Krasin, M.J.; Srivastava, D.K.; et al. Anterior hypopituitarism in adult survivors of childhood cancers treated with cranial radiotherapy: A report from the ST jude lifetime cohort study. J. Clin. Oncol. 2015, 33, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Cyran, E. Hypophysenschädigung durch schädelbasisfraktur. Deutsche Medizinische Wochenschrift 1918, 44, 1261. [Google Scholar]
- Behan, L.A.; Phillips, J.; Thompson, C.J.; Agha, A. Neuroendocrine disorders after traumatic brain injury. J. Neurol. Neurosurg. Psychiatry 2008, 79, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, F.; Kelestimur, F. Pituitary dysfunction following traumatic brain injury: Clinical perspectives. Neuropsychiatr. Dis. Treat. 2015, 11, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- Kokshoorn, N.E.; Wassenaar, M.J.; Biermasz, N.R.; Roelfsema, F.; Smit, J.W.; Romijn, J.A.; Pereira, A.M. Hypopituitarism following traumatic brain injury: Prevalence is affected by the use of different dynamic tests and different normal values. Eur. J. Endocrinol. 2010, 162, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Pekic, S.; Popovic, V. Diagnosis of endocrine disease: Expanding the cause of hypopituitarism. Eur. J. Endocrinol. 2017, 176, R269–R282. [Google Scholar] [CrossRef] [PubMed]
- Bazarian, J.J.; McClung, J.; Shah, M.N.; Cheng, Y.T.; Flesher, W.; Kraus, J. Mild traumatic brain injury in the United States, 1998–2000. Brain Inj. 2005, 19, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Langlois, J.A.; Rutland-Brown, W.; Wald, M.M. The epidemiology and impact of traumatic brain injury: A brief overview. J. Head Trauma Rehabil. 2006, 21, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.J.; Kreitschmann-Andermahr, I.; Ghigo, E.; Stalla, G.K.; Agha, A. Hypothalamopituitary dysfunction following traumatic brain injury and aneurysmal subarachnoid hemorrhage: A systematic review. JAMA 2007, 298, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Report to Congress on Mild Traumativ Brain Injury in the United States: Steps to Prevent a Serious Public Health Problem; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2003.
- American Congress of Rehabilitation Medicine. Definition of Mild Traumatic Brain Injury; American Congress of Rehabilitation Medicine: Reston, VA, USA, 1993. [Google Scholar]
- Tanriverdi, F.; Unluhizarci, K.; Coksevim, B.; Selcuklu, A.; Casanueva, F.F.; Kelestimur, F. Kickboxing sport as a new cause of traumatic brain injury-mediated hypopituitarism. Clin. Endocrinol. (Oxf.) 2007, 66, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Tanriverdi, F.; Unluhizarci, K.; Kocyigit, I.; Tuna, I.S.; Karaca, Z.; Durak, A.C.; Selcuklu, A.; Casanueva, F.F.; Kelestimur, F. Brief communication: Pituitary volume and function in competing and retired male boxers. Ann. Intern. Med. 2008, 148, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Kelestimur, F.; Tanriverdi, F.; Atmaca, H.; Unluhizarci, K.; Selcuklu, A.; Casanueva, F.F. Boxing as a sport activity associated with isolated GH deficiency. J. Endocrinol. Investig. 2004, 27, rc28–rc32. [Google Scholar] [CrossRef] [PubMed]
- Ives, J.C.; Alderman, M.; Stred, S.E. Hypopituitarism after multiple concussions: A retrospective case study in an adolescent male. J. Athl. Train. 2007, 42, 431–439. [Google Scholar] [PubMed]
