CERA Attenuates Kidney Fibrogenesis in the db/db Mouse by Influencing the Renal Myofibroblast Generation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Picrosirius Red Staining
2.3. Immunohistochemical Staining
2.4. RNA Isolation and Real-Time PCR
2.5. Analysis of Renal TGF-β1 Protein Expression
2.6. Statistical Analyses
3. Results and Discussion
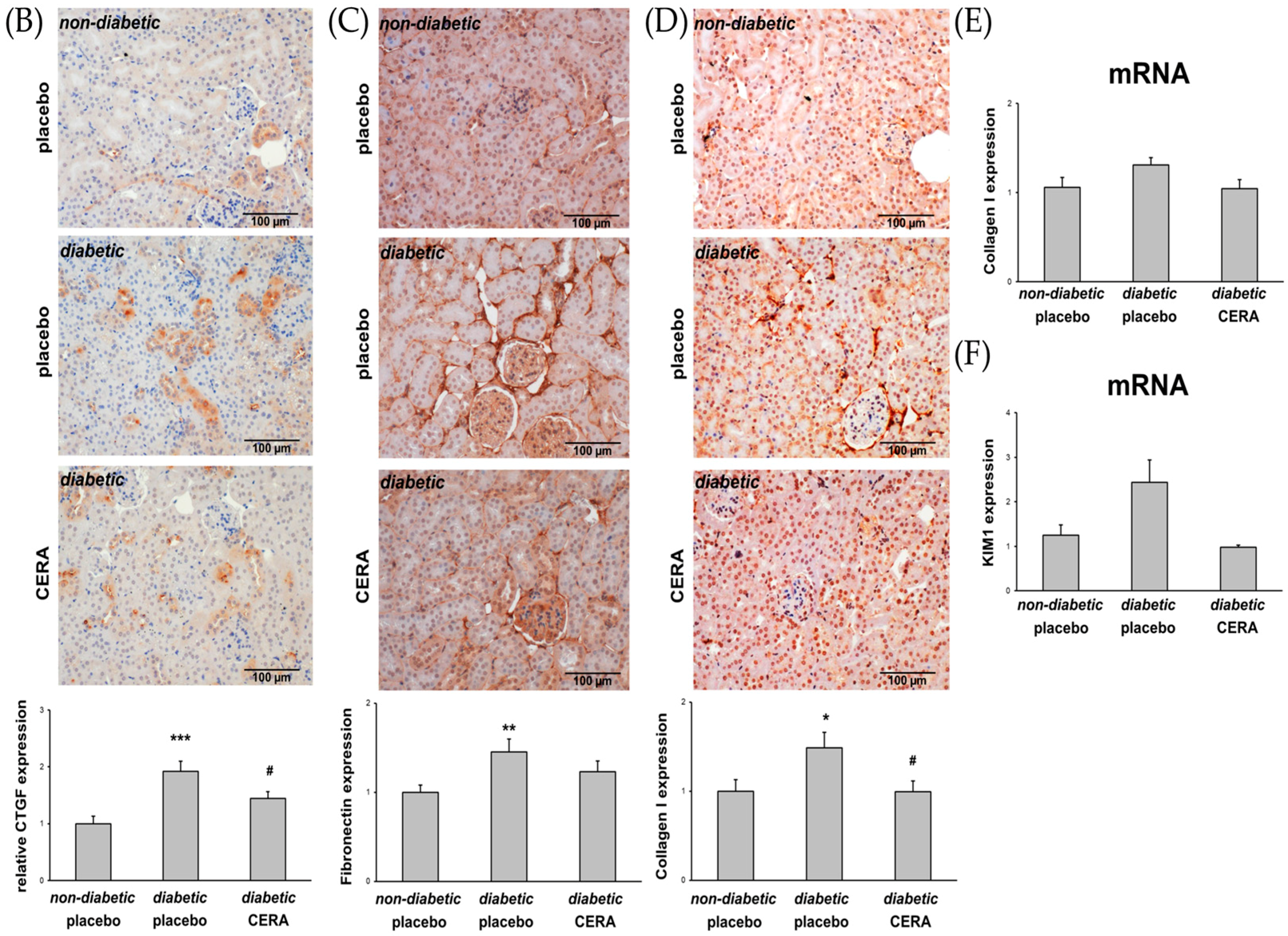
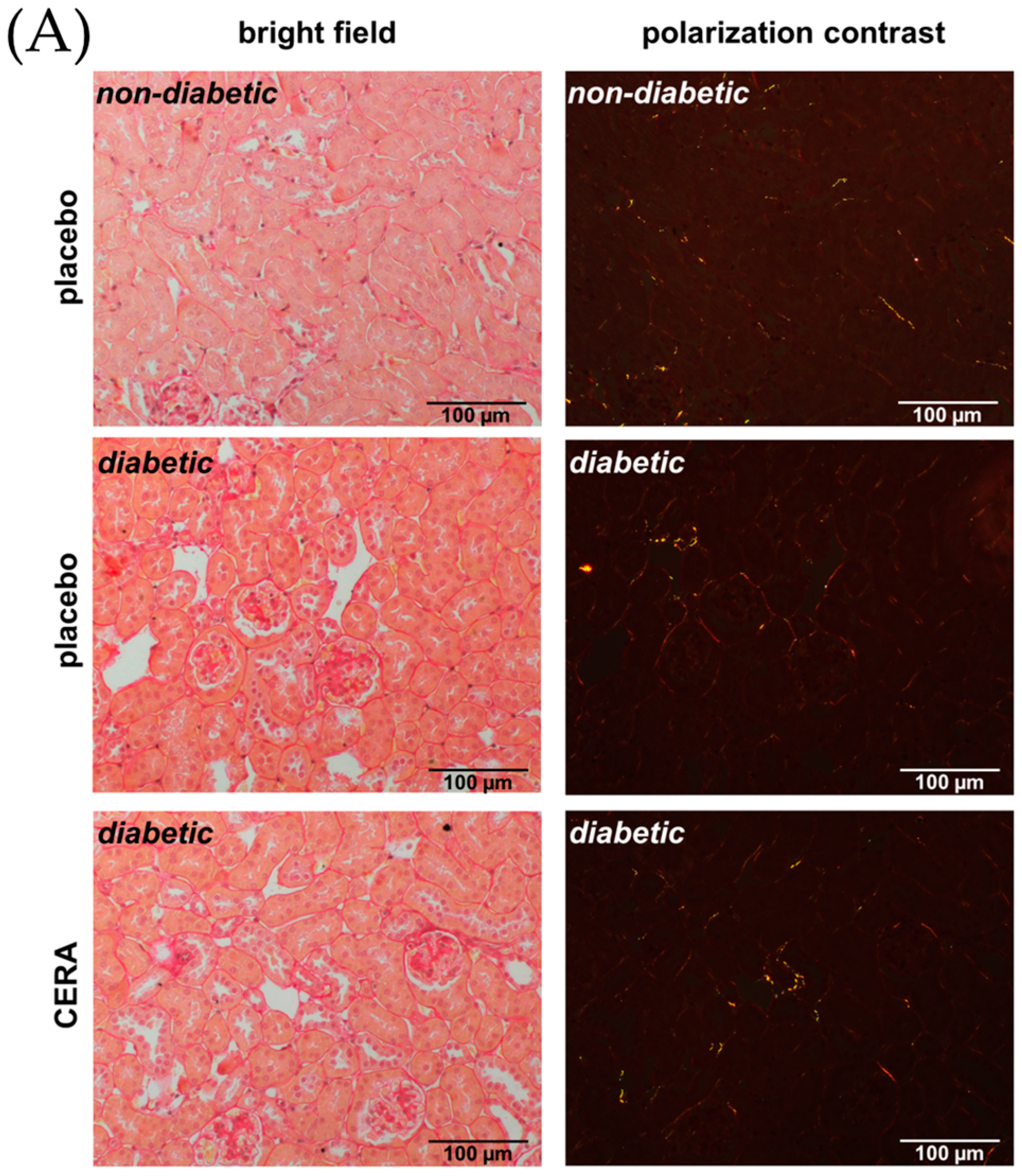
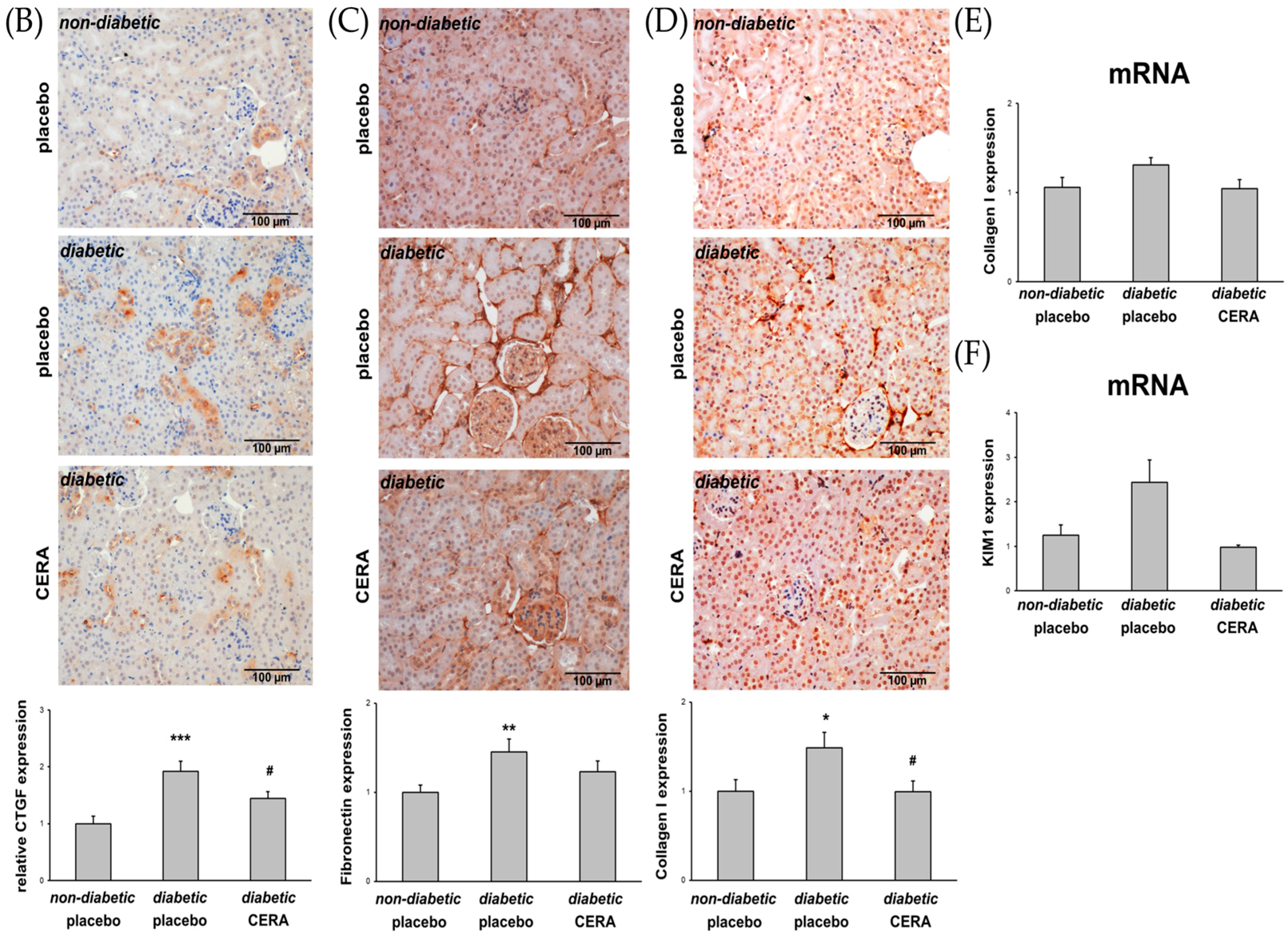
3.1. CERA Inhibits Tubulointerstitial Fibrosis in Mice with Diabetes Type 2
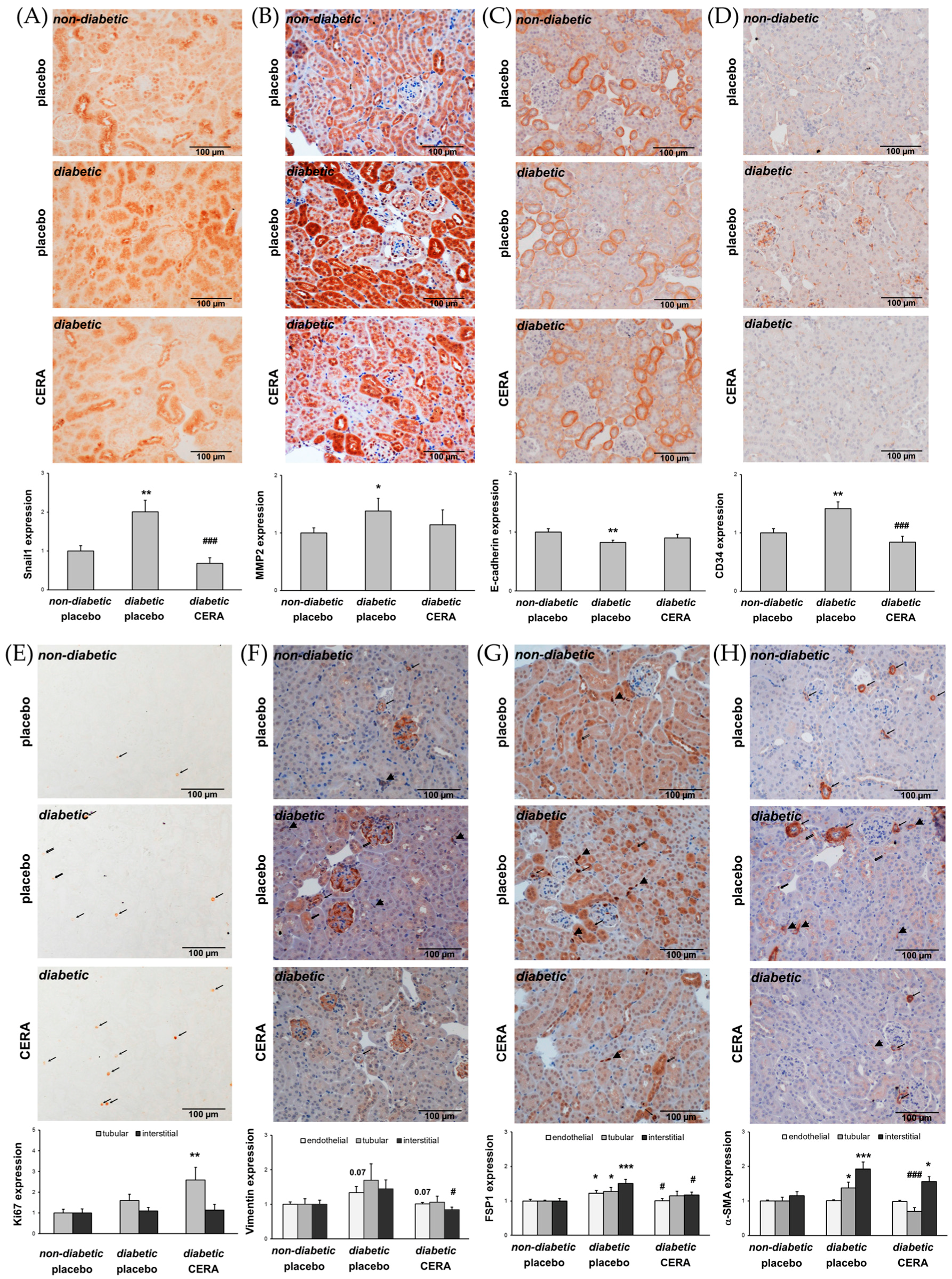
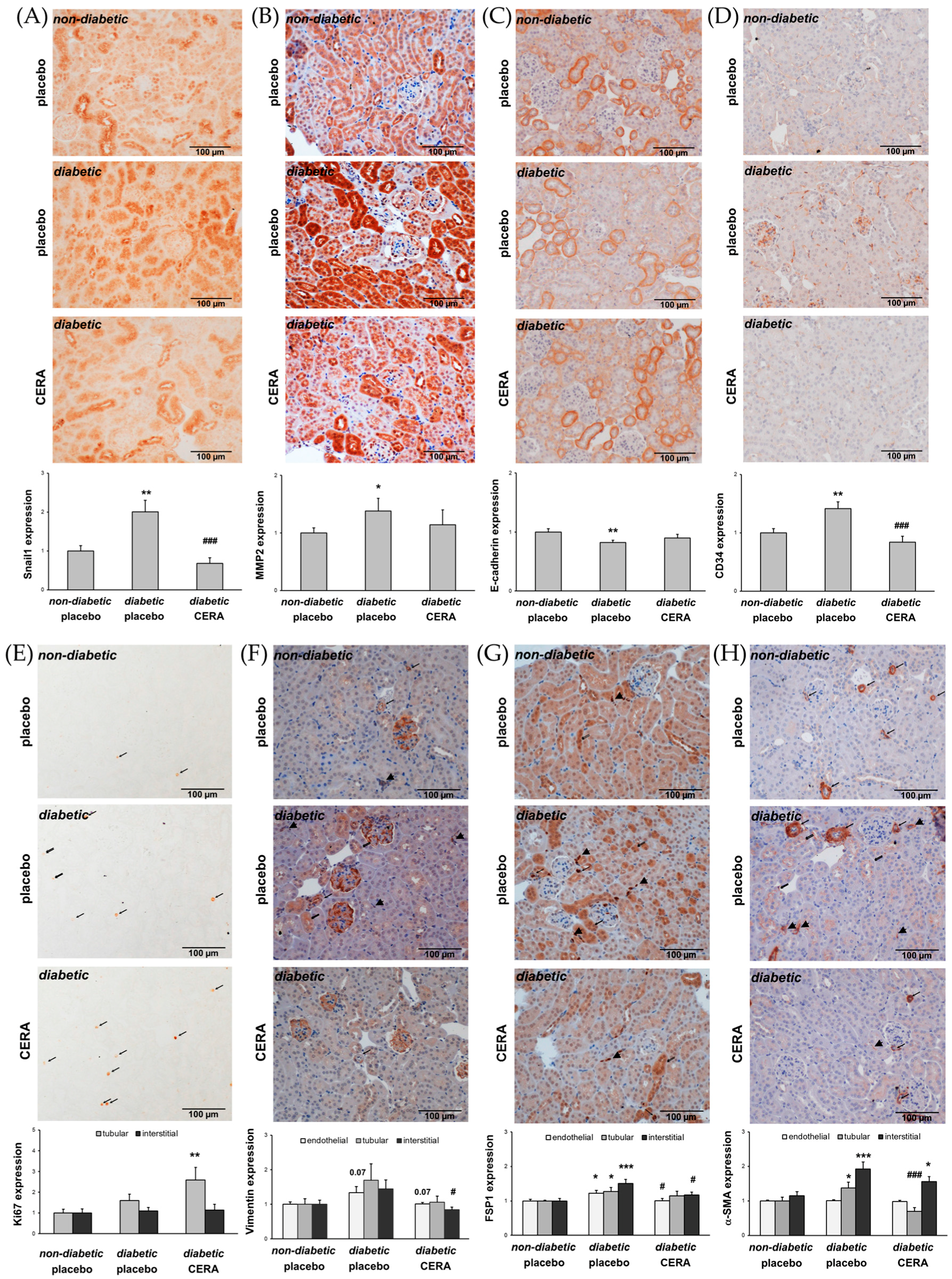
3.2. CERA Influences Myofibroblast Population in Renal Interstitium of db/db Mice
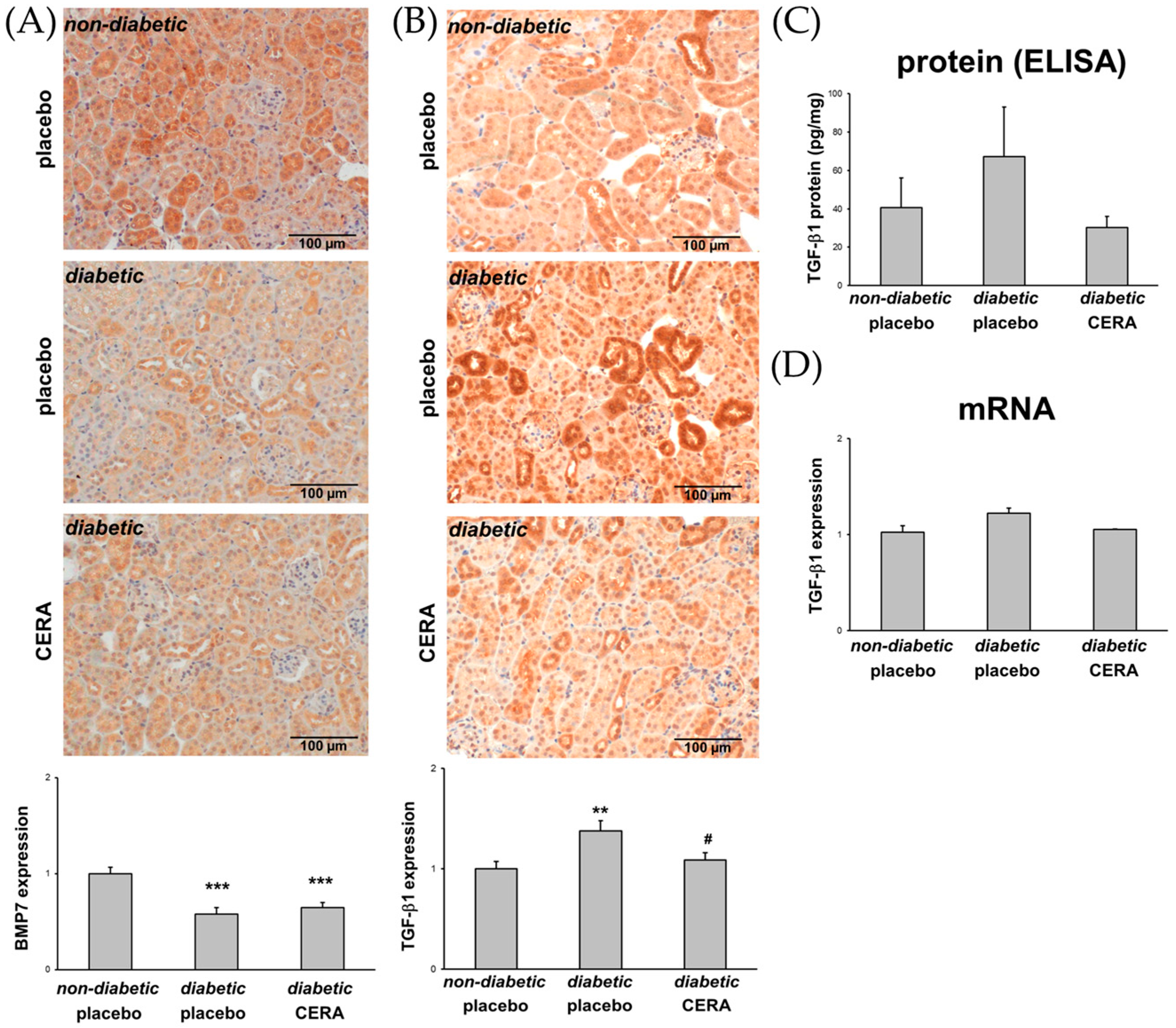
3.3. The Expression of TGF-β1 Is Reduced by CERA in Diabetic Kidneys
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wu, Y.; Ding, Y.; Tanaka, Y.; Zhang, W. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int. J. Med. Sci. 2014, 11, 1185–1200. [Google Scholar] [CrossRef] [PubMed]
- Molitch, M.E.; DeFronzo, R.A.; Franz, M.J.; Keane, W.F.; Mogensen, C.E.; Parving, H.H.; Steffes, M.W.; American Diabetes Association. Nephropathy in diabetes. Diabetes Care 2004, 27 (Suppl. 1), S79–S83. [Google Scholar] [PubMed]
- Nauta, F.L.; Boertien, W.E.; Bakker, S.J.; van Goor, H.; van Oeveren, W.; de Jong, P.E.; Bilo, H.; Gansevoort, R.T. Glomerular and tubular damage markers are elevated in patients with diabetes. Diabetes Care 2011, 34, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, T.D. Renal tubulointerstitial fibrosis: Common but never simple. Am. J. Physiol. Renal Physiol. 2009, 296, F1239–F1244. [Google Scholar] [CrossRef] [PubMed]
- Simonson, M.S. Phenotypic transitions and fibrosis in diabetic nephropathy. Kidney Int. 2007, 71, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Zeisberg, M. Renal fibroblasts and myofibroblasts in chronic kidney disease. J. Am. Soc. Nephrol. 2006, 17, 2992–2998. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Chen, X.; Poronnik, P.; Pollock, C.A. The renal cortical fibroblast in renal tubulointerstitial fibrosis. Int. J. Biochem. Cell Biol. 2006, 38, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am. J. Pathol. 2001, 159, 1465–1475. [Google Scholar] [CrossRef]
- Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanism, and therapeutic intervention. J. Am. Soc. Nephrol. 2004, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kanasaki, K.; Taduri, G.; Koya, D. Diabetic nephropathy: The role of inflammation in fibroblast activation and kidney fibrosis. Front. Endocrinol. (Lausanne) 2013, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, I.; Wolf, G. Epithelial-to-mesenchymal transition in diabetic nephropathy: Fact or fiction? Cells 2015, 4, 631–652. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qu, X.; Bertram, J.F. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 2009, 175, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Kizu, A.; Medici, D.; Kalluri, R. Endothelial-mesenchymal transition as a novel mechanism for generating myofibroblasts during diabetic nephropathy. Am. J. Pathol. 2009, 175, 1371–1373. [Google Scholar] [CrossRef] [PubMed]
- Alpers, C.E.; Hudkins, K.L. Mouse models of diabetic nephropathy. Curr. Opin. Nephrol. Hypertens. 2011, 20, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, I.; Liebisch, M.; Daniel, C.; Amann, K.; Wolf, G. Heterozygosity of mitogen-activated protein kinase organizer 1 ameliorates diabetic nephropathy and suppresses epithelial-to-mesenchymal transition-like changes in db/db mice. Nephrol. Dial. Transplant. 2017, 32, 2017–2034. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, I.; Wolf, G. The role of hypoxia and Morg1 in renal injury. Eur. J. Clin. Investig. 2015, 45, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, I.; Ruster, C.; Franke, S.; Liebisch, M.; Wolf, G. Erythropoietin ameliorates podocyte injury in advanced diabetic nephropathy in the db/db mouse. Am. J. Physiol. Renal Physiol. 2013, 305, F911–F918. [Google Scholar] [CrossRef] [PubMed]
