
Oxidative Stress in COPD: Sources, Markers, and Potential Mechanisms

Abstract
:
1. Introduction
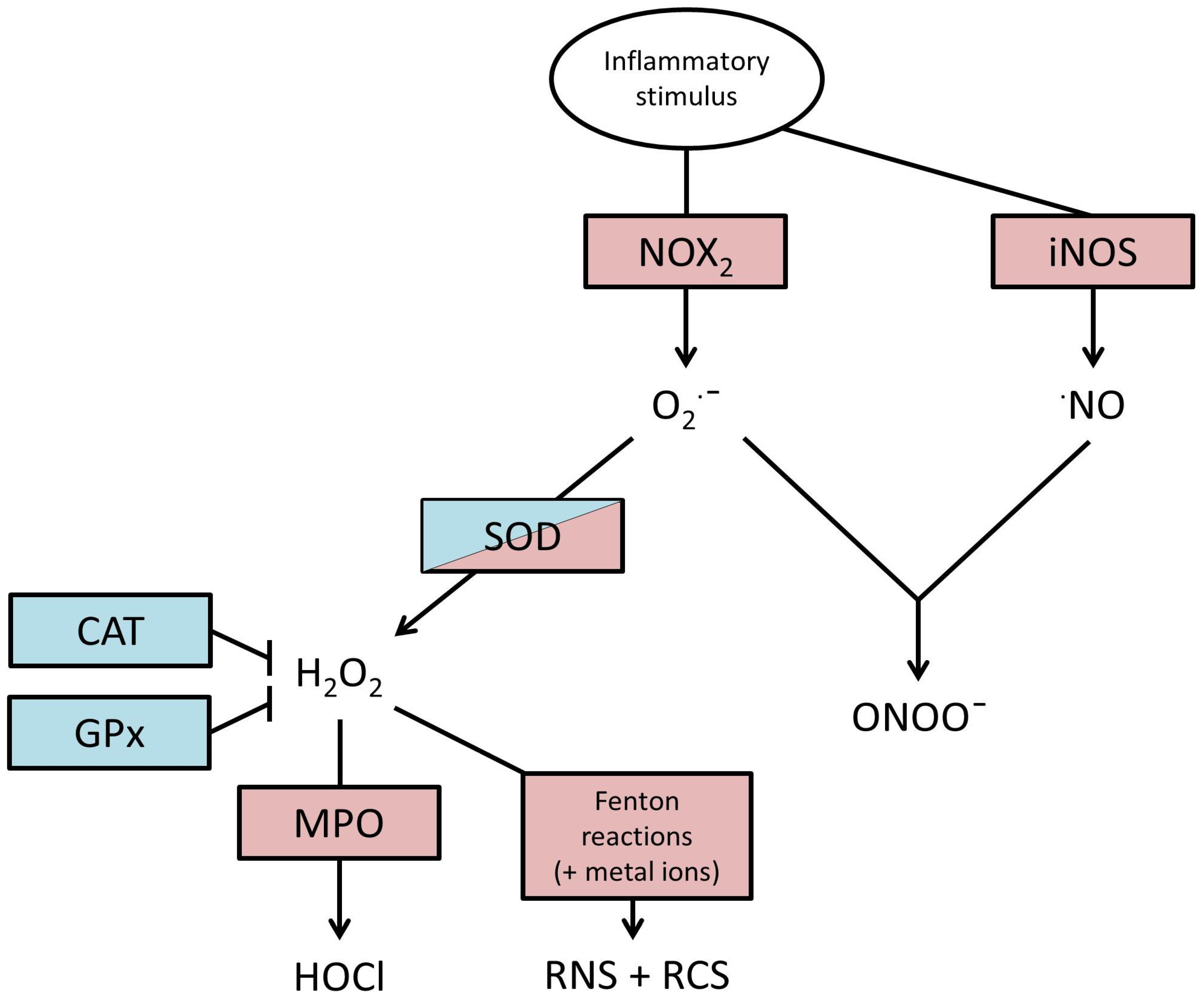
2. Production of ROS
3. Actions of ROS
4. Endogenous Defences against ROS
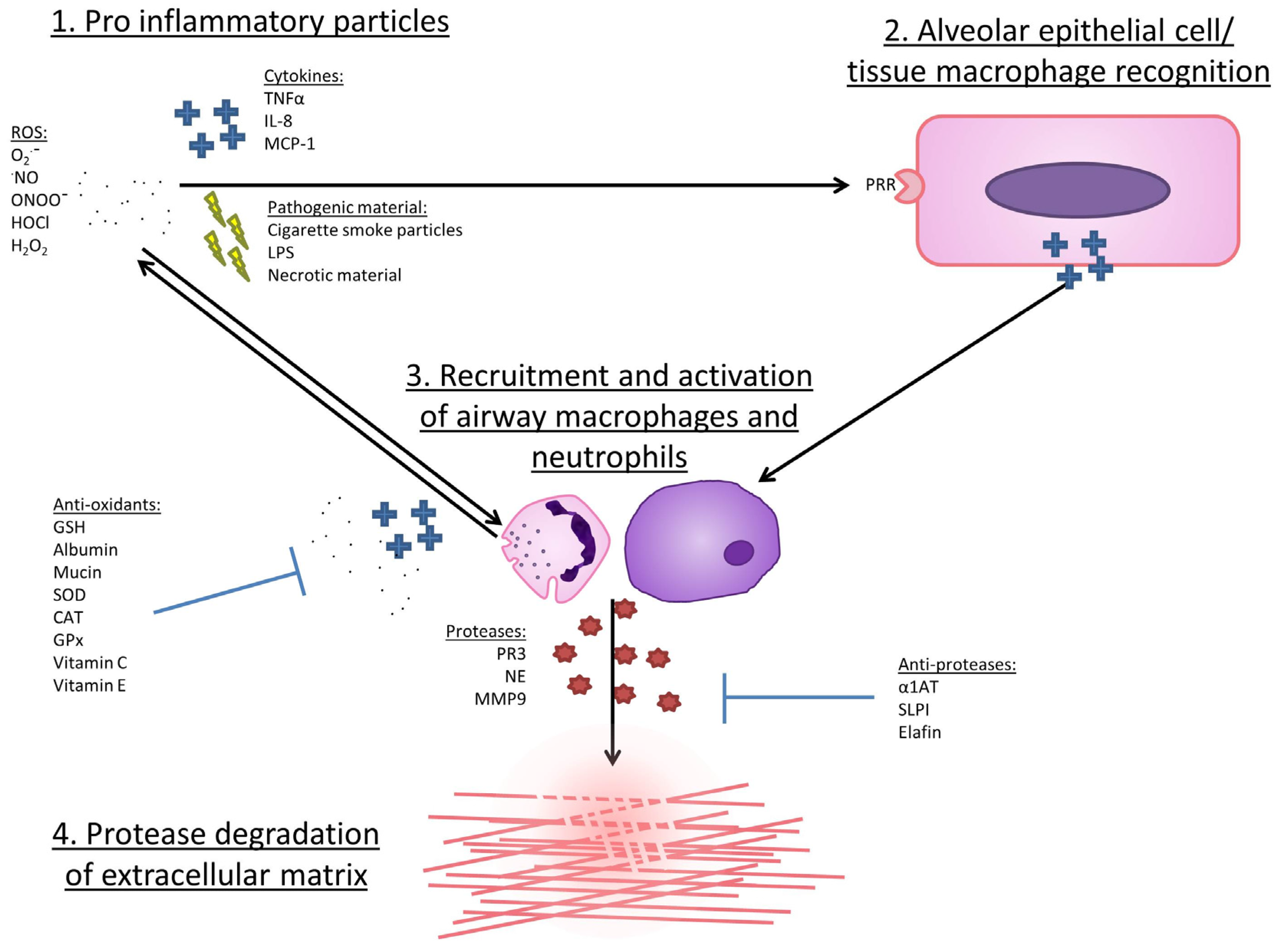
5. Evidence for an Association between Oxidative Stress and COPD
6. The Cellular Sources of ROS in COPD
6.1. Neutrophils
6.2. Monocytes/Macrophages
6.3. Lung Epithelial Cells
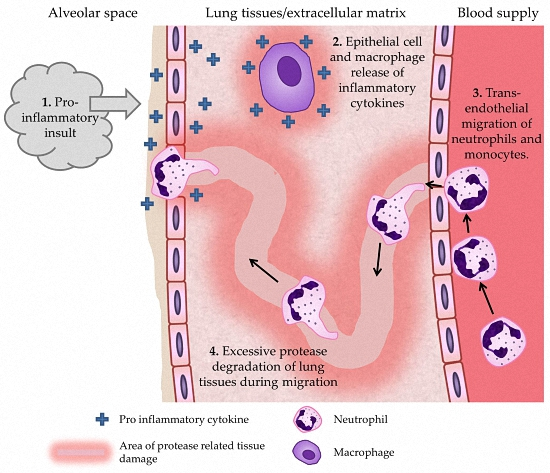
7. Mechanisms by Which ROS May Lead to COPD Development or Progression
8. Why Might ROS Damage Be Heightened in COPD?
8.1. Reduced Anti-Inflammatory Defence in COPD
8.2. Increased Pro-Inflammatory Enzyme Expression in COPD
9. Therapeutic Studies
9.1. N-Acetyl-Cysteine (NAC)
9.2. Glutathione
9.3. Nrf2 and Nrf2-Activators
9.4. Vitamins C and E
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Murray, C.J.; Lopez, A.D. Measuring the global burden of disease. N. Engl. J. Med. 2013, 369, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Nacul, L.; Soljak, M.; Samarasundera, E.; Hopkinson, N.S.; Lacerda, E.; Indulkar, T.; Flowers, J.; Walford, H.; Majeed, A. Copd in england: A comparison of expected, model-based prevalence and observed prevalence from general practice data. J. Public Health 2011, 33, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Nice Chronic Obstructive Pulmonary Disease: Costing Report. Nice Clinical Guideline 101. National Institute for Health and Clinical Excellence: London, UK, 2011. Available online: https://www.Nice.Org.Uk/guidance/cg101/resources/costing-report-134511805 (accessed on 10 September 2016).
- WHO World Health Statistics 2008: Full Report. Switzerland. World Health Organisation, 2008. Available online: http://www.Who.Int/whosis/whostat/2008/en/ (accessed on 10 September 2016).
- Cohen, B.H.; Ball, W.C., Jr.; Brashears, S.; Diamond, E.L.; Kreiss, P.; Levy, D.A.; Menkes, H.A.; Permutt, S.; Tockman, M.S. Risk factors in Chronic Obstructive Pulmonary Disease (COPD). Am. J. Epidemiol. 1977, 105, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Louhelainen, N.; Rytila, P.; Haahtela, T.; Kinnula, V.L.; Djukanovic, R. Persistence of oxidant and protease burden in the airways after smoking cessation. BMC Pulm. Med. 2009, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Rutgers, S.R.; Postma, D.S.; ten Hacken, N.H.; Kauffman, H.F.; van Der Mark, T.W.; Koeter, G.H.; Timens, W. Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax 2000, 55, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Taraseviciene-Stewart, L.; Douglas, I.S.; Nana-Sinkam, P.S.; Lee, J.D.; Tuder, R.M.; Nicolls, M.R.; Voelkel, N.F. Is alveolar destruction and emphysema in chronic obstructive pulmonary disease an immune disease? Proc. Am. Thorac Soc. 2006, 3, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Stone, H.; McNab, G.; Wood, A.M.; Stockley, R.A.; Sapey, E. Variability of sputum inflammatory mediators in COPD and α1-antitrypsin deficiency. Eur. Respir. J. 2012, 40, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.E.; Thorley, A.; Culpitt, S.V.; Dodd, S.; Donnelly, L.E.; Demattos, C.; Fitzgerald, M.; Barnes, P.J. Alveolar macrophage-mediated elastolysis: Roles of matrix metalloproteinases, cysteine, and serine proteases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L867–L873. [Google Scholar] [CrossRef] [PubMed]
- Voynow, J.A.; Young, L.R.; Wang, Y.; Horger, T.; Rose, M.C.; Fischer, B.M. Neutrophil elastase increases muc5ac mrna and protein expression in respiratory epithelial cells. Am. J. Physiol. 1999, 276, L835–L843. [Google Scholar] [PubMed]
- Pryor, W.A.; Stone, K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann. N. Y. Acad. Sci. 1993, 686, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. Nadph oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, Y.; Morisaki, T.; Kojima, M.; Uchiyama, A.; Tanaka, M. Inos expression by activated neutrophils from patients with sepsis. ANZ J. Surg. 2001, 71, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Quijano, C.; Alvarez, B.; Gatti, R.M.; Augusto, O.; Radi, R. Pathways of peroxynitrite oxidation of thiol groups. Biochem. J. 1997, 322, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Graham, A.; Hogg, N.; Kalyanaraman, B.; O’Leary, V.; Darley-Usmar, V.; Moncada, S. Peroxynitrite modification of low-density lipoprotein leads to recognition by the macrophage scavenger receptor. FEBS Lett. 1993, 330, 181–185. [Google Scholar] [CrossRef]