- Tanriverdi, F.; Unluhizarci, K.; Kelestimur, F. Pituitary function in subjects with mild traumatic brain injury: A review of literature and proposal of a screening strategy. Pituitary 2010, 13, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Agha, A.; Rogers, B.; Mylotte, D.; Taleb, F.; Tormey, W.; Phillips, J.; Thompson, C.J. Neuroendocrine dysfunction in the acute phase of traumatic brain injury. Clin. Endocrinol. (Oxf.) 2004, 60, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Bavisetty, S.; Bavisetty, S.; McArthur, D.L.; Dusick, J.R.; Wang, C.; Cohan, P.; Boscardin, W.J.; Swerdloff, R.; Levin, H.; Chang, D.J.; et al. Chronic hypopituitarism after traumatic brain injury: Risk assessment and relationship to outcome. Neurosurgery 2008, 62, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, S.A.; Oberoi, A.L.; Gilkison, C.R.; Masel, B.E.; Urban, R.J. Prevalence of neuroendocrine dysfunction in patients recovering from traumatic brain injury. J. Clin. Endocrinol. Metab. 2001, 86, 2752–2756. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Schneider, H.J.; Yassouridis, A.; Saller, B.; von Rosen, F.; Stalla, G.K. Predictors of anterior pituitary insufficiency after traumatic brain injury. Clin. Endocrinol. (Oxf.) 2008, 68, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.; Juul, A.; Poulsgaard, L.; Kosteljanetz, M.; Brennum, J.; Feldt-Rasmussen, U. Prevalence and predictive factors of post-traumatic hypopituitarism. Clin. Endocrinol. (Oxf.) 2007, 67, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Dusick, J.R.; Wang, C.; Cohan, P.; Swerdloff, R.; Kelly, D.F. Pathophysiology of hypopituitarism in the setting of brain injury. Pituitary 2012, 15, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Hellawell, D.J.; Taylor, R.T.; Pentland, B. Cognitive and psychosocial outcome following moderate or severe traumatic brain injury. Brain Inj. 1999, 13, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.F.; McArthur, D.L.; Levin, H.; Swimmer, S.; Dusick, J.R.; Cohan, P.; Wang, C.; Swerdloff, R. Neurobehavioral and quality of life changes associated with growth hormone insufficiency after complicated mild, moderate, or severe traumatic brain injury. J. Neurotrauma 2006, 23, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Englander, J.; Bushnik, T.; Oggins, J.; Katznelson, L. Fatigue after traumatic brain injury: Association with neuroendocrine, sleep, depression and other factors. Brain Inj. 2010, 24, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Verweij, B.H.; Muizelaar, J.P.; Vinas, F.C.; Peterson, P.L.; Xiong, Y.; Lee, C.P. Impaired cerebral mitochondrial function after traumatic brain injury in humans. J. Neurosurg. 2000, 93, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Fiskum, G. Mitochondrial participation in ischemic and traumatic neural cell death. J. Neurotrauma 2000, 17, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Glenn, T.C.; Kelly, D.F.; Boscardin, W.J.; McArthur, D.L.; Vespa, P.; Oertel, M.; Hovda, D.A.; Bergsneider, M.; Hillered, L.; Martin, N.A. Energy dysfunction as a predictor of outcome after moderate or severe head injury: Indices of oxygen, glucose, and lactate metabolism. J. Cereb. Blood Flow Metab. 2003, 23, 1239–1250. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Bingham, E.M.; Hopkins, D.; Smith, D.; Pernet, A.; Hallett, W.; Reed, L.; Marsden, P.K.; Amiel, S.A. The role of insulin in human brain glucose metabolism: An 18fluoro-deoxyglucose positron emission tomography study. Diabetes 2002, 51, 3384–3390. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.M.; Meijer, R.I.; Barrett, E.J. Insulin regulates brain function, but how does it get there? Diabetes 2014, 63, 3992–3997. [Google Scholar] [CrossRef] [PubMed]
- Hausen, A.C.; Ruud, J.; Jiang, H.; Hess, S.; Varbanov, H.; Kloppenburg, P.; Bruning, J.C. Insulin-dependent activation of MCH neurons impairs locomotor activity and insulin sensitivity in obesity. Cell Rep. 2016, 17, 2512–2521. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Caceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.X.; et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Konner, A.C.; Hess, S.; Tovar, S.; Mesaros, A.; Sanchez-Lasheras, C.; Evers, N.; Verhagen, L.A.; Bronneke, H.S.; Kleinridders, A.; Hampel, B.; et al. Role for insulin signaling in catecholaminergic neurons in control of energy homeostasis. Cell Metab. 