- Eren, Z.; Gunal, M.Y.; Ari, E.; Coban, J.; Cakalagaoglu, F.; Caglayan, B.; Beker, M.C.; Akdeniz, T.; Yanikkaya, G.; Kilic, E.; et al. Pleiotropic and renoprotective effects of erythropoietin beta on experimental diabetic nephropathy model. Nephron 2016, 132, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Menne, J.; Park, J.K.; Shushakova, N.; Mengel, M.; Meier, M.; Fliser, D. The continuous erythropoietin receptor activator affects different pathways of diabetic renal injury. J. Am. Soc. Nephrol. 2007, 18, 2046–2053. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, M.; Park, J.K.; Tossidou, I.; Bartels, J.; Shushakova, N.; Menne, J.; Fliser, D. Erythropoietin prevents diabetes-induced podocyte damage. Kidney Blood Press. Res. 2008, 31, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Puchtler, H.; Waldrop, F.S.; Valentine, L.S. Polarization microscopic studies of connective tissue stained with picro-sirius red FBA. Beiträge zur Pathologie 1973, 150, 174–187. [Google Scholar] [CrossRef]
- Junqueira, L.C.; Bignolas, G.; Brentani, R.R. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem. J. 1979, 11, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Rich, L.; Whittaker, P. Collagen and picrosirius red staining: A polarized light assessment of fibrillary hue and spatial distribution. Braz. J. Morphol. Sci. 2005, 22, 8. [Google Scholar]
- Van Timmeren, M.M.; van den Heuvel, M.C.; Bailly, V.; Bakker, S.J.; van Goor, H.; Stegeman, C.A. Tubular kidney injury molecule-1 (KIM-1) in human renal disease. J. Pathol. 2007, 212, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Xu-Dubois, Y.C.; Galichon, P.; Brocheriou, I.; Baugey, E.; Morichon, R.; Jouanneau, C.; Ouali, N.; Rondeau, E.; Hertig, A. Expression of the transcriptional regulator snail1 in kidney transplants displaying epithelial-to-mesenchymal transition features. Nephrol. Dial. Transplant. 2014, 29, 2136–2144. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, E.; Loeffler, I.; Wolf, G. Morg1 heterozygous mice are protected from acute renal ischemia-reperfusion injury. Am. J. Physiol. Renal Physiol. 2009, 297, F1273–F1287. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Long, Y.; Li, S.; Liu, Z.; Zhu, F.; Hou, F.F.; Nie, J. Homocysteine induces collagen I expression by downregulating histone methyltransferase G9a. PLoS ONE 2015, 10, e0130421. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Xu, F.; Sabbisetti, V.; Grgic, I.; Movahedi Naini, S.; Wang, N.; Chen, G.; Xiao, S.; Patel, D.; Henderson, J.M.; et al. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J. Clin. Investig. 2013, 123, 4023–4035. [Google Scholar] [CrossRef] [PubMed]