- Kinkade, J.M., Jr.; Pember, S.O.; Barnes, K.C.; Shapira, R.; Spitznagel, J.K.; Martin, L.E. Differential distribution of distinct forms of myeloperoxidase in different azurophilic granule subpopulations from human neutrophils. Biochem. Biophys. Res. Commun. 1983, 114, 296–303. [Google Scholar] [CrossRef]
- Church, D.F.; Pryor, W.A. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ. Health Perspect. 1985, 64, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, D.K. Ozone-induced lung inflammation and mucosal barrier disruption: Toxicology, mechanisms, and implications. J. Toxicol. Environ. Health B Crit. Rev. 1999, 2, 31–86. [Google Scholar] [CrossRef] [PubMed]
- Nichols, B.G.; Woods, J.S.; Luchtel, D.L.; Corral, J.; Koenig, J.Q. Effects of ozone exposure on nuclear factor-kappab activation and tumor necrosis factor-alpha expression in human nasal epithelial cells. Toxicol. Sci. 2001, 60, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Hu, M.L.; Louie, S.; Duvall, T.R.; Tarkington, B.K.; Motchnik, P.; Cross, C.E. Interaction of nitrogen dioxide with human plasma. Antioxidant depletion and oxidative damage. FEBS Lett. 1992, 313, 62–66. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Comparative reactivities of various biological compounds with myeloperoxidase hydrogen peroxide-chloride, and similarity of the oxidant to hypochlorite. Biochim. Biophys. Acta 1985, 840, 204–210. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Hypochlorite-induced oxidation of proteins in plasma: Formation of chloramines and nitrogen-centred radicals and their role in protein fragmentation. Biochem. J. 1999, 340, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Van der Vliet, A.; Eiserich, J.P.; O’Neill, C.A.; Halliwell, B.; Cross, C.E. Tyrosine modification by reactive nitrogen species: A closer look. Arch. Biochem. Biophys. 1995, 319, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Kelly, F.J.; Mudway, I.S. Protein oxidation at the air-lung interface. Amino Acids 2003, 25, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.A.; Buchanan, J.D. Reactions of O−2, H2O2 and other oxidants with sulfhydryl enzymes. Photochem. Photobiol. 1978, 28, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Carp, H.; Miller, F.; Hoidal, J.R.; Janoff, A. Potential mechanism of emphysema: α1-Proteinase inhibitor recovered from lungs of cigarette smokers contains oxidized methionine and has decreased elastase inhibitory capacity. Proc. Natl. Acad. Sci. USA 1982, 79, 2041–2045. [Google Scholar] [CrossRef] [PubMed]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar] [PubMed]
- Heinecke, J.W.; Li, W.; Mueller, D.M.; Bohrer, A.; Turk, J. Cholesterol chlorohydrin synthesis by the myeloperoxidase-hydrogen peroxide-chloride system: Potential markers for lipoproteins oxidatively damaged by phagocytes. Biochemistry 1994, 33, 10127–10136. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; van den Berg, J.J.; Winterbourn, C.C. Chlorination of cholesterol in cell membranes by hypochlorous acid. Arch. Biochem. Biophys. 1996, 332, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Prutz, W.A. Hypochlorous acid interactions with thiols, nucleotides, DNA, and other biological substrates. Arch. Biochem. Biophys. 1996, 332, 110–120. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Denu, J.M.; Tanner, K.G. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, J.W.; Sun, C.; Magalhaes, M.A.; Glogauer, M. Rac regulates ptdinsp(3) signaling and the chemotactic compass through a redox-mediated feedback loop. Blood 2011, 118, 6164–6171. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med. 2010, 14, 840–860. [Google Scholar] [CrossRef] [PubMed]