2011, 13, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Guthoff, M.; Grichisch, Y.; Canova, C.; Tschritter, O.; Veit, R.; Hallschmid, M.; Haring, H.U.; Preissl, H.; Hennige, A.M.; Fritsche, A. Insulin modulates food-related activity in the central nervous system. J. Clin. Endocrinol. Metab. 2010, 95, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Guthoff, M.; Stingl, K.T.; Tschritter, O.; Rogic, M.; Heni, M.; Stingl, K.; Hallschmid, M.; Haring, H.U.; Fritsche, A.; Preissl, H.; et al. The insulin-mediated modulation of visually evoked magnetic fields is reduced in obese subjects. PLoS ONE 2011, 6, e19482. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Eisenberg, J.; Yang, J. Human blood-brain barrier insulin receptor. J. Neurochem. 1985, 44, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Dodd, G.T.; Tiganis, T. Insulin action in the brain: Roles in energy and glucose homeostasis. J. Neuroendocrinol. 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Chari, M.; Yang, C.S.; Lam, C.K.; Lee, K.; Mighiu, P.; Kokorovic, A.; Cheung, G.W.; Lai, T.Y.; Wang, P.Y.; Lam, T.K. Glucose transporter-1 in the hypothalamic glial cells mediates glucose sensing to regulate glucose production in vivo. Diabetes 2011, 60, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Morgello, S.; Uson, R.R.; Schwartz, E.J.; Haber, R.S. The human blood-brain barrier glucose transporter (Glut1) is a glucose transporter of gray matter astrocytes. Glia 1995, 14, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Ley, E.J.; Srour, M.K.; Clond, M.A.; Barnajian, M.; Tillou, A.; Mirocha, J.; Salim, A. Diabetic patients with traumatic brain injury: Insulin deficiency is associated with increased mortality. J. Trauma 2011, 70, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Nov, O.; Kohl, A.; Lewis, E.C.; Bashan, N.; Dvir, I.; Ben-Shlomo, S.; Fishman, S.; Wueest, S.; Konrad, D.; Rudich, A. Interleukin-1beta may mediate insulin resistance in liver-derived cells in response to adipocyte inflammation. Endocrinology 2010, 151, 4247–4256. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Vanhorebeek, I.; Langouche, L.; Van den Berghe, G. Endocrine aspects of acute and prolonged critical illness. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Salim, A.; Hadjizacharia, P.; Dubose, J.; Brown, C.; Inaba, K.; Chan, L.S.; Margulies, D. Persistent hyperglycemia in severe traumatic brain injury: An independent predictor of outcome. Am. Surg. 2009, 75, 25–29. [Google Scholar] [PubMed]
- Jeremitsky, E.; Omert, L.A.; Dunham, C.M.; Wilberger, J.; Rodriguez, A. The impact of hyperglycemia on patients with severe brain injury. J. Trauma 2005, 58, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Rovlias, A.; Kotsou, S. The influence of hyperglycemia on neurological outcome in patients with severe head injury. Neurosurgery 2000, 46, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Zhang, S.; Wang, M.L. Clinical significance of admission hyperglycemia and factors related to it in patients with acute severe head injury. Surg. Neurol. 1995, 44, 373–377. [Google Scholar] [CrossRef]
- Van den Berghe, G. Novel insights into the neuroendocrinology of critical illness. Eur. J. Endocrinol. 2000, 143, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Godoy, D.A.; Behrouz, R.; Di Napoli, M. Glucose control in acute brain injury: Does it matter? Curr. Opin. Crit. Care 2016, 22, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Investigators, N.-S.S.; Finfer, S.; Chittock, D.R.; Su, S.Y.; Blair, D.; Foster, D.; Dhingra, V.; Bellomo, R.; Cook, D.; Dodek, P.; et al. Intensive versus conventional glucose control in critically ill patients. N. Engl. J. Med. 2009, 360, 1283–1297. [Google Scholar]
