
CVD and Oxidative Stress



Abstract
:1. Introduction
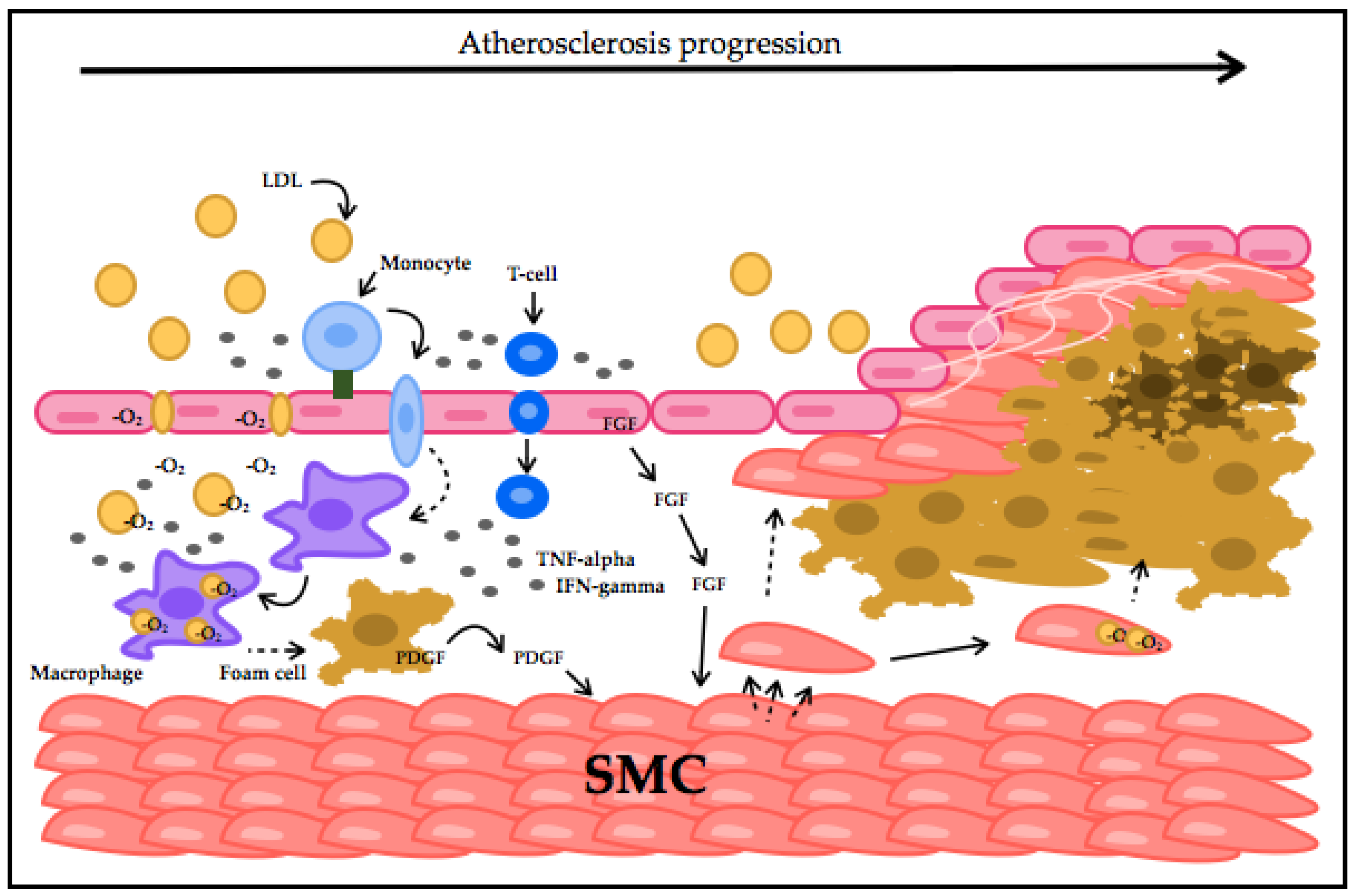
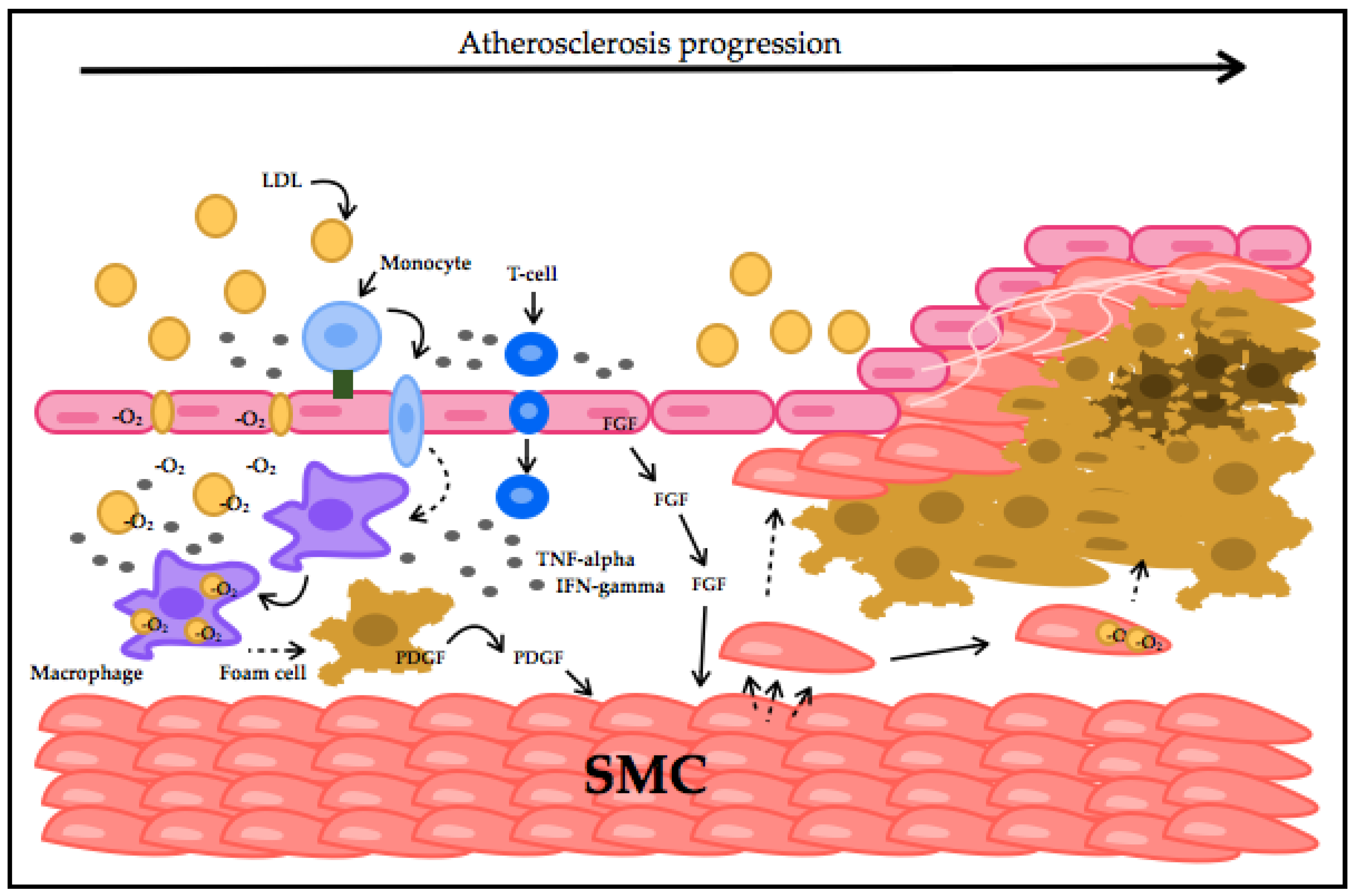
2. CVD Highlights and Their Relationship with Oxidative Stress
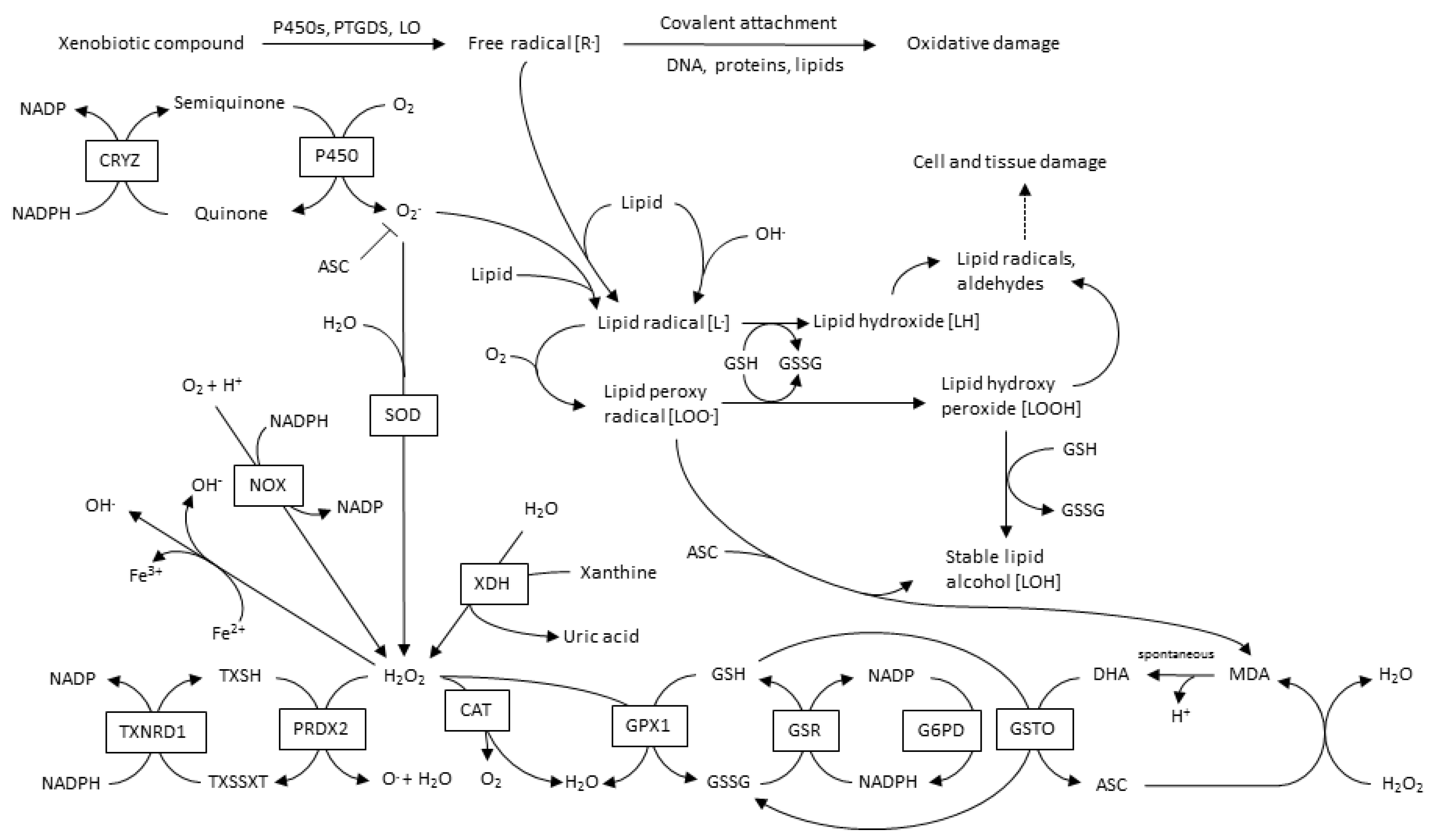
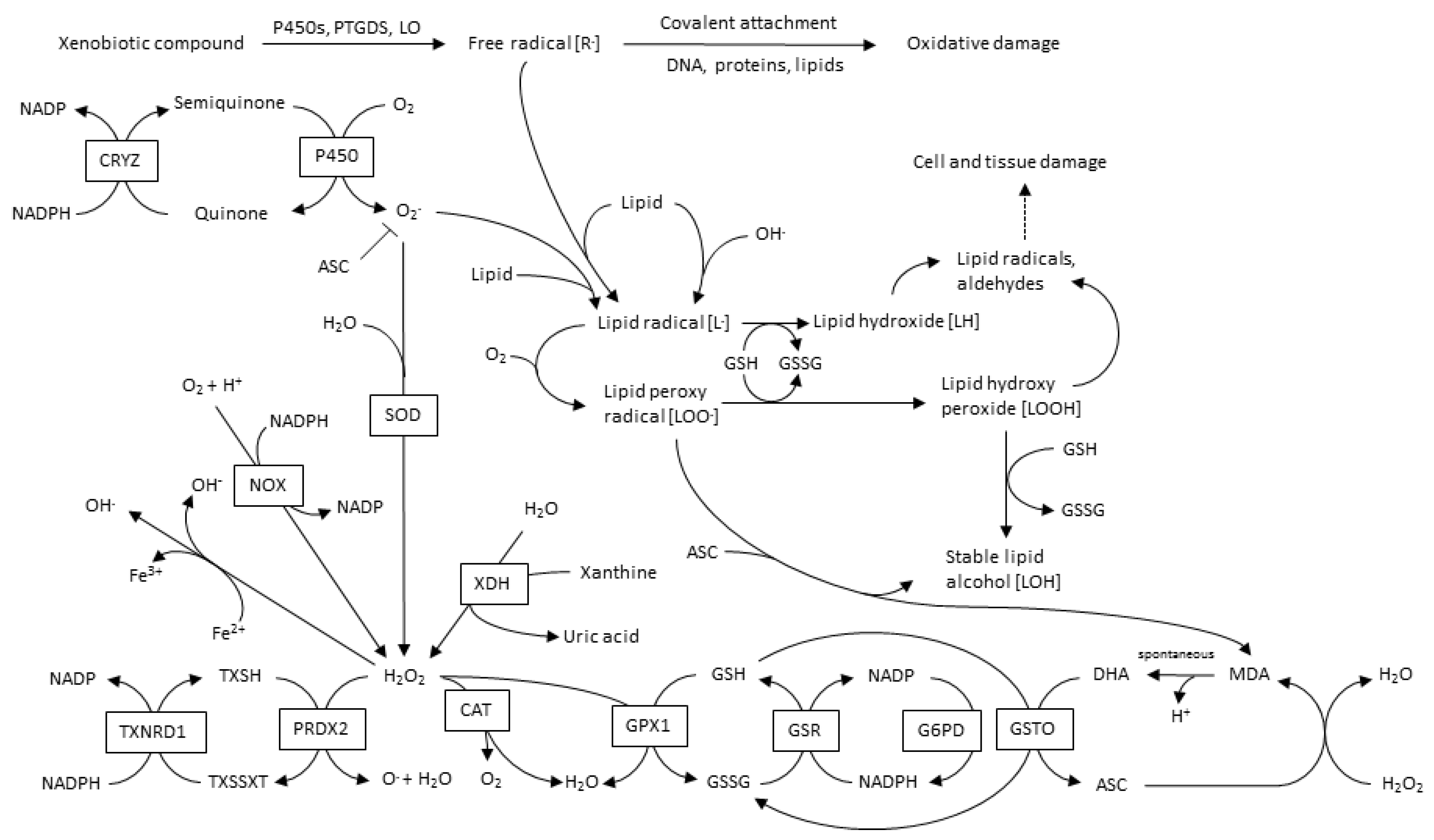
3. ROS Types and Principal Sources in CVD
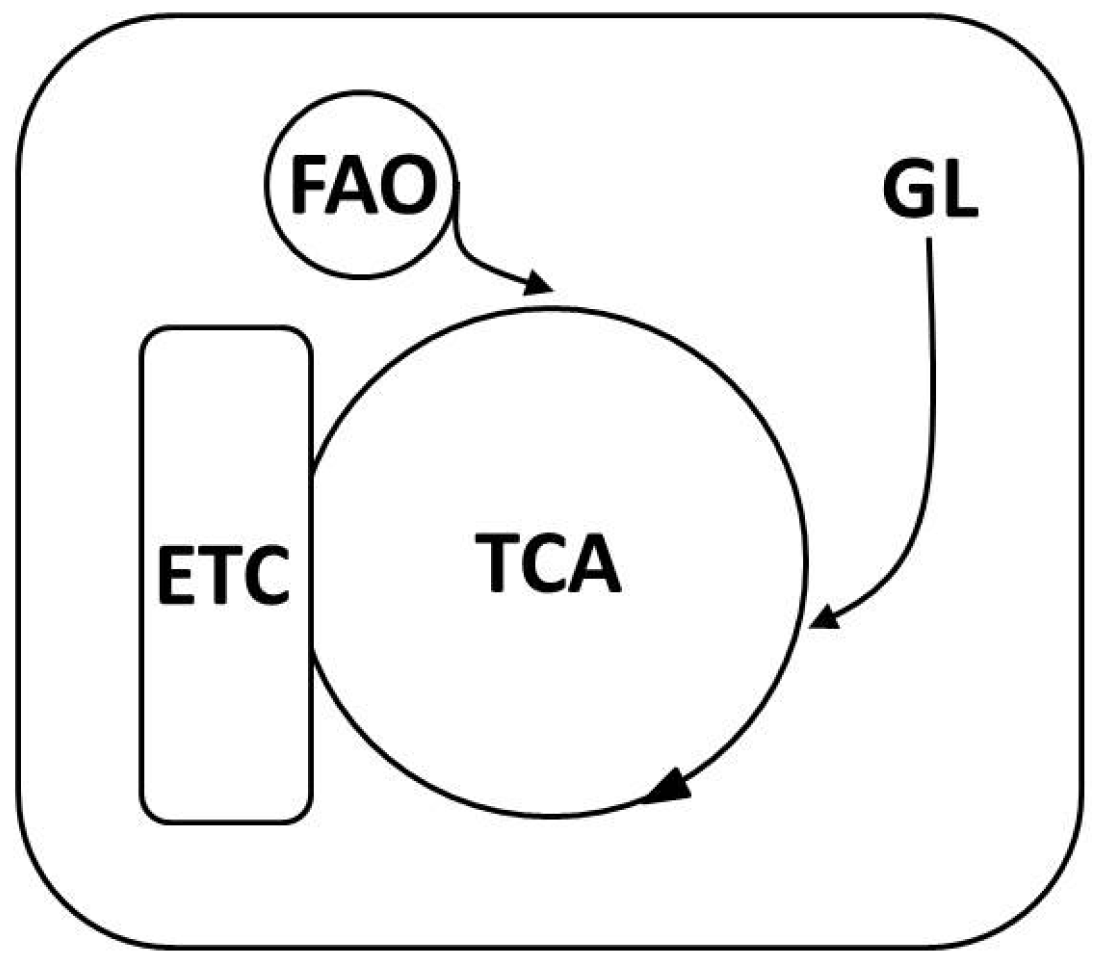
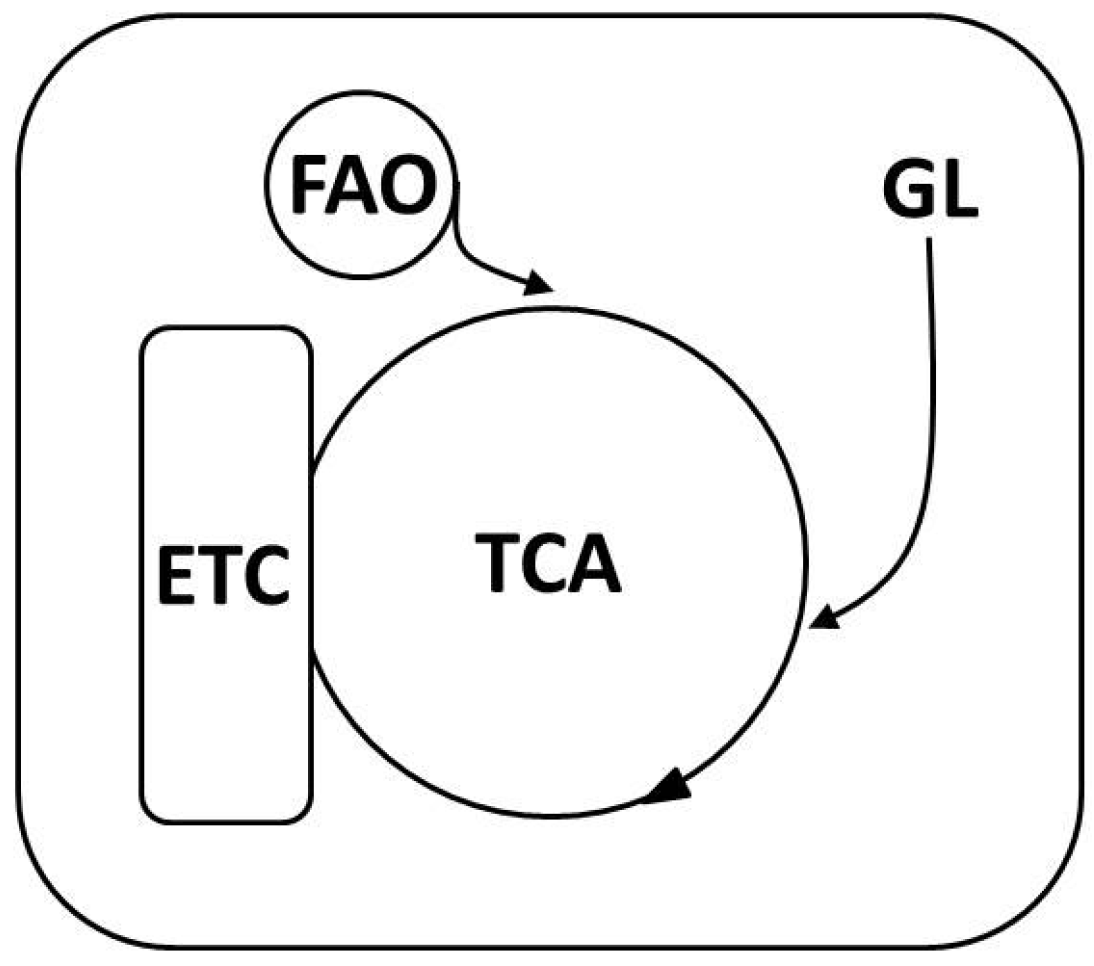
3.1. Mitochondrial-ROS
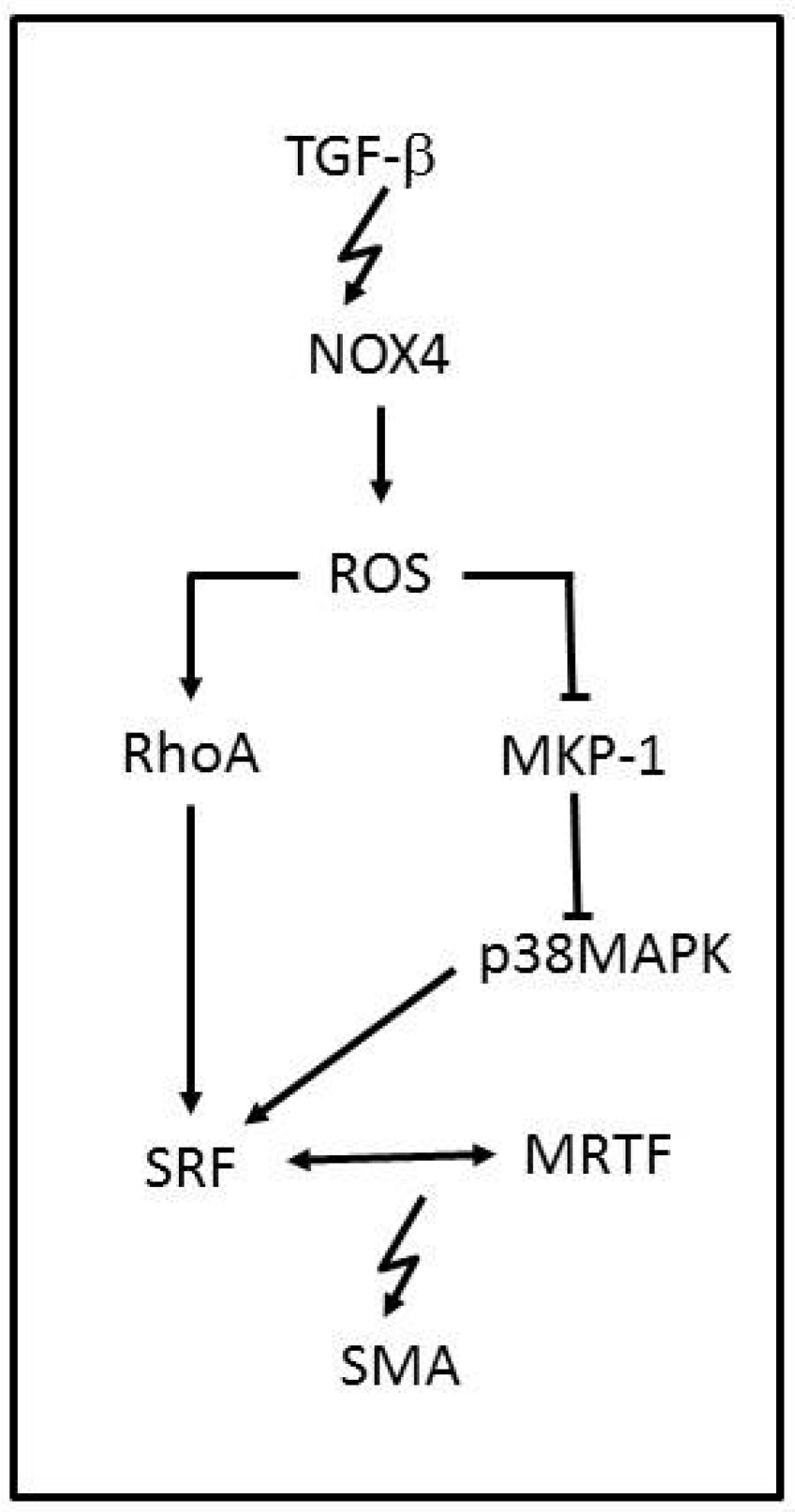
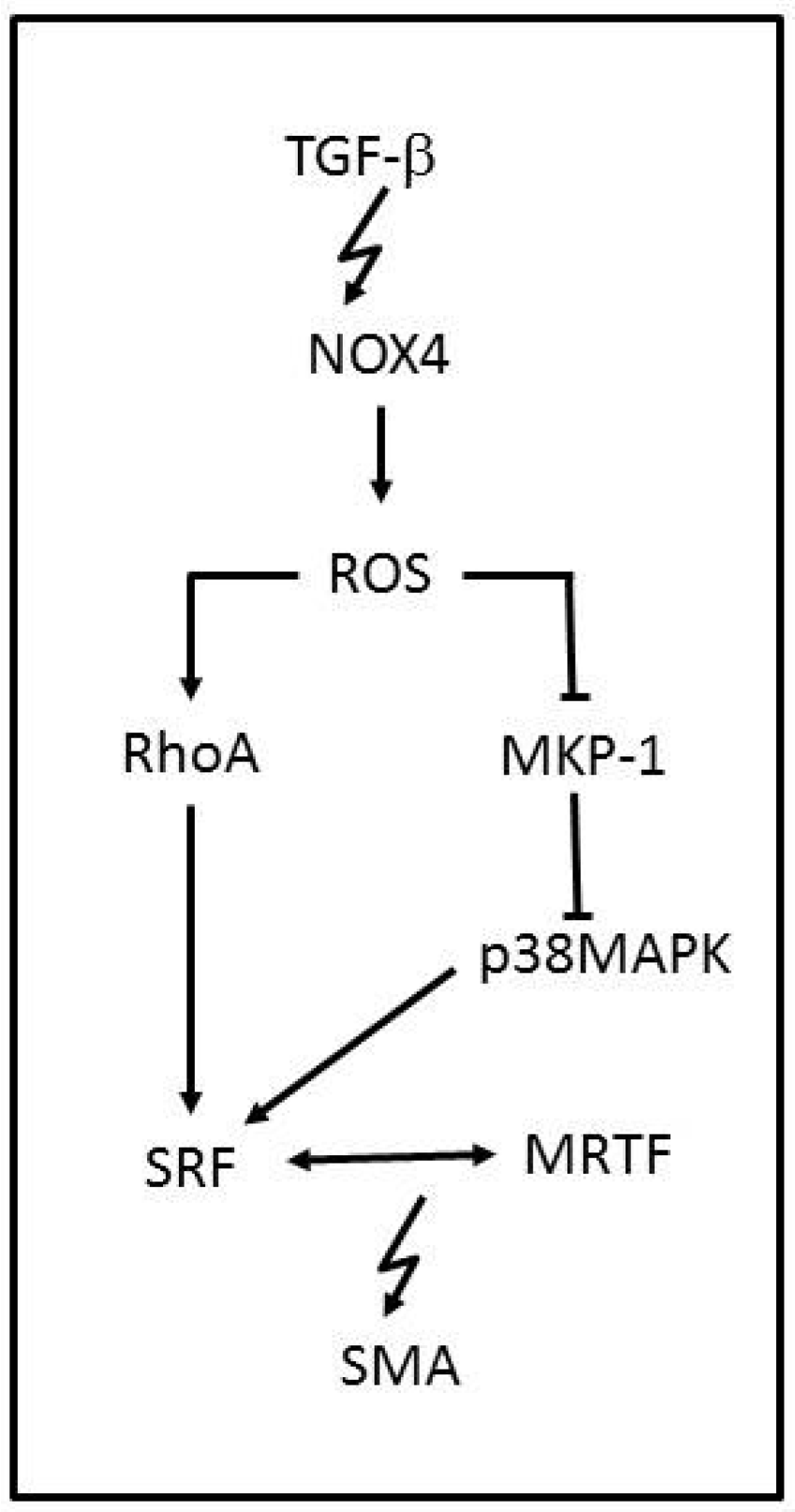
3.2. NADPH Oxidases
3.3. Xanthine Oxidase
3.4. Lipoxygenase
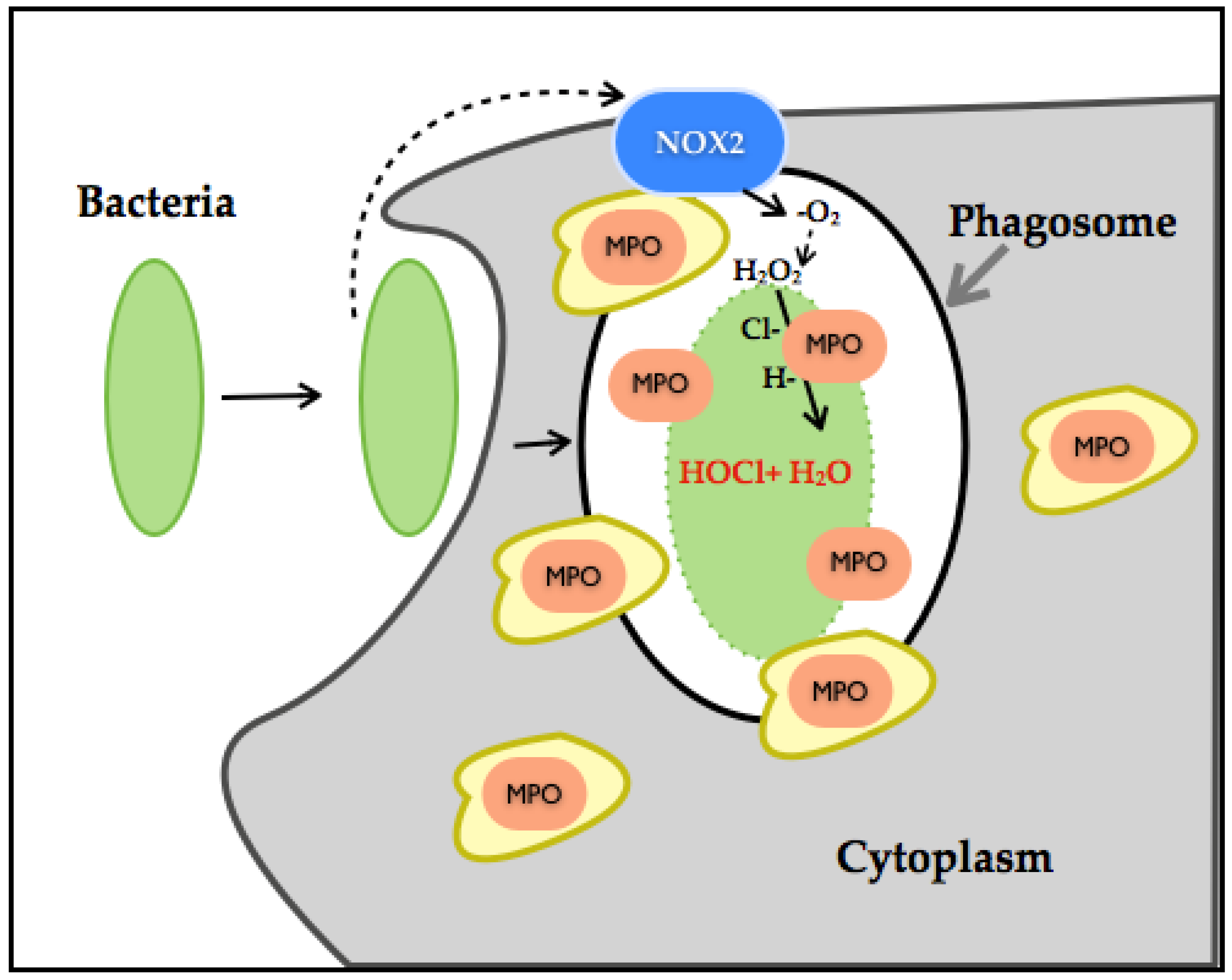
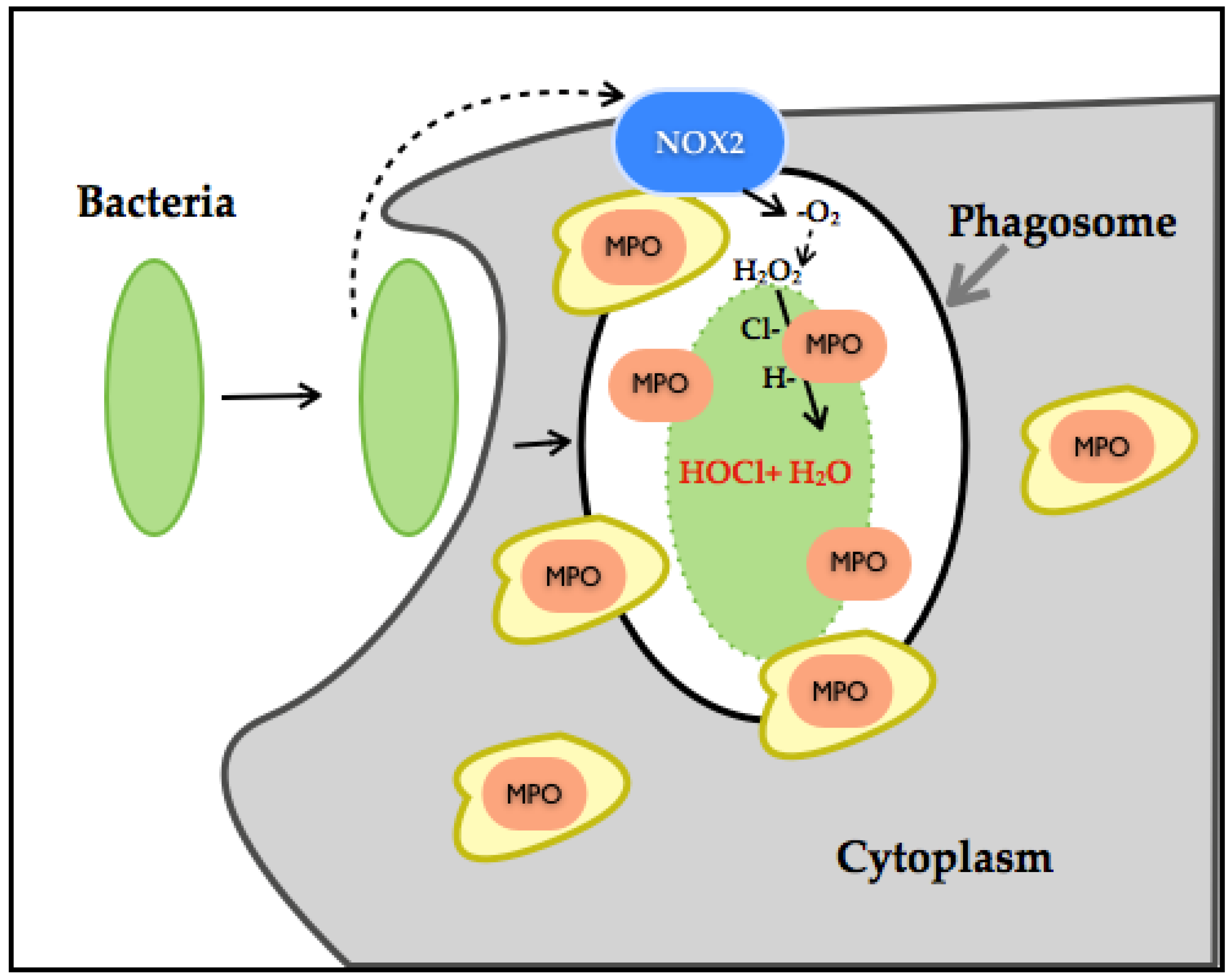
3.5. Myeloperoxidase
4. Common Pharmacological Approaches for CVD and Their Relationship with ROS
5. ROS Biomarkers, Novel Therapies and Challenges
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Mittal, C.K.; Murad, F. Activation of guanylate cyclase by superoxide dismutase and hydroxyl radical: A physiological regulator of guanosine 3′,5′-monophosphate formation. Proc. Natl. Acad. Sci. USA 1977, 74, 4360–4364. [Google Scholar] [CrossRef] [PubMed]
- Olguin-Albuerne, M.; Moran, J. Ros produced by NOX2 control in vitro development of cerebellar granule neurons development. ASN Neuro 2015. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Fu, P.; Levine, A.D. REDOX regulation of IL-13 signaling in intestinal epithelial cells: Usage of alternate pathways mediates distinct gene expression patterns. Cell. Signal. 2010, 22, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Abimannan, T.; Peroumal, D.; Parida, J.R.; Barik, P.K.; Padhan, P.; Devadas, S. Oxidative stress modulates the cytokine response of differentiated Th17 and Th1 cells. Free Radic. Biol. Med. 2016, 99, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Fujino, G.; Noguchi, T.; Matsuzawa, A.; Yamauchi, S.; Saitoh, M.; Takeda, K.; Ichijo, H. Thioredoxin and TRAF family proteins regulate reactive oxygen species-dependent activation of ASK1 through reciprocal modulation of the N-terminal homophilic interaction of ASK1. Mol. Cell. Biol. 2007, 27, 8152–8163. [Google Scholar] [CrossRef] [PubMed]
- Heppner, D.E.; Hristova, M.; Dustin, C.M.; Danyal, K.; Habibovic, A.; van der Vliet, A. The NADPH oxidases DUOX1 and NOX2 play distinct roles in redox regulation of epidermal growth factor receptor signaling. J. Biol. Chem. 2016, 291, 23282–23293. [Google Scholar] [CrossRef] [PubMed]
- Sart, S.; Song, L.; Li, Y. Controlling redox status for stem cell survival, expansion, and differentiation. Oxid. Med. Cell. Longev. 2015, 2015, 105135. [Google Scholar] [CrossRef] [PubMed]
- Krumova, K.; Cosa, G. Overview of reactive oxygen species. In Singlet Oxygen: Applications in Biosciences and Nanosciences; The Royal Society of Chemistry: London, UK, 2016; Chapter 1; Volume 1, pp. 1–21. [Google Scholar]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Austin, V.; Crack, P.J.; Bozinovski, S.; Miller, A.A.; Vlahos, R. Copd and stroke: Are systemic inflammation and oxidative stress the missing links? Clin. Sci. 2016, 130, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Mendis, S.; Puska, P.; Norrving, B. Global Atlas on Cardiovascular Disease Prevention and Control; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; et al. Heart disease and stroke statistics—2016 update: A report from the american heart association. Circulation 2016, 133, e38–e360. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Blaha, M.J.; Dai, S.; Ford, E.S.; Fox, C.S.; Franco, S.; et al. Heart disease and stroke statistics—2014 update: A report from the american heart association. Circulation 2014, 129, e28–e292. [Google Scholar] [CrossRef]
- Mehta, J.L.; Saldeen, T.G.; Rand, K. Interactive role of infection, inflammation and traditional risk factors in atherosclerosis and coronary artery disease. J. Am. Coll. Cardiol. 1998, 31, 1217–1225. [Google Scholar] [CrossRef]
- Zwaka, T.P.; Hombach, V.; Torzewski, J. C-reactive protein-mediated low density lipoprotein uptake by macrophages: Implications for atherosclerosis. Circulation 2001, 103, 1194–1197. [Google Scholar] [CrossRef] [PubMed]
- Schuh, J.; Fairclough, G.F.; Haschemeyer, R.H. Oxygen-mediated heterogeneity of apo-low-density lipoprotein. Proc. Natl. Acad. Sci. USA 1978, 75, 3173–3177. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H. The AGE-receptor in the pathogenesis of diabetic complications. Diabetes Metab. Res. Rev. 2001, 17, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Ho, Y.K.; Basu, S.K.; Brown, M.S. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. USA 1979, 76, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Steinbrecher, U.P.; Fisher, M.; Witztum, J.L.; Curtiss, L.K. Immunogenicity of homologous low density lipoprotein after methylation, ethylation, acetylation, or carbamylation: Generation of antibodies specific for derivatized lysine. J. Lipid Res. 1984, 25, 1109–1116. [Google Scholar] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Avogaro, P.; Cazzolato, G.; Bittolo-Bon, G. Some questions concerning a small, more electronegative LDL circulating in human plasma. Atherosclerosis 1991, 91, 163–171. [Google Scholar] [CrossRef]
- Brownlee, M.; Cerami, A.; Vlassara, H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N. Engl. J. Med. 1988, 318, 1315–1321. [Google Scholar] [PubMed]
- Witztum, J.L.; Mahoney, E.M.; Branks, M.J.; Fisher, M.; Elam, R.; Steinberg, D. Nonenzymatic glucosylation of low-density lipoprotein alters its biologic activity. Diabetes 1982, 31, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Bucala, R.; Makita, Z.; Koschinsky, T.; Cerami, A.; Vlassara, H. Lipid advanced glycosylation: Pathway for lipid oxidation in vivo. Proc. Natl. Acad. Sci. USA 1993, 90, 6434–6438. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.L.; Laimins, M.; Lopes-Virella, M.F. Isolation, characterization, and metabolism of the glycated and nonglycated subfractions of low-density lipoproteins isolated from type I diabetic patients and nondiabetic subjects. Diabetes 1995, 44, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Sobal, G.; Menzel, J.; Sinzinger, H. Why is glycated LDL more sensitive to oxidation than native LDL? A comparative study. Prostaglandins Leukot. Essent. Fatty Acids 2000, 63, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and atherosclerosis. Mediat. Inflam. 2013, 2013, 152786. [Google Scholar] [CrossRef] [PubMed]