- Vogt, W. Oxidation of methionyl residues in proteins: Tools, targets, and reversal. Free Radic. Biol. Med. 1995, 18, 93–105. [Google Scholar] [CrossRef]
- Kim, H.J.; Ha, S.; Lee, H.Y.; Lee, K.J. Rosics: Chemistry and proteomics of cysteine modifications in redox biology. Mass Spectrom Rev. 2015, 34, 184–208. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Mosoni, L.; Berlett, B.S.; Stadtman, E.R. Methionine residues as endogenous antioxidants in proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 15036–15040. [Google Scholar] [CrossRef]
- Iwao, Y.; Ishima, Y.; Yamada, J.; Noguchi, T.; Kragh-Hansen, U.; Mera, K.; Honda, D.; Suenaga, A.; Maruyama, T.; Otagiri, M. Quantitative evaluation of the role of cysteine and methionine residues in the antioxidant activity of human serum albumin using recombinant mutants. IUBMB Life 2012, 64, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.Y.; Desorchers, P.E.; Pizzo, S.V.; Gonias, S.L.; Sahakian, J.A.; Levine, R.L.; Weiss, S.J. Oxidative dissociation of human alpha 2-macroglobulin tetramers into dysfunctional dimers. J. Biol. Chem. 1994, 269, 4683–4691. [Google Scholar] [PubMed]
- Rosen, H.; Klebanoff, S.J.; Wang, Y.; Brot, N.; Heinecke, J.W.; Fu, X. Methionine oxidation contributes to bacterial killing by the myeloperoxidase system of neutrophils. Proc. Natl. Acad. Sci. USA 2009, 106, 18686–18691. [Google Scholar] [CrossRef] [PubMed]
- Horvath, I.; Hunt, J.; Barnes, P.J.; Alving, K.; Antczak, A.; Baraldi, E.; Becher, G.; van Beurden, W.J.; Corradi, M.; Dekhuijzen, R.; et al. Exhaled breath condensate: Methodological recommendations and unresolved questions. Eur. Respir. J. 2005, 26, 523–548. [Google Scholar] [CrossRef] [PubMed]
- Dekhuijzen, P.N.; Aben, K.K.; Dekker, I.; Aarts, L.P.; Wielders, P.L.; van Herwaarden, C.L.; Bast, A. Increased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1996, 154, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Nowak, D.; Kasielski, M.; Antczak, A.; Pietras, T.; Bialasiewicz, P. Increased content of thiobarbituric acid-reactive substances and hydrogen peroxide in the expired breath condensate of patients with stable chronic obstructive pulmonary disease: No significant effect of cigarette smoking. Respir. Med. 1999, 93, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.D.; Hill, K.E.; Burk, R.F.; Nammour, T.M.; Badr, K.F.; Roberts, L.J., 2nd. A series of prostaglandin f2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. USA 1990, 87, 9383–9387. [Google Scholar] [CrossRef] [PubMed]
- Montuschi, P.; Collins, J.V.; Ciabattoni, G.; Lazzeri, N.; Corradi, M.; Kharitonov, S.A.; Barnes, P.J. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am. J. Respir. Crit. Care Med. 2000, 162, 1175–1177. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, S.; Carraro, S.; Corradi, M.; Alinovi, R.; Pasquale, M.F.; Piacentini, G.; Zacchello, F.; Baraldi, E. Leukotrienes and 8-isoprostane in exhaled breath condensate of children with stable and unstable asthma. J. Allergy Clin. Immunol. 2004, 113, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, E.; Carraro, S.; Alinovi, R.; Pesci, A.; Ghiro, L.; Bodini, A.; Piacentini, G.; Zacchello, F.; Zanconato, S. Cysteinyl leukotrienes and 8-isoprostane in exhaled breath condensate of children with asthma exacerbations. Thorax 2003, 58, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, M.L.; Novelli, F.; Costa, F.; Malagrino, L.; Melosini, L.; Bacci, E.; Cianchetti, S.; Dente, F.L.; Di Franco, A.; Vagaggini, B.; et al. Malondialdehyde in exhaled breath condensate as a marker of oxidative stress in different pulmonary diseases. Mediat. Inflamm. 2011, 2011, 891752. [Google Scholar] [CrossRef] [PubMed]
- Kluchova, Z.; Petrasova, D.; Joppa, P.; Dorkova, Z.; Tkacova, R. The association between oxidative stress and obstructive lung impairment in patients with COPD. Physiol. Res. 2007, 56, 51–56. [Google Scholar] [PubMed]