- Van den Berghe, G.; Schoonheydt, K.; Becx, P.; Bruyninckx, F.; Wouters, P.J. Insulin therapy protects the central and peripheral nervous system of intensive care patients. Neurology 2005, 64, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, G.; Wouters, P.; Weekers, F.; Verwaest, C.; Bruyninckx, F.; Schetz, M.; Vlasselaers, D.; Ferdinande, P.; Lauwers, P.; Bouillon, R. Intensive insulin therapy in critically ill patients. N. Engl. J. Med. 2001, 345, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Bilotta, F.; Caramia, R.; Cernak, I.; Paoloni, F.P.; Doronzio, A.; Cuzzone, V.; Santoro, A.; Rosa, G. Intensive insulin therapy after severe traumatic brain injury: A randomized clinical trial. Neurocrit. Care 2008, 9, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Magnoni, S.; Tedesco, C.; Carbonara, M.; Pluderi, M.; Colombo, A.; Stocchetti, N. Relationship between systemic glucose and cerebral glucose is preserved in patients with severe traumatic brain injury, but glucose delivery to the brain may become limited when oxidative metabolism is impaired: Implications for glycemic control. Crit. Care Med. 2012, 40, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Pittas, A.G.; Siegel, R.D.; Lau, J. Insulin therapy for critically ill hospitalized patients: A meta-analysis of randomized controlled trials. Arch. Intern. Med. 2004, 164, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Coester, A.; Neumann, C.R.; Schmidt, M.I. Intensive insulin therapy in severe traumatic brain injury: A randomized trial. J. Trauma 2010, 68, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Wiener, R.S.; Wiener, D.C.; Larson, R.J. Benefits and risks of tight glucose control in critically ill adults: A meta-analysis. JAMA 2008, 300, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, J.R.; Lima, E.R.; Cossetti, R.J.; Azevedo, R.P. Intensive insulin therapy versus conventional glycemic control in patients with acute neurological injury: A prospective controlled trial. Arquivos de Neuro-Psiquiatria 2007, 65, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Langley, J.; Adams, G. Insulin-based regimens decrease mortality rates in critically ill patients: A systematic review. Diabetes Metab. Res. Rev. 2007, 23, 184–192. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Report on Diabetes; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Hoane, M.R.; Swan, A.A.; Heck, S.E. The effects of a high-fat sucrose diet on functional outcome following cortical contusion injury in the rat. Behav. Brain Res. 2011, 223, 119–124. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Benarroch, E.E. Brain-derived neurotrophic factor: Regulation, effects, and potential clinical relevance. Neurology 2015, 84, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Nagayach, A.; Patro, N.; Patro, I. Astrocytic and microglial response in experimentally induced diabetic rat brain. Metab. Brain Dis. 2014, 29, 747–761. [Google Scholar] [CrossRef] [PubMed]
- Giaume, C.; Kirchhoff, F.; Matute, C.; Reichenbach, A.; Verkhratsky, A. Glia: The fulcrum of brain diseases. Cell Death Differ. 2007, 14, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Duelli, R.; Maurer, M.H.; Staudt, R.; Heiland, S.; Duembgen, L.; Kuschinsky, W. Increased cerebral glucose utilization and decreased glucose transporter Glut1 during chronic hyperglycemia in rat brain. Brain Res. 2000, 858, 338–347. [Google Scholar] [CrossRef]
- Singh, P.; Jain, A.; Kaur, G. Impact of hypoglycemia and diabetes on CNS: Correlation of mitochondrial oxidative stress with DNA damage. Mol. Cell. Biochem. 2004, 260, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Wang, H.D.; Cong, Z.X.; Zhou, X.M.; Xu, J.G.; Jia, Y.; Ding, Y. Puerarin ameliorates oxidative stress in a rodent model of traumatic brain injury. J. Surg. Res. 2014, 186, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Satapati, S.; Fu, X.; He, T.; Mehdibeigi, R.; Spring-Robinson, C.; Duarte, J.; Potthoff, M.J.; Browning, J.D.; Burgess, S.C. Progressive adaptation of hepatic ketogenesis in mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1226–E1235. [Google Scholar] [CrossRef] [PubMed]