- Raggi, P. Inflammation, depression and atherosclerosis or depression, inflammation and atherosclerosis? Atherosclerosis 2016, 251, 542–543. [Google Scholar] [CrossRef] [PubMed]
- Assies, J.; Mocking, R.J.; Lok, A.; Ruhe, H.G.; Pouwer, F.; Schene, A.H. Effects of oxidative stress on fatty acid- and one-carbon-metabolism in psychiatric and cardiovascular disease comorbidity. Acta Psychiatr. Scand. 2014, 130, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive oxygen species: A key hallmark of cardiovascular disease. Adv. Med. 2016, 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Griendling, K.K. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ. Res. 2015, 116, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial oxidative stress, mitochondrial DNA damage and their role in age-related vascular dysfunction. Int. J. Mol. Sci. 2015, 16, 15918–15953. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.K. Antimycin-insensitive oxidation of succinate and reduced nicotinamide-adenine dinucleotide in electron-transport particles. I. Ph dependency and hydrogen peroxide formation. Biochim. Biophys. Acta 1966, 122, 157–166. [Google Scholar] [CrossRef]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [PubMed]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, P.; Mollnau, H.; Oelze, M.; Schulz, E.; Wickramanayake, J.M.; Muller, J.; Schuhmacher, S.; Hortmann, M.; Baldus, S.; Gori, T.; et al. First evidence for a crosstalk between mitochondrial and NADPH oxidase-derived reactive oxygen species in nitroglycerin-triggered vascular dysfunction. Antioxid. Redox Signal. 2008, 10, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin ii-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of NOX4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Nazarewicz, R.R.; Dikalova, A.E.; Bikineyeva, A.; Dikalov, S.I. NOX2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1131–H1140. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Nazarewicz, R.R.; Bikineyeva, A.; Hilenski, L.; Lassegue, B.; Griendling, K.K.; Harrison, D.G.; Dikalova, A.E. NOX2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid. Redox Signal. 2014, 20, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I.; Fisslthaler, B.; Dimmeler, S.; Kemp, B.E.; Busse, R. Phosphorylation of Thr495 regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 2001, 88, E68–E75. [Google Scholar] [CrossRef]
- Lin, M.I.; Fulton, D.; Babbitt, R.; Fleming, I.; Busse, R.; Pritchard, K.A., Jr.; Sessa, W.C. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of l-arginine metabolism to efficient nitric oxide production. J. Biol. Chem. 2003, 278, 44719–44726. [Google Scholar] [CrossRef] [PubMed]