- Zeng, M.; Li, Y.; Jiang, Y.J.; Lu, G.F.; Huang, X.M.; Guan, K.P. Local and systemic oxidative stress and glucocorticoid receptor levels in chronic obstructive pulmonary disease patients. Can. Respir. J. 2013, 20, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, M.; Sugiura, H.; Yamagata, S.; Koarai, A.; Shirato, K. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am. J. Respir. Crit. Care Med. 2000, 162, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Ricciardolo, F.L.; Caramori, G.; Ito, K.; Capelli, A.; Brun, P.; Abatangelo, G.; Papi, A.; Chung, K.F.; Adcock, I.; Barnes, P.J.; et al. Nitrosative stress in the bronchial mucosa of severe chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2005, 116, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Pesci, A.; Majori, M.; Cuomo, A.; Borciani, N.; Bertacco, S.; Cacciani, G.; Gabrielli, M. Neutrophils infiltrating bronchial epithelium in chronic obstructive pulmonary disease. Respir. Med. 1998, 92, 863–870. [Google Scholar] [CrossRef]
- Stockley, J.A.; Walton, G.M.; Lord, J.M.; Sapey, E. Aberrant neutrophil functions in stable chronic obstructive pulmonary disease: The neutrophil as an immunotherapeutic target. Int. Immunopharmacol. 2013, 17, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Sapey, E.; Stockley, J.A.; Greenwood, H.; Ahmad, A.; Bayley, D.; Lord, J.M.; Insall, R.H.; Stockley, R.A. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 183, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Walton, G.M.; Begg, M.; Amour, A.; Hessel, E.M.; Usher, A.K.H.; Pottle, M.; Chadwick, C.; Stockley, R.; Stockley, J.A.; Sapey, E. Targeting phosphoinositide 3 kinase reduces damaging neutrophil functions without impairing bacterial phagocytosis in COPD. In Proceedings of the American Thoracic Society International Conference, Denver, CO, USA, 2015; p. A2740.
- Sapey, E.; Greenwood, H.; Walton, G.; Mann, E.; Love, A.; Aaronson, N.; Insall, R.H.; Stockley, R.A.; Lord, J.M. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: Toward targeted treatments for immunosenescence. Blood 2014, 123, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Noguera, A.; Batle, S.; Miralles, C.; Iglesias, J.; Busquets, X.; MacNee, W.; Agusti, A.G. Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax 2001, 56, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Morrison, D.; Donaldson, K.; MacNee, W. Systemic oxidative stress in asthma, COPD, and smokers. Am. J. Respir. Crit. Care Med. 1996, 154, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Renkema, T.E.; Postma, D.S.; Noordhoek, J.A.; Sluiter, H.J.; Kauffman, H.F. Influence of in vivo prednisolone on increased in vitro O2− generation by neutrophils in emphysema. Eur. Respir. J. 1993, 6, 90–95. [Google Scholar] [PubMed]
- Paone, G.; Conti, V.; Vestri, A.; Leone, A.; Puglisi, G.; Benassi, F.; Brunetti, G.; Schmid, G.; Cammarella, I.; Terzano, C. Analysis of sputum markers in the evaluation of lung inflammation and functional impairment in symptomatic smokers and COPD patients. Dis. Mark. 2011, 31, 91–100. [Google Scholar] [CrossRef]
- Gupta, V.; Khan, A.; Higham, A.; Lemon, J.; Sriskantharajah, S.; Amour, A.; Hessel, E.M.; Southworth, T.; Singh, D. The effect of phosphatidylinositol-3 kinase inhibition on matrix metalloproteinase-9 and reactive oxygen species release from chronic obstructive pulmonary disease neutrophils. Int. Immunopharmacol. 2016, 35, 155–162. [Google Scholar] [CrossRef]
- Korkmaz, B.; Moreau, T.; Gauthier, F. Neutrophil elastase, proteinase 3 and cathepsin g: Physicochemical properties, activity and physiopathological functions. Biochimie 2008, 90, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Ginzberg, H.H.; Cherapanov, V.; Dong, Q.; Cantin, A.; McCulloch, C.A.; Shannon, P.T.; Downey, G.P. Neutrophil-mediated epithelial injury during transmigration: Role of elastase. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G705–G717. [Google Scholar] [PubMed]
- Takeyama, K.; Agusti, C.; Ueki, I.; Lausier, J.; Cardell, L.O.; Nadel, J.A. Neutrophil-dependent goblet cell degranulation: Role of membrane-bound elastase and adhesion molecules. Am. J. Physiol. 1998, 275, L294–L302. [Google Scholar] [PubMed]