- Schachtrup, C.; Ryu, J.K.; Helmrick, M.J.; Vagena, E.; Galanakis, D.K.; Degen, J.L.; Margolis, R.U.; Akassoglou, K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J. Neurosci. 2010, 30, 5843–5854. [Google Scholar] [CrossRef] [PubMed]
- Götz, M.; Sirko, S. Potential of glial cells. In Stem Cells Handbook, 2nd ed.; Sell, S., Ed.; Springer: New York, NY, USA, 2013; pp. 347–361. [Google Scholar]
- Sirko, S.; Behrendt, G.; Johansson, P.A.; Tripathi, P.; Costa, M.; Bek, S.; Heinrich, C.; Tiedt, S.; Colak, D.; Dichgans, M.; et al. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. Cell Stem Cell 2013, 12, 426–439. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Obesity and Overweight; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Flegal, K.M.; Carroll, M.D.; Ogden, C.L.; Curtin, L.R. Prevalence and trends in obesity among us adults, 1999–2008. JAMA 2010, 303, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Winfield, R.D.; Bochicchio, G.V. The critically injured obese patient: A review and a look ahead. J. Am. Coll. Surg. 2013, 216, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Festa, A.; D’gostino, R., Jr.; Williams, K.; Karter, A.J.; Mayer-Davis, E.J.; Tracy, R.P.; Haffner, S.M. The relation of body fat mass and distribution to markers of chronic inflammation. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Nov, O.; Shapiro, H.; Ovadia, H.; Tarnovscki, T.; Dvir, I.; Shemesh, E.; Kovsan, J.; Shelef, I.; Carmi, Y.; Voronov, E.; et al. Interleukin-1beta regulates fat-liver crosstalk in obesity by auto-paracrine modulation of adipose tissue inflammation and expandability. PLoS ONE 2013, 8, e53626. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Dixit, V.D. Immunological complications of obesity. Nat. Immunol. 2012, 13, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Mohamed-Ali, V.; Goodrick, S.; Rawesh, A.; Katz, D.R.; Miles, J.M.; Yudkin, J.S.; Klein, S.; Coppack, S.W. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J. Clin. Endocrinol. Metab. 1997, 82, 4196–4200. [Google Scholar] [PubMed]
- Fantuzzi, G. Adipose tissue, adipokines, and inflammation. J. Allergy Clin. Immunol. 2005, 115, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E. Novel insights into the pathophysiology of nonalcoholic fatty liver disease. Semin. Liver Dis. 2010, 30, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Hubert, H.B.; Feinleib, M.; McNamara, P.M.; Castelli, W.P. Obesity as an independent risk factor for cardiovascular disease: A 26-year follow-up of participants in the framingham heart study. Circulation 1983, 67, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Malur, A.; Marshall, I.; Huizar, I.; Barna, B.P.; Pories, W.; Dohm, L.; Kavuru, M.S.; Thomassen, M.J. Alveolar macrophage activation in obese patients with obstructive sleep apnea. Surgery 2012, 151, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Aleman, M.M.; Walton, B.L.; Byrnes, J.R.; Wolberg, A.S. Fibrinogen and red blood cells in venous thrombosis. Thromb. Res. 2014, 133 (Suppl. 1), S38–S40. [Google Scholar] [CrossRef] [PubMed]
- Goldhaber, S.Z.; Bounameaux, H. Pulmonary embolism and deep vein thrombosis. Lancet 2012, 379, 1835–1846. [Google Scholar] [CrossRef]
- Borg, M.L.; Omran, S.F.; Weir, J.; Meikle, P.J.; Watt, M.J. Consumption of a high-fat diet, but not regular endurance exercise training, regulates hypothalamic lipid accumulation in mice. J. Physiol. 2012, 590, 4377–4389. [Google Scholar] [CrossRef] [PubMed]
- Schur, E.A.; Melhorn, S.J.; Oh, S.K.; Lacy, J.M.; Berkseth, K.E.; Guyenet, S.J.; Sonnen, J.A.; Tyagi, V.; Rosalynn, M.; De Leon, B.; et al. Radiologic evidence that hypothalamic gliosis is associated with obesity and insulin resistance in humans. Obesity 2015, 23, 2142–2148. [Google Scholar] [CrossRef] [PubMed]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Guillemot-Legris, O.; Muccioli, G.G. Obesity-induced neuroinflammation: Beyond the hypothalamus. Trends Neurosci. 2017, 40, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, M.D.; Thaler, J.P. Hypothalamic inflammation and gliosis in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Posey, K.A.; Clegg, D.J.; Printz, R.L.; Byun, J.; Morton, G.J.; Vivekanandan-Giri, A.; Pennathur, S.; Baskin, D.G.; Heinecke, J.W.; Woods, S.C.; et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1003–E1012. [Google Scholar] [CrossRef] [PubMed]