- Loot, A.E.; Schreiber, J.G.; Fisslthaler, B.; Fleming, I. Angiotensin II impairs endothelial function via tyrosine phosphorylation of the endothelial nitric oxide synthase. J. Exp. Med. 2009, 206, 2889–2896. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Mann, G.E. Vascular NAD(p)h oxidase activation in diabetes: A double-edged sword in redox signalling. Cardiovasc. Res. 2009, 82, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Abel, E.D. Mitochondria in the diabetic heart. Cardiovasc. Res. 2010, 88, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Kayama, Y.; Raaz, U.; Jagger, A.; Adam, M.; Schellinger, I.N.; Sakamoto, M.; Suzuki, H.; Toyama, K.; Spin, J.M.; Tsao, P.S. Diabetic cardiovascular disease induced by oxidative stress. Int. J. Mol. Sci. 2015, 16, 25234–25263. [Google Scholar] [CrossRef]
- Quast, U.; Stephan, D.; Bieger, S.; Russ, U. The impact of ATP-sensitive k+ channel subtype selectivity of insulin secretagogues for the coronary vasculature and the myocardium. Diabetes 2004, 53, S156–S164. [Google Scholar] [CrossRef] [PubMed]
- Madungwe, N.B.; Zilberstein, N.F.; Feng, Y.; Bopassa, J.C. Critical role of mitochondrial ROS is dependent on their site of production on the electron transport chain in ischemic heart. Am. J. Cardiovasc. Dis. 2016, 6, 93–108. [Google Scholar] [PubMed]
- Mozaffari, M.S.; Baban, B.; Liu, J.Y.; Abebe, W.; Sullivan, J.C.; El-Marakby, A. Mitochondrial complex I and NAD(P)H oxidase are major sources of exacerbated oxidative stress in pressure-overloaded ischemic-reperfused hearts. Basic Res. Cardiol. 2011, 106, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Bernardi, P. A capful of mechanisms regulating the mitochondrial permeability transition. J. Mol. Cell. Cardiol. 2009, 46, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Martin-Padura, I.; de Nigris, F.; Giorgio, M.; Mansueto, G.; Somma, P.; Condorelli, M.; Sica, G.; De Rosa, G.; Pelicci, P. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc. Natl. Acad. Sci. USA 2003, 100, 2112–2116. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, P.; Kunduzova, O.; Masini, E.; Cambon, C.; Bani, D.; Raimondi, L.; Seguelas, M.H.; Nistri, S.; Colucci, W.; Leducq, N.; et al. Oxidative stress by monoamine oxidase mediates receptor-independent cardiomyocyte apoptosis by serotonin and postischemic myocardial injury. Circulation 2005, 112, 3297–3305. [Google Scholar] [CrossRef]
- Carpi, A.; Menabo, R.; Kaludercic, N.; Pelicci, P.; Di Lisa, F.; Giorgio, M. The cardioprotective effects elicited by p66Shc ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim. Biophys. Acta 2009, 1787, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Kaludercic, N.; Takimoto, E.; Nagayama, T.; Feng, N.; Lai, E.W.; Bedja, D.; Chen, K.; Gabrielson, K.L.; Blakely, R.D.; Shih, J.C.; et al. Monoamine oxidase a-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ. Res. 2010, 106, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed]
- Amanso, A.M.; Griendling, K.K. Differential roles of NADPH oxidases in vascular physiology and pathophysiology. Front. Biosci. 2012, 4, 1044–1064. [Google Scholar]
- Li, J.; Stouffs, M.; Serrander, L.; Banfi, B.; Bettiol, E.; Charnay, Y.; Steger, K.; Krause, K.H.; Jaconi, M.E. The NADPH oxidase NOX4 drives cardiac differentiation: Role in regulating cardiac transcription factors and map kinase activation. Mol. Biol. Cell 2006, 17, 3978–3988. [Google Scholar] [CrossRef] [PubMed]