- Gadek, J.E.; Fells, G.A.; Zimmerman, R.L.; Rennard, S.I.; Crystal, R.G. Antielastases of the human alveolar structures. Implications for the protease-antiprotease theory of emphysema. J. Clin Investig. 1981, 68, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Liou, T.G.; Campbell, E.J. Nonisotropic enzyme—Inhibitor interactions: A novel nonoxidative mechanism for quantum proteolysis by human neutrophils. Biochemistry 1995, 34, 16171–16177. [Google Scholar] [CrossRef] [PubMed]
- Churg, A.; Wang, R.D.; Xie, C.; Wright, J.L. Alpha-1-antitrypsin ameliorates cigarette smoke-induced emphysema in the mouse. Am. J. Respir. Crit. Care Med. 2003, 168, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Petrache, I.; Fijalkowska, I.; Medler, T.R.; Skirball, J.; Cruz, P.; Zhen, L.; Petrache, H.I.; Flotte, T.R.; Tuder, R.M. Alpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am. J. Pathol. 2006, 169, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Matheson, N.R.; Wong, P.S.; Travis, J. Enzymatic inactivation of human alpha-1-proteinase inhibitor by neutrophil myeloperoxidase. Biochem. Biophys. Res. Commun. 1979, 88, 402–409. [Google Scholar] [CrossRef]
- Shapiro, S.D. The macrophage in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999, 160, S29–32. [Google Scholar] [CrossRef] [PubMed]
- Aldonyte, R.; Jansson, L.; Piitulainen, E.; Janciauskiene, S. Circulating monocytes from healthy individuals and COPD patients. Respir. Res. 2003, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Tager, M.; Piecyk, A.; Kohnlein, T.; Thiel, U.; Ansorge, S.; Welte, T. Evidence of a defective thiol status of alveolar macrophages from COPD patients and smokers. Chronic obstructive pulmonary disease. Free Radic Biol. Med. 2000, 29, 1160–1165. [Google Scholar] [CrossRef]
- Tomaki, M.; Sugiura, H.; Koarai, A.; Komaki, Y.; Akita, T.; Matsumoto, T.; Nakanishi, A.; Ogawa, H.; Hattori, T.; Ichinose, M. Decreased expression of antioxidant enzymes and increased expression of chemokines in COPD lung. Pulm. Pharmacol. Ther. 2007, 20, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Capelli, A.; Lusuardi, M.; Balbo, P.; Vecchio, C.; Maestrelli, P.; Mapp, C.E.; Fabbri, L.M.; Donner, C.F.; Saetta, M. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am. J. Respir. Crit. Care Med. 1998, 158, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Hodge, G.; Scicchitano, R.; Reynolds, P.N.; Holmes, M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol. Cell. Biol. 2003, 81, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Hodge, S.; Matthews, G.; Mukaro, V.; Ahern, J.; Shivam, A.; Hodge, G.; Holmes, M.; Jersmann, H.; Reynolds, P.N. Cigarette smoke-induced changes to alveolar macrophage phenotype and function are improved by treatment with procysteine. Am. J. Respir. Cell. Mol. Biol. 2011, 44, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Lofdahl, J.M.; Wahlstrom, J.; Skold, C.M. Different inflammatory cell pattern and macrophage phenotype in chronic obstructive pulmonary disease patients, smokers and non-smokers. Clin. Exp. Immunol. 2006, 145, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Shaykhiev, R.; Krause, A.; Salit, J.; Strulovici-Barel, Y.; Harvey, B.G.; O’Connor, T.P.; Crystal, R.G. Smoking-dependent reprogramming of alveolar macrophage polarization: Implication for pathogenesis of chronic obstructive pulmonary disease. J. Immunol. 2009, 183, 2867–2883. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Ito, M.; Elliott, W.M.; Cosio, B.; Caramori, G.; Kon, O.M.; Barczyk, A.; Hayashi, S.; Adcock, I.M.; Hogg, J.C.; et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N. Engl. J. Med. 2005, 352, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Vandivier, R.W.; Fadok, V.A.; Hoffmann, P.R.; Bratton, D.L.; Penvari, C.; Brown, K.K.; Brain, J.D.; Accurso, F.J.; Henson, P.M. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J. Clin. Investig. 2002, 109, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Leikauf, G.D.; Simpson, L.G.; Santrock, J.; Zhao, Q.; Abbinante-Nissen, J.; Zhou, S.; Driscoll, K.E. Airway epithelial cell responses to ozone injury. Environ. Health Perspect. 1995, 103, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Bagdonas, E.; Raudoniute, J.; Bruzauskaite, I.; Aldonyte, R. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int. J. Chronic Obstruct. Pulm. Dis. 2015, 10, 995–1013. [Google Scholar]