- Valdearcos, M.; Robblee, M.M.; Benjamin, D.I.; Nomura, D.K.; Xu, A.W.; Koliwad, S.K. Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 2014, 9, 2124–2138. [Google Scholar] [CrossRef] [PubMed]
- Guillemot-Legris, O.; Masquelier, J.; Everard, A.; Cani, P.D.; Alhouayek, M.; Muccioli, G.G. High-fat diet feeding differentially affects the development of inflammation in the central nervous system. J. Neuroinflamm. 2016, 13, 206. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ottaway, N.; Schriever, S.C.; Legutko, B.; Garcia-Caceres, C.; de la Fuente, E.; Mergen, C.; Bour, S.; Thaler, J.P.; Seeley, R.J.; et al. Hormones and diet, but not body weight, control hypothalamic microglial activity. Glia 2014, 62, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Knight, A.G.; Gupta, S.; Keller, J.N.; Bruce-Keller, A.J. Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J. Neurochem. 2012, 120, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Jastroch, M.; Morin, S.; Tschop, M.H.; Yi, C.X. The hypothalamic neural-glial network and the metabolic syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Hao, S.; Dey, A.; Yu, X.; Baban, B. Blood-brain barrier breakdown promotes macrophage infiltration and cognitive impairment in leptin receptor-deficient mice. J. Cereb. Blood Flow Metab. 2016, 36, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial inflammatory signaling orchestrates the hypothalamic immune response to dietary excess and mediates obesity susceptibility. Cell Metab. 2017, 26, 185–197.e183. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Horvath, T.L.; Sarman, B.; Garcia-Caceres, C.; Enriori, P.J.; Sotonyi, P.; Shanabrough, M.; Borok, E.; Argente, J.; Chowen, J.A.; Perez-Tilve, D.; et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 14875–14880. [Google Scholar] [CrossRef] [PubMed]
- Fuente-Martin, E.; Garcia-Caceres, C.; Argente-Arizon, P.; Diaz, F.; Granado, M.; Freire-Regatillo, A.; Castro-Gonzalez, D.; Ceballos, M.L.; Frago, L.M.; Dickson, S.L.; et al. Ghrelin regulates glucose and glutamate transporters in hypothalamic astrocytes. Sci. Rep. 2016, 6, 23673. [Google Scholar] [CrossRef] [PubMed]
- McNay, D.E.; Briancon, N.; Kokoeva, M.V.; Maratos-Flier, E.; Flier, J.S. Remodeling of the arcuate nucleus energy-balance circuit is inhibited in obese mice. J. Clin. Investig. 2012, 122, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Boitard, C.; Etchamendy, N.; Sauvant, J.; Aubert, A.; Tronel, S.; Marighetto, A.; Laye, S.; Ferreira, G. Juvenile, but not adult exposure to high-fat diet impairs relational memory and hippocampal neurogenesis in mice. Hippocampus 2012, 22, 2095–2100. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.; Liu, M.M.; Birnbaum, S.; Wolf, S.E.; Minei, J.P.; Gatson, J.W. Adult obese mice suffer from chronic secondary brain injury after mild TBI. J. Neuroinflamm. 2016, 13, 171. [Google Scholar] [CrossRef] [PubMed]
- Christmas, A.B.; Reynolds, J.; Wilson, A.K.; Franklin, G.A.; Miller, F.B.; Richardson, J.D.; Rodriguez, J.L. Morbid obesity impacts mortality in blunt trauma. Am. Surg. 2007, 73, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Ditillo, M.; Pandit, V.; Rhee, P.; Aziz, H.; Hadeed, S.; Bhattacharya, B.; Friese, R.S.; Davis, K.; Joseph, B. Morbid obesity predisposes trauma patients to worse outcomes: A national trauma data bank analysis. J. Trauma Acute Care Surg. 2014, 76, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Flegal, K.M.; Kit, B.K.; Orpana, H.; Graubard, B.I. Association