- Martin-Garrido, A.; Brown, D.I.; Lyle, A.N.; Dikalova, A.; Seidel-Rogol, B.; Lassegue, B.; San Martin, A.; Griendling, K.K. NADPH oxidase 4 mediates TGF-β-induced smooth muscle α-actin via p38MAPK and serum response factor. Free Radic. Biol. Med. 2011, 50, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Szocs, K.; Lassegue, B.; Sorescu, D.; Hilenski, L.L.; Valppu, L.; Couse, T.L.; Wilcox, J.N.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Upregulation of NOX-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Takac, I.; Schröder, K. No superoxide—No stress? Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1255–1257. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. NOX4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, L.A.; Kovacic, L.; Rodriguez, J.; Gosemann, J.H.; Kubica, M.; Pircalabioru, G.G.; Friedmacher, F.; Cean, A.; Ghise, A.; Sarandan, M.B.; et al. NADPH oxidase-derived H2O2 subverts pathogen signaling by oxidative phosphotyrosine conversion to PB-DOPA. Proc. Natl. Acad. Sci. USA 2016, 113, 10406–10411. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Hampton, M.B.; Livesey, J.H.; Kettle, A.J. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: Implications for microbial killing. J. Biol. Chem. 2006, 281, 39860–39869. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Tian, W.; Stull, N.D.; Grinstein, S.; Atkinson, S.; Dinauer, M.C. A fluorescently tagged c-terminal fragment of p47 phox detects NADPH oxidase dynamics during phagocytosis. Mol. Biol. Cell 2009, 20, 1520–1532. [Google Scholar] [CrossRef] [PubMed]
- Rosen, H.; Klebanoff, S.J.; Wang, Y.; Brot, N.; Heinecke, J.W.; Fu, X. Methionine oxidation contributes to bacterial killing by the myeloperoxidase system of neutrophils. Proc. Natl. Acad. Sci. USA 2009, 106, 18686–18691. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Kettle, A.J.; Hampton, M.B. Reactive oxygen species and neutrophil function. Ann. Rev. Biochem. 2016, 85, 765–792. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66 Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Pagano, P.J.; Clark, J.K.; Cifuentes-Pagano, M.E.; Clark, S.M.; Callis, G.M.; Quinn, M.T. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: Enhancement by angiotensin II. Proc. Natl. Acad. Sci. USA 1997, 94, 14483–14488. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Jaquet, V.; Krause, K.H. NOX5: From basic biology to signaling and disease. Free Radic. Biol. Med. 2012, 52, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. NOX4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Lassegue, B.; Sorescu, D.; Szocs, K.; Yin, Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel GP91(phox) homologues in vascular smooth muscle cells: NOX1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 2001, 88, 888–894. [Google Scholar] [CrossRef] [PubMed]