- Smit-de Vries, M.P.; van der Toorn, M.; Bischoff, R.; Kauffman, H.F. Resistance of quiescent and proliferating airway epithelial cells to H2O2 challenge. Eur. Respir. J. 2007, 29, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Hampton, M.B.; Stamenkovic, I.; Winterbourn, C.C. Interaction with substrate sensitises caspase-3 to inactivation by hydrogen peroxide. FEBS Lett. 2002, 517, 229–232. [Google Scholar] [CrossRef]
- Baker, A.; Santos, B.D.; Powis, G. Redox control of caspase-3 activity by thioredoxin and other reduced proteins. Biochem. Biophys. Res. Commun. 2000, 268, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Thornton, D.J.; Rousseau, K.; McGuckin, M.A. Structure and function of the polymeric mucins in airways mucus. Annu. Rev. Physiol. 2008, 70, 459–486. [Google Scholar] [CrossRef] [PubMed]
- Sapey, E.; Bayley, D.; Ahmad, A.; Newbold, P.; Snell, N.; Stockley, R.A. Inter-relationships between inflammatory markers in patients with stable COPD with bronchitis: Intra-patient and inter-patient variability. Thorax 2008, 63, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wiegman, C.; Seiffert, J.M.; Zhu, J.; Clarke, C.; Chang, Y.; Bhavsar, P.; Adcock, I.; Zhang, J.; Zhou, X.; et al. Effects of N-acetylcysteine in ozone-induced chronic obstructive pulmonary disease model. PLoS ONE 2013, 8, e80782. [Google Scholar] [CrossRef] [PubMed]
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.; Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Russell, K.E.; Chung, K.F.; Clarke, C.J.; Durham, A.L.; Mallia, P.; Footitt, J.; Johnston, S.L.; Barnes, P.J.; Hall, S.R.; Simpson, K.D.; et al. The mif antagonist iso-1 attenuates corticosteroid-insensitive inflammation and airways hyperresponsiveness in an ozone-induced model of COPD. PLoS ONE 2016, 11, e0146102. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Hoshino, Y.; Hara, T.; Muro, S.; Nakamura, H.; Mishima, M.; Yodoi, J. Thioredoxin-1 ameliorates cigarette smoke-induced lung inflammation and emphysema in mice. J. Pharmacol. Exp. Ther. 2008, 325, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Belchamber, K.B.; Singh, R.; Wedzicha, J.A.; Barnes, P.J.; Donnelly, L.E. Elevated mitochondrial reactive oxygen species in COPD macrophages at exacerbation and with bacterial phagocytosis. Am. J. Respir. Crit. Care 2015, 191, A6377. [Google Scholar]
- Hara, H.; Araya, J.; Ito, S.; Kobayashi, K.; Takasaka, N.; Yoshii, Y.; Wakui, H.; Kojima, J.; Shimizu, K.; Numata, T.; et al. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L737–L746. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; de Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res. 2013, 14, 97. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Shameem, M.; Husain, Q. Altered oxidant-antioxidant levels in the disease prognosis of chronic obstructive pulmonary disease. Int. J. Tuberc. Lung Dis. 2013, 17, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Tavilani, H.; Nadi, E.; Karimi, J.; Goodarzi, M.T. Oxidative stress in COPD patients, smokers, and non-smokers. Respir. Care 2012, 57, 2090–2094. [Google Scholar] [CrossRef] [PubMed]
- Betsuyaku, T.; Fuke, S.; Inomata, T.; Kaga, K.; Morikawa, T.; Odajima, N.; Adair-Kirk, T.; Nishimura, M. Bronchiolar epithelial catalase is diminished in smokers with mild COPD. Eur. Respir. J. 2013, 42, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.C.; Oliveira, A.L.; Viegas-Crespo, A.M.; Vicente, L.; Barreiros, A.; Monteiro, P.; Pinheiro, T.; Bugalho De Almeida, A. Systemic markers of the redox balance in chronic obstructive pulmonary disease. Biomarkers 2004, 9, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Maestrelli, P.; Páska, C.; Saetta, M.; Turato, G.; Nowicki, Y.; Monti, S.; Formichi, B.; Miniati, M.; Fabbri, L.M. Decreased haem oxygenase-1 and increased inducible nitric oxide synthase in the lung of severe COPD patients. European Respir. J. 2003, 21, 971–976. [Google Scholar] [CrossRef]