- Sahlberg, C.; Peltonen, E.; Lukinmaa, P.L.; Alaluusua, S. Dioxin alters gene expression in mouse embryonic tooth explants. J. Dent. Res. 2007, 86, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Rauthan, M.; Pilon, M. A chemical screen to identify inducers of the mitochondrial unfolded protein response in C. elegans. Worm 2015, 4, e1096490. [Google Scholar] [CrossRef] [PubMed]
- Costamagna, D.; Quattrocelli, M.; van Tienen, F.; Umans, L.; de Coo, I.F.; Zwijsen, A.; Huylebroeck, D.; Sampaolesi, M. Smad1/5/8 are myogenic regulators of murine and human mesoangioblasts. J. Mol. Cell Biol. 2016, 8, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.O.; Steadman, R. Diabetic nephropathy: The central role of renal proximal tubular cells in tubulointerstitial injury. Histol. Histopathol. 2002, 17, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Farris, A.B.; Adams, C.D.; Brousaides, N.; Della Pelle, P.A.; Collins, A.B.; Moradi, E.; Smith, R.N.; Grimm, P.C.; Colvin, R.B. Morphometric and visual evaluation of fibrosis in renal biopsies. J. Am. Soc. Nephrol. 2011, 22, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Street, J.M.; Souza, A.C.; Alvarez-Prats, A.; Horino, T.; Hu, X.; Yuen, P.S.; Star, R.A. Automated quantification of renal fibrosis with Sirius Red and polarization contrast microscopy. Physiol. Rep. 2014, 2, e12088. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.M.; Wahab, N.A. Extracellular matrix metabolism in diabetic nephropathy. J. Am. Soc. Nephrol. 2003, 14, 1358–1373. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V. Can we target tubular damage to prevent renal function decline in diabetes? Semin. Nephrol. 2012, 32, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Twigg, S.; Chen, X.; Polhill, T.S.; Poronnik, P.; Gilbert, R.E.; Pollock, C.A. Integrated actions of transforming growth factor-beta1 and connective tissue growth factor in renal fibrosis. Am. J. Physiol. Renal Physiol. 2005, 288, F800–F809. [Google Scholar] [CrossRef] [PubMed]