- San Jose, G.; Bidegain, J.; Robador, P.A.; Diez, J.; Fortuno, A.; Zalba, G. Insulin-induced NADPH oxidase activation promotes proliferation and matrix metalloproteinase activation in monocytes/macrophages. Free Radic. Biol. Med. 2009, 46, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimian, T.; Li, M.W.; Lemarie, C.A.; Simeone, S.M.; Pagano, P.J.; Gaestel, M.; Paradis, P.; Wassmann, S.; Schiffrin, E.L. Mitogen-activated protein kinase-activated protein kinase 2 in angiotensin II-induced inflammation and hypertension: Regulation of oxidative stress. Hypertension 2011, 57, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Meijles, D.N.; Pagano, P.J. NADPH oxidases: Key modulators in aging and age-related cardiovascular diseases? Clin. Sci. 2016, 130, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; West, N.E.; Black, E.; McDonald, D.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Vascular superoxide production by NAD(P)H oxidase: Association with endothelial dysfunction and clinical risk factors. Circ. Res. 2000, 86, E85–E90. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of NOX family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol. 2008, 52, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Barry-Lane, P.A.; Patterson, C.; van der Merwe, M.; Hu, Z.; Holland, S.M.; Yeh, E.T.; Runge, M.S. P47 phox is required for atherosclerotic lesion progression in ApoE(−/−) mice. J. Clin. Investig. 2001, 108, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Judkins, C.P.; Diep, H.; Broughton, B.R.; Mast, A.E.; Hooker, E.U.; Miller, A.A.; Selemidis, S.; Dusting, G.J.; Sobey, C.G.; Drummond, G.R. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE−/− mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H24–H32. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, A.L.; Carrell, S.; Johnson, B.; Stanic, B.; Banfi, B.; Miller, F.J. Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis 2011, 216, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Vendrov, A.E.; Vendrov, K.C.; Smith, A.; Yuan, J.; Sumida, A.; Robidoux, J.; Runge, M.S.; Madamanchi, N.R. NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid. Redox Signal. 2015, 23, 1389–1409. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, R.M.; Eid, A.A.; Gorin, Y.C.; Block, K.; Escobar, G.P.; Bailey, S.; Abboud, H.E. NOX4-derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes. Am. J. Physiol. Cell Physiol. 2012, 302, C597–C604. [Google Scholar] [CrossRef] [PubMed]
- Green, D.E.; Murphy, T.C.; Kang, B.Y.; Kleinhenz, J.M.; Szyndralewiez, C.; Page, P.; Sutliff, R.L.; Hart, C.M. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am. J. Respir. Cell Mol. Biol. 2012, 47, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Norton, C.E.; Broughton, B.R.; Jernigan, N.L.; Walker, B.R.; Resta, T.C. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires EGFR-dependent activation of NAD(P)H oxidase 2. Antioxid. Redox Signal. 2013, 18, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.N.; Jayaram, R.; Nahar, K.; Antoniades, C.; Verheule, S.; Channon, K.M.; Alp, N.J.; Schotten, U.; Casadei, B. Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: Implications for the antiarrhythmic effect of statins. Circulation 2011, 124, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase-mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. The reduction of cytochrome c by milk xanthine oxidase. J. Biol. Chem. 1968, 243, 5753–5760. [Google Scholar] [PubMed]