- Drost, E.M.; Skwarski, K.M.; Sauleda, J.; Soler, N.; Roca, J.; Agusti, A.; MacNee, W. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax 2005, 60, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Siedlinski, M.; van Diemen, C.C.; Postma, D.S.; Vonk, J.M.; Boezen, H.M. Superoxide dismutases, lung function and bronchial responsiveness in a general population. Eur. Respir. J. 2009, 33, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Kinnula, V.L.; Crapo, J.D. Superoxide dismutases in the lung and human lung diseases. Am. J. Respir Crit. Care Med. 2003, 167, 1600–1619. [Google Scholar] [CrossRef]
- Juul, K.; Tybjaerg-Hansen, A.; Marklund, S.; Heegaard, N.H.; Steffensen, R.; Sillesen, H.; Jensen, G.; Nordestgaard, B.G. Genetically reduced antioxidative protection and increased ischemic heart disease risk: The copenhagen city heart study. Circulation 2004, 109, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Bowler, R.P.; Nicks, M.; Olsen, D.A.; Thogersen, I.B.; Valnickova, Z.; Hojrup, P.; Franzusoff, A.; Enghild, J.J.; Crapo, J.D. Furin proteolytically processes the heparin-binding region of extracellular superoxide dismutase. J. Biol. Chem. 2002, 277, 16505–16511. [Google Scholar] [CrossRef] [PubMed]
- Dahl, M.; Bowler, R.P.; Juul, K.; Crapo, J.D.; Levy, S.; Nordestgaard, B.G. Superoxide dismutase 3 polymorphism associated with reduced lung function in two large populations. Am. J. Respir. Crit. Care Med. 2008, 178, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Vibhuti, A.; Arif, E.; Deepak, D.; Singh, B.; Qadar Pasha, M.A. Correlation of oxidative status with bmi and lung function in COPD. Clin. Biochem. 2007, 40, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Aruoma, O.I.; Halliwell, B.; Hoey, B.M.; Butler, J. The antioxidant action of N-acetylcysteine: Its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med. 1989, 6, 593–597. [Google Scholar] [CrossRef]
- Decramer, M.; Rutten-van Molken, M.; Dekhuijzen, P.N.; Troosters, T.; van Herwaarden, C.; Pellegrino, R.; van Schayck, C.P.; Olivieri, D.; Del Donno, M.; De Backer, W.; et al. Effects of n-acetylcysteine on outcomes in chronic obstructive pulmonary disease (bronchitis randomized on nac cost-utility study, broncus): A randomised placebo-controlled trial. Lancet 2005, 365, 1552–1560. [Google Scholar] [CrossRef]
- Zheng, J.-P.; Wen, F.-Q.; Bai, C.-X.; Wan, H.-Y.; Kang, J.; Chen, P.; Yao, W.-Z.; Ma, L.-J.; Li, X.; Raiteri, L.; et al. Twice daily n-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (pantheon): A randomised, double-blind placebo-controlled trial. Lancet Respir. Med. 2014, 2, 187–194. [Google Scholar] [CrossRef]
- Cazzola, M.; Calzetta, L.; Page, C.; Jardim, J.; Chuchalin, A.G.; Rogliani, P.; Matera, M.G. Influence of n-acetylcysteine on chronic bronchitis or COPD exacerbations: A meta-analysis. Eur. Respir. Rev. 2015, 24, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Borok, Z.; Buhl, R.; Grimes, G.J.; Bokser, A.D.; Hubbard, R.C.; Holroyd, K.J.; Roum, J.H.; Czerski, D.B.; Cantin, A.M.; Crystal, R.G. Effect of glutathione aerosol on oxidant-antioxidant imbalance in idiopathic pulmonary fibrosis. Lancet 1991, 338, 215–216. [Google Scholar] [CrossRef]