- Kusaba, T.; Lalli, M.; Kramann, R.; Kobayashi, A.; Humphreys, B.D. Differentiated kidney epithelial cells repair injured proximal tubule. Proc. Natl. Acad. Sci. USA 2014, 111, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.B.; Qu, X.; Caruana, G.; Li, J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 2016, 92, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Deane, J.A.; Campanale, N.V.; Bertram, J.F.; Ricardo, S.D. The contribution of bone marrow-derived cells to the development of renal interstitial fibrosis. Stem Cells 2007, 25, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Simon-Tillaux, N.; Hertig, A. Snail and kidney fibrosis. Nephrol. Dial. Transplant. 2017, 32, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Geng, J.; Zhou, Z.; Tian, J.; Li, X. Microrna-130b improves renal tubulointerstitial fibrosis via repression of snail-induced epithelial-mesenchymal transition in diabetic nephropathy. Sci. Rep. 2016, 6, 20475. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Johnson, N.W.; Gao, J. Epithelial-mesenchymal transition in oral squamous cell carcinoma triggered by transforming growth factor-beta1 is snail family-dependent and correlates with matrix metalloproteinase-2 and -9 expressions. Int. J. Oncol. 2010, 37, 663–668. [Google Scholar] [PubMed]
- Wang, S.; Chen, Q.; Simon, T.C.; Strebeck, F.; Chaudhary, L.; Morrissey, J.; Liapis, H.; Klahr, S.; Hruska, K.A. Bone morphogenic protein-7 (BMP-7), a novel therapy for diabetic nephropathy. Kidney Int. 2003, 63, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.; Qi, Y.; Du, J. Akt2 mediates TGF-β1-induced epithelial to mesenchymal transition by deactivating GSK3β/snail signaling pathway in renal tubular epithelial cells. Cell. Physiol. Biochem. 2014, 34, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Kamata, N.; Fujimoto, R.; Tsutsumi, S.; Tomonari, M.; Taki, M.; Hosokawa, H.; Nagayama, M. Increased invasion and matrix metalloproteinase-2 expression by snail-induced mesenchymal transition in squamous cell carcinomas. Int. J. Oncol. 2003, 22, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Allaire, A.D.; Ballenger, K.A.; Wells, S.R.; McMahon, M.J.; Lessey, B.A. Placental apoptosis in preeclampsia. Obstet. Gynecol. 2000, 96, 271–276. [Google Scholar] [PubMed]