- Maxwell, A.J.; Bruinsma, K.A. Uric acid is closely linked to vascular nitric oxide activity. Evidence for mechanism of association with cardiovascular disease. J. Am. Coll. Cardiol. 2001, 38, 1850–1858. [Google Scholar] [CrossRef]
- Doehner, W.; Anker, S.D. Xanthine oxidase inhibition for chronic heart failure: Is allopurinol the next therapeutic advance in heart failure? Heart 2005, 91, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Fahey, T.; Struthers, A.D.; MacDonald, T.M. Association between allopurinol and mortality in heart failure patients: A long-term follow-up study. Int. J. Clin. Pract. 2009, 63, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Noman, A.; Ang, D.S.; Ogston, S.; Lang, C.C.; Struthers, A.D. Effect of high-dose allopurinol on exercise in patients with chronic stable angina: A randomised, placebo controlled crossover trial. Lancet 2010, 375, 2161–2167. [Google Scholar] [CrossRef]
- Higgins, P.; Dawson, J.; Lees, K.R.; McArthur, K.; Quinn, T.J.; Walters, M.R. Xanthine oxidase inhibition for the treatment of cardiovascular disease: A systematic review and meta-analysis. Cardiovasc. Ther. 2012, 30, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Jankov, R.P.; Kantores, C.; Pan, J.; Belik, J. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L233–L245. [Google Scholar] [CrossRef] [PubMed]
- Banks, M.F.; Gerasimovskaya, E.V.; Tucker, D.A.; Frid, M.G.; Carpenter, T.C.; Stenmark, K.R. Egr-1 antisense oligonucleotides inhibit hypoxia-induced proliferation of pulmonary artery adventitial fibroblasts. J. Appl. Physiol. 2005, 98, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Ning, W.; Matthay, M.A.; Feghali-Bostwick, C.A.; Choi, A.M. Mapk pathway mediates egr-1-hsp70-dependent cigarette smoke-induced chemokine production. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L1297–L1303. [Google Scholar] [CrossRef] [PubMed]
- Givertz, M.M.; Mann, D.L.; Lee, K.L.; Ibarra, J.C.; Velazquez, E.J.; Hernandez, A.F.; Mascette, A.M.; Braunwald, E. Xanthine oxidase inhibition for hyperuricemic heart failure patients. Circ. Heart Fail. 2013, 6, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Van Leyen, K. Lipoxygenase: An emerging target for stroke therapy. CNS Neurol. Disord. Drug Targets 2013, 12, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.D.; Cyrus, T. 12/15-lipoxygenase, oxidative modification of LDL and atherogenesis. Trends Cardiovasc. Med. 2001, 11, 116–124. [Google Scholar] [PubMed]