- Marrades, R.M.; Roca, J.; Barbera, J.A.; de Jover, L.; MacNee, W.; Rodriguez-Roisin, R. Nebulized glutathione induces bronchoconstriction in patients with mild asthma. Am. J. Respir. Crit. Care Med. 1997, 156, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2 small maf heterodimer mediates the induction of phase ii detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Goven, D.; Boutten, A.; Lecon-Malas, V.; Marchal-Somme, J.; Amara, N.; Crestani, B.; Fournier, M.; Leseche, G.; Soler, P.; Boczkowski, J.; et al. Altered Nrf2/keap1-bach1 equilibrium in pulmonary emphysema. Thorax 2008, 63, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Betsuyaku, T.; Ito, Y.; Nagai, K.; Nasuhara, Y.; Kaga, K.; Kondo, S.; Nishimura, M. Down-regulated Nf-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Cell. Mol. Biol. 2008, 39, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Ishii, Y.; Itoh, K.; Kiwamoto, T.; Kimura, T.; Matsuno, Y.; Morishima, Y.; Hegab, A.E.; Homma, S.; Nomura, A.; et al. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 2005, 10, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Sussan, T.E.; Rangasamy, T.; Blake, D.J.; Malhotra, D.; El-Haddad, H.; Bedja, D.; Yates, M.S.; Kombairaju, P.; Yamamoto, M.; Liby, K.T.; et al. Targeting Nrf2 with the triterpenoid cddo-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Sethi, S.; Kong, X.; Yarmus, L.; Brown, R.H.; Feller-Kopman, D.; Wise, R.; Biswal, S. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci. Transl. Med. 2011, 3, 78ra32. [Google Scholar] [CrossRef] [PubMed]
- Kesic, M.J.; Simmons, S.O.; Bauer, R.; Jaspers, I. Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free Radic. Biol. Med. 2011, 51, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Sargeant, L.A.; Jaeckel, A.; Wareham, N.J. Interaction of vitamin c with the relation between smoking and obstructive airways disease in epic norfolk. European prospective investigation into cancer and nutrition. Eur. Respir. J. 2000, 16, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Tsiligianni, I.G.; van der Molen, T. A systematic review of the role of vitamin insufficiencies and supplementation in COPD. Respir. Res. 2010, 11, 171. [Google Scholar] [CrossRef] [PubMed]
- Agler, A.H.; Kurth, T.; Gaziano, J.M.; Buring, J.E.; Cassano, P.A. Randomised vitamin e supplementation and risk of chronic lung disease in the women’s health study. Thorax 2011, 66, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Rautalahti, M.; Virtamo, J.; Haukka, J.; Heinonen, O.P.; Sundvall, J.; Albanes, D.; Huttunen, J.K. The effect of alpha-tocopherol and beta-carotene supplementation on COPD symptoms. Am. J. Respir. Crit. Care Med. 1997, 156, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2012, 3, CD007176. [Google Scholar]
- Rahman, I. Pharmacological antioxidant strategies as therapeutic interventions for COPD. Biochim. Biophys. Acta 2012, 1822, 714–728. [Google Scholar] [CrossRef] [PubMed]
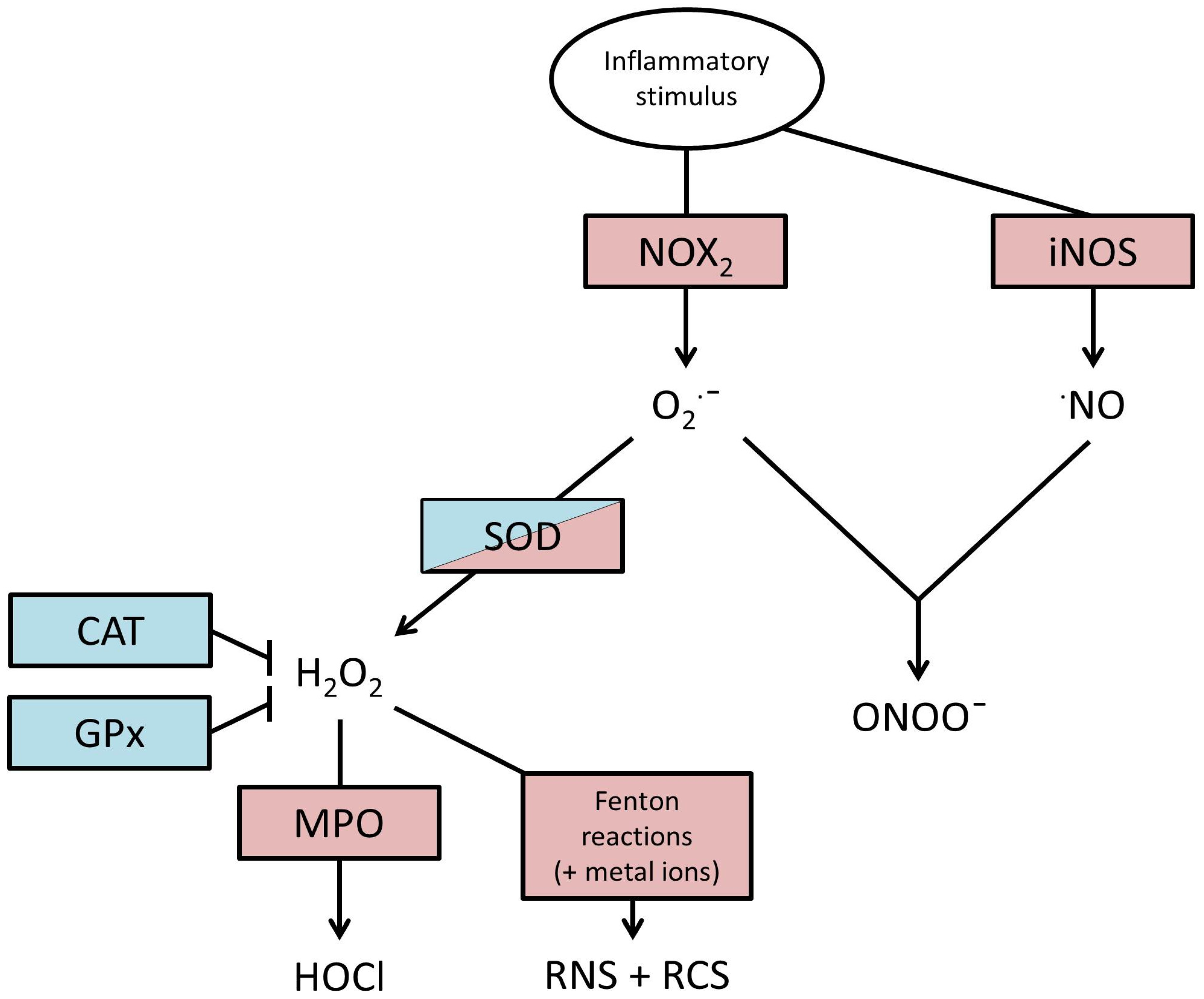
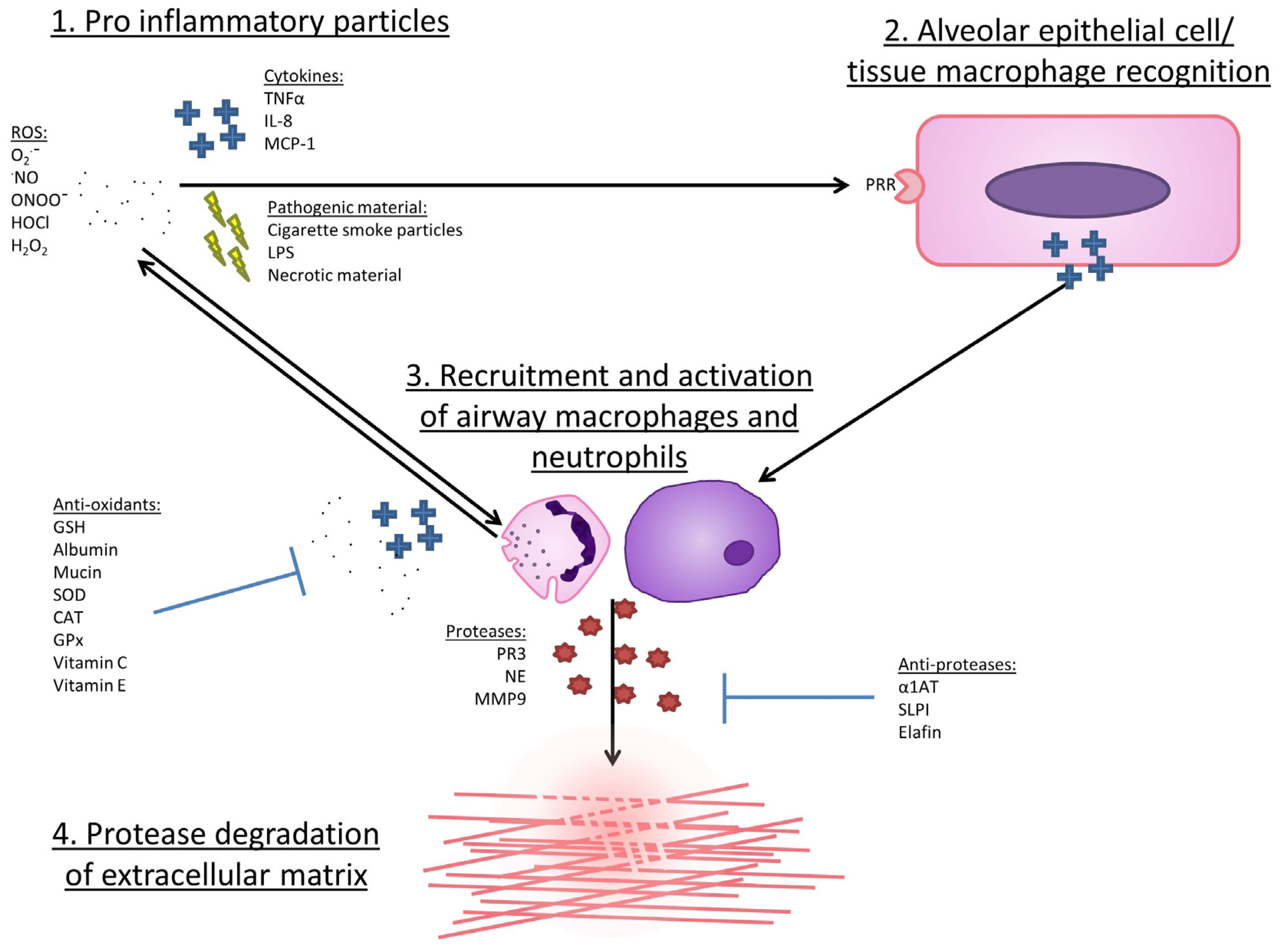

{kind=link}
{kind=link}
{kind=link}
| Functions of ROS | Potentially Related Features of COPD |
|---|---|
| Alteration of biological molecules: Thiols [22] Amines [23] Amino acid residues [24] Charge profiles and disulphide bond formation [25,26] DNA/RNA [31] | Reduced antioxidant and antiprotease enzyme activity: SOD [98,99] Catalase [98,99] GPx [98] α1AT [27,28] |
| Altered expression of ROS related enzymes: ↓ Catalase [75,100] ↓ GPx [50,101] ↓ SOD [98] ↑ iNOS [102] | |
| Mitochondrial respiration [32] | Altered mitochondrial function [92,95,96,97] |
| Intracellular signalling [33,34] | Altered expression of ROS related enzymes: ↓ Catalase [75,100] ↓ GPx [50,101] ↓ SOD [98] ↑ iNOS [102] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGuinness, A.J.A.; Sapey, E. Oxidative Stress in COPD: Sources, Markers, and Potential Mechanisms. J. Clin. Med. 2017, 6, 21. https://doi.org/10.3390/jcm6020021
McGuinness AJA, Sapey E. Oxidative Stress in COPD: Sources, Markers, and Potential Mechanisms. Journal of Clinical Medicine. 2017; 6(2):21. https://doi.org/10.3390/jcm6020021
Chicago/Turabian StyleMcGuinness, Adam John Anthony, and Elizabeth Sapey. 2017. "Oxidative Stress in COPD: Sources, Markers, and Potential Mechanisms" Journal of Clinical Medicine 6, no. 2: 21. https://doi.org/10.3390/jcm6020021
APA StyleMcGuinness, A. J. A., & Sapey, E. (2017). Oxidative Stress in COPD: Sources, Markers, and Potential Mechanisms. Journal of Clinical Medicine, 6(2), 21. https://doi.org/10.3390/jcm6020021