- Cheng, S.; Pollock, A.S.; Mahimkar, R.; Olson, J.L.; Lovett, D.H. Matrix metalloproteinase 2 and basement membrane integrity: A unifying mechanism for progressive renal injury. FASEB J. 2006, 20, 1898–1900. [Google Scholar] [CrossRef] [PubMed]
- Tveitaras, M.K.; Skogstrand, T.; Leh, S.; Helle, F.; Iversen, B.M.; Chatziantoniou, C.; Reed, R.K.; Hultstrom, M. Matrix metalloproteinase-2 knockout and heterozygote mice are protected from hydronephrosis and kidney fibrosis after unilateral ureteral obstruction. PLoS ONE 2015, 10, e0143390. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Galichon, P.; Hertig, A. Epithelial to mesenchymal transition as a biomarker in renal fibrosis: Are we ready for the bedside? Fibrogenes. Tissue Repair 2011, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.C.; Ross, M.J. Epithelial-to-mesenchymal transition of tubular epithelial cells in renal fibrosis: A new twist on an old tale. Kidney Int. 2016, 89, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Chen, H.; Wang, C.; Peng, Y.; Sun, L.; Liu, H.; Liu, F. Rapamycin ameliorates kidney fibrosis by inhibiting the activation of mTOR signaling in interstitial macrophages and myofibroblasts. PLoS ONE 2012, 7, e33626. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, J.A.; Mertens, P.R. Myofibroblasts, regeneration or renal fibrosis—Is there a decisive hint? Nephrol. Dial. Transplant. 2013, 28, 2678–2681. [Google Scholar] [CrossRef] [PubMed]
- Falke, L.L.; Gholizadeh, S.; Goldschmeding, R.; Kok, R.J.; Nguyen, T.Q. Diverse origins of the myofibroblast-implications for kidney fibrosis. Nat. Rev. Nephrol. 2015, 11, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G. New insights into the pathophysiology of diabetic nephropathy: From haemodynamics to molecular pathology. Eur. J. Clin. Investig. 2004, 34, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nakamura, T.; Noble, N.A.; Ruoslahti, E.; Border, W.A. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1993, 90, 1814–1818. [Google Scholar] [CrossRef] [PubMed]