of all-cause mortality with overweight and obesity using standard body mass index categories: A systematic review and meta-analysis. JAMA 2013, 309, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferri, F.; Compagnone, C.; Yoganandan, N.; Gennarelli, T.A. Traumatic brain injury after frontal crashes: Relationship with body mass index. J. Trauma 2009, 66, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.V.; Rhee, P.; Neville, A.L.; Sangthong, B.; Salim, A.; Demetriades, D. Obesity and traumatic brain injury. J. Trauma 2006, 61, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Arbabi, S.; Wahl, W.L.; Hemmila, M.R.; Kohoyda-Inglis, C.; Taheri, P.A.; Wang, S.C. The cushion effect. J. Trauma 2003, 54, 1090–1093. [Google Scholar] [CrossRef] [PubMed]
- Bostman, O.M. Body mass index of patients with elbow and ankle fractures requiring surgical treatment. J. Trauma 1994, 37, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.R.; Michalsky, M.P.; Teich, S.; Groner, J.I.; Caniano, D.A.; Schuster, D.P. Childhood obesity: A risk factor for injuries observed at a level-1 trauma center. J. Pediatr. Surg. 2009, 44, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Anagnostis, P.; Athyros, V.G.; Tziomalos, K.; Karagiannis, A.; Mikhailidis, D.P. Clinical review: The pathogenetic role of cortisol in the metabolic syndrome: A hypothesis. J. Clin. Endocrinol. Metab. 2009, 94, 2692–2701. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the international diabetes federation task force on epidemiology and prevention; national heart, lung, and blood institute; american heart association; world heart federation; international atherosclerosis society; and international association for the study of obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [PubMed]
- Schlaich, M.; Straznicky, N.; Lambert, E.; Lambert, G. Metabolic syndrome: A sympathetic disease? Lancet Diabetes Endocrinol. 2015, 3, 148–157. [Google Scholar] [CrossRef]
- Kelly, D.F.; Chaloner, C.; Evans, D.; Mathews, A.; Cohan, P.; Wang, C.; Swerdloff, R.; Sim, M.S.; Lee, J.; Wright, M.J.; et al. Prevalence of pituitary hormone dysfunction, metabolic syndrome, and impaired quality of life in retired professional football players: A prospective study. J. Neurotrauma 2014, 31, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Ingebrigtsen, T.; Romner, B. Biochemical serum markers for brain damage: A short review with emphasis on clinical utility in mild head injury. Restor. Neurol. Neurosci. 2003, 21, 171–176. [Google Scholar] [PubMed]
- Woertgen, C.; Rothoerl, R.D.; Holzschuh, M.; Metz, C.; Brawanski, A. Comparison of serial S-100 and NSE serum measurements after severe head injury. Acta Neurochir. (Wien) 1997, 139, 1161–1164; discussion 1165. [Google Scholar] [CrossRef] [PubMed]
- Tenedieva, V.D.; Potapov, A.A.; Gaitur, E.I.; Amcheslavski, V.G.; Micrikova, L.V.; Tenedieva, N.D.; Voronov, V.G. Thyroid hormones in comatose patients with traumatic brain injury. Acta Neurochir. Suppl. 2000, 76, 385–391. [Google Scholar] [PubMed]
- Woolf, P.D.; Lee, L.A.; Hamill, R.W.; McDonald, J.V. Thyroid test abnormalities in traumatic brain injury: Correlation with neurologic impairment and sympathetic nervous system activation. Am. J. Med. 1988, 84, 201–208. [Google Scholar] [CrossRef]
- Dimopoulou, I.; Tsagarakis, S.; Korfias, S.; Zervakis, D.; Douka, E.; Thalassinos, N.; Sakas, D.E.; Roussos, C. Relationship of thyroid function to post-traumatic S-100b serum levels in survivors of severe head injury: Preliminary results. Intensive Care Med. 2004, 30, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Jost, S.; Kutz, S.; Ebert, A.D.; Kratz, T.; Wunderlich, M.T.; Synowitz, H. Temporal profile of release of neurobiochemical markers of brain damage after traumatic brain injury is associated with intracranial pathology as demonstrated in cranial computerized tomography. J. Neurotrauma 2000, 17, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Pleines, U.E.; Morganti-Kossmann, M.C.; Rancan, M.; Joller, H.; Trentz, O.; Kossmann, T. S-100β reflects the extent of injury and outcome, whereas neuronal specific enolase is a better indicator of neuroinflammation in patients with severe traumatic brain injury. J. Neurotrauma 2001, 18, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Kaptein, E.M.; Weiner, J.M.; Robinson, W.J.; Wheeler, W.S.; Nicoloff, J.T. Relationship of altered thyroid hormone indices to survival in nonthyroidal illnesses. Clin. Endocrinol. (Oxf.) 1982, 16, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Garmendia Madariaga, A.; Santos Palacios, S.; Guillen-Grima, F.; Galofre, J.C. The incidence and prevalence of thyroid dysfunction in Europe: A meta-analysis. J. Clin. Endocrinol. Metab. 2014, 99, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Rovet, J.F. The role of thyroid hormones for brain development and cognitive function. Endocr. Dev. 2014, 26, 26–43. [Google Scholar] [PubMed]
- Li, J.; Donangelo, I.; Abe, K.; Scremin, O.; Ke, S.; Li, F.; Milanesi, A.; Liu, Y.Y.; Brent, G.A. Thyroid hormone treatment activates protective pathways in both in vivo and in vitro models of neuronal injury. Mol. Cell. Endocrinol. 2017, 452, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Genovese, T.; Impellizzeri, D.; Ahmad, A.; Cornelius, C.; Campolo, M.; Cuzzocrea, S.; Esposito, E. Post-ischaemic thyroid hormone treatment in a rat model of acute stroke. Brain Res. 2013, 1513, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.D.; Coleman, M.L.; Pugh, C.W. Hypoxia, hypoxia-inducible factors (HIF), HIF hydroxylases and oxygen sensing. Cell. Mol. Life Sci. 2009, 66, 3539–3554. [Google Scholar] [CrossRef] [PubMed]
- Crupi, R.; Paterniti, I.; Campolo, M.; Di Paola, R.; Cuzzocrea, S.; Esposito, E. Exogenous T3 administration provides neuroprotection in a murine model of traumatic brain injury. Pharmacol. Res. 2013, 70, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Sadana, P.; Coughlin, L.; Burke, J.; Woods, R.; Mdzinarishvili, A. Anti-edema action of thyroid hormone in MCAO model of ischemic brain stroke: Possible association with AQP4 modulation. J. Neurol. Sci. 2015, 354, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Ambrosius, W.; Kazmierski, R.; Gupta, V.; Warot, A.W.; Adamczewska-Kocialkowska, D.; Blazejewska, A.; Ziemnicka, K.; Nowinski, W.L. Low free triiodothyronine levels are related to poor prognosis in acute ischemic stroke. Exp. Clin. Endocrinol. Diabetes 2011, 119, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Dressler, J.; Mueller, E. High thyroglobulin (Tg) concentrations in fatal traumatic brain injuries. Am. J. Forensic Med. Pathol. 2006, 27, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, G.; Griesmaier, E.; He, X.; Kapelari, K.; Urbanek, M.; Simbruner, G.; Gressens, P.; Keller, M. T3 replacement does not prevent excitotoxic cell death but reduces developmental neuronal apoptosis in newborn mice. Eur. J. Paediatr. Neurol. 2007, 11, 129–135. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Sirko, S. Traumatic Brain Injury: At the Crossroads of Neuropathology and Common Metabolic Endocrinopathies. J. Clin. Med. 2018, 7, 59. https://doi.org/10.3390/jcm7030059
Li M, Sirko S. Traumatic Brain Injury: At the Crossroads of Neuropathology and Common Metabolic Endocrinopathies. Journal of Clinical Medicine. 2018; 7(3):59. https://doi.org/10.3390/jcm7030059
Chicago/Turabian StyleLi, Melanie, and Swetlana Sirko. 2018. "Traumatic Brain Injury: At the Crossroads of Neuropathology and Common Metabolic Endocrinopathies" Journal of Clinical Medicine 7, no. 3: 59. https://doi.org/10.3390/jcm7030059
APA StyleLi, M., & Sirko, S. (2018). Traumatic Brain Injury: At the Crossroads of Neuropathology and Common Metabolic Endocrinopathies. Journal of Clinical Medicine, 7(3), 59. https://doi.org/10.3390/jcm7030059