- Hills, C.E.; Bland, R.; Bennett, J.; Ronco, P.M.; Squires, P.E. TGF-beta1 mediates glucose-evoked up-regulation of connexin-43 cell-to-cell communication in HCD-cells. Cell. Physiol. Biochem. 2009, 24, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottinger, E.P.; Bitzer, M. TGF-beta signaling in renal disease. J. Am. Soc. Nephrol. 2002, 13, 2600–2610. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, I.; Wolf, G. Transforming growth factor-β and the progression of renal disease. Nephrol. Dial. Transplant. 2014, 29, i37–i45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Q. Bone morphogenetic protein-7 and Gremlin: New emerging therapeutic targets for diabetic nephropathy. Biochem. Biophys. Res. Commun. 2009, 383, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.N.; Lapage, J.; Hirschberg, R. Loss of tubular bone morphogenetic protein-7 in diabetic nephropathy. J. Am. Soc. Nephrol. 2001, 12, 2392–2399. [Google Scholar] [PubMed]
- De Petris, L.; Hruska, K.A.; Chiechio, S.; Liapis, H. Bone morphogenetic protein-7 delays podocyte injury due to high glucose. Nephrol. Dial. Transplant. 2007, 22, 3442–3450. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.L.; Gorin, Y. Myofibroblast differentiation during fibrosis: Role of NAD(P)H oxidases. Kidney Int. 2011, 79, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Hills, C.E.; Squires, P.E. TGF-beta1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am. J. Nephrol. 2010, 31, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Umezawa, A.; Takenaka, T.; Suzuki, H.; Okada, H. The contribution of epithelial-mesenchymal transition to renal fibrosis differs among kidney disease models. Kidney Int. 2015, 87, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Chou, K.J.; Lee, P.T.; Chen, Y.S.; Chang, T.Y.; Hsu, C.Y.; Huang, W.C.; Chung, H.M.; Fang, H.C. Erythropoietin suppresses epithelial to mesenchymal transition and intercepts Smad signal transduction through a MEK-dependent mechanism in pig kidney (LLC-PK1) cell lines. Exp. Cell Res. 2010, 316, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Fraser, D.; Phillips, A. Erk, p38, and Smad signaling pathways differentially regulate transforming growth factor-beta1 autoinduction in proximal tubular epithelial cells. Am. J. Pathol. 2006, 169, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M. Tissue protection by erythropoietin: New findings in a moving field. Kidney Int. 2013, 84, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Coldewey, S.M.; Khan, A.I.; Kapoor, A.; Collino, M.; Rogazzo, M.; Brines, M.; Cerami, A.; Hall, P.; Sheaff, M.; Kieswich, J.E.; et al. Erythropoietin attenuates acute kidney dysfunction in murine experimental sepsis by activation of the β-common receptor. Kidney Int. 2013, 84, 482–490. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Sence Primer | Antisense Primer | TAnn. | Reference |
|---|---|---|---|---|
| collagen I | 5′-GAGAGGTGAACAAGGTCCCG-3′ | 5′-AAACCTCTCTCGCCTCTTGC-3′ | 64 °C | [28] |
| KIM1 | 5′-ATGAATCAGATTCAAGTCTTC-3′ | 5′-TCTGGTTTGTGAGTCCATGTG-3′ | 55 °C | [29] |
| TGF-β1 | 5′-AAGGGCTACCATGCCAACTT-3′ | 5′-CGGGTTGTGTTGGTTGTAGA-3′ | 62 °C | [30] |
| GUSB | 5′-CCGATTATCCAGAGCGAGTATG-3′ | 5′-CTCAGCGGTGACTGGTTCG-3′ | 59 °C | [31] |
| HPRT | 5′-TGGATACAGGCCAGACTTTGTT-3′ | 5′-CAGATTCAACTTGCGCTCATC-3′ | 59 °C | [32] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, C.; Deininger, N.; Wolf, G.; Loeffler, I. CERA Attenuates Kidney Fibrogenesis in the db/db Mouse by Influencing the Renal Myofibroblast Generation. J. Clin. Med. 2018, 7, 15. https://doi.org/10.3390/jcm7020015
Fischer C, Deininger N, Wolf G, Loeffler I. CERA Attenuates Kidney Fibrogenesis in the db/db Mouse by Influencing the Renal Myofibroblast Generation. Journal of Clinical Medicine. 2018; 7(2):15. https://doi.org/10.3390/jcm7020015
Chicago/Turabian StyleFischer, Christin, Natalie Deininger, Gunter Wolf, and Ivonne Loeffler. 2018. "CERA Attenuates Kidney Fibrogenesis in the db/db Mouse by Influencing the Renal Myofibroblast Generation" Journal of Clinical Medicine 7, no. 2: 15. https://doi.org/10.3390/jcm7020015
APA StyleFischer, C., Deininger, N., Wolf, G., & Loeffler, I. (2018). CERA Attenuates Kidney Fibrogenesis in the db/db Mouse by Influencing the Renal Myofibroblast Generation. Journal of Clinical Medicine, 7(2), 15. https://doi.org/10.3390/jcm7020015