- Lotzer, K.; Funk, C.D.; Habenicht, A.J. The 5-lipoxygenase pathway in arterial wall biology and atherosclerosis. Biochim. Biophys. Acta 2005, 1736, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Back, M.; Hansson, G.K. Leukotriene receptors in atherosclerosis. Ann. Med. 2006, 38, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.D. Leukotriene modifiers as potential therapeutics for cardiovascular disease. Nat. Rev. Drug Discov. 2005, 4, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Flamand, N.; Mancuso, P.; Serezani, C.H.; Brock, T.G. Leukotrienes: Mediators that have been typecast as villains. Cell. Mol. Life Sci. CMLS 2007, 64, 2657–2670. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Poeckel, D.; Funk, C.D. The 5-lipoxygenase/leukotriene pathway in preclinical models of cardiovascular disease. Cardiovasc. Res. 2010, 86, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Shekher, A.; Singh, M. Role of eicosanoid inhibition of ischemia reperfusion injury: Intact and isolated rat heart studies. Methods Find. Exp. Clin. Pharmacol. 1997, 19, 223–229. [Google Scholar] [PubMed]
- Adamek, A.; Jung, S.; Dienesch, C.; Laser, M.; Ertl, G.; Bauersachs, J.; Frantz, S. Role of 5-lipoxygenase in myocardial ischemia-reperfusion injury in mice. Eur. J. Pharmacol. 2007, 571, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Baron, M.; Bouhlel, M.A.; Vanhoutte, J.; Copin, C.; Sebti, Y.; Derudas, B.; Mayi, T.; Bories, G.; Tailleux, A.; et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARγ and LXRα pathways. Circ. Res. 2011, 108, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Levick, S.P.; Loch, D.C.; Taylor, S.M.; Janicki, J.S. Arachidonic acid metabolism as a potential mediator of cardiac fibrosis associated with inflammation. J. Immunol. 2007, 178, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Gu, J.; Chakrabarti, S.K.; Aylor, K.; Marshall, J.; Takahashi, Y.; Yoshimoto, T.; Nadler, J.L. The role of 12/15-lipoxygenase in the expression of interleukin-6 and tumor necrosis factor-α in macrophages. Endocrinology 2007, 148, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Robertson, A.K.; Soderberg-Naucler, C. Inflammation and atherosclerosis. Ann. Rev. Pathol. 2006, 1, 297–329. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kayama, Y.; Sakamoto, M.; Iuchi, H.; Shimizu, I.; Yoshino, T.; Katoh, D.; Nagoshi, T.; Tojo, K.; Minamino, T.; et al. Arachidonate 12/15-lipoxygenase-induced inflammation and oxidative stress are involved in the development of diabetic cardiomyopathy. Diabetes 2015, 64, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Baldus, S.; Rudolph, V.; Roiss, M.; Ito, W.D.; Rudolph, T.K.; Eiserich, J.P.; Sydow, K.; Lau, D.; Szocs, K.; Klinke, A.; et al. Heparins increase endothelial nitric oxide bioavailability by liberating vessel-immobilized myeloperoxidase. Circulation 2006, 113, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Anatoliotakis, N.; Deftereos, S.; Bouras, G.; Giannopoulos, G.; Tsounis, D.; Angelidis, C.; Kaoukis, A.; Stefanadis, C. Myeloperoxidase: Expressing inflammation and oxidative stress in cardiovascular disease. Curr. Top. Med. Chem. 2013, 13, 115–138. [Google Scholar] [CrossRef] [PubMed]
- Van der Veen, B.S.; de Winther, M.P.; Heeringa, P. Myeloperoxidase: Molecular mechanisms of action and their relevance to human health and disease. Antioxid. Redox Signal. 2009, 11, 2899–2937. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Kassim, S.Y.; Parks, W.C.; Heinecke, J.W. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J. Biol. Chem. 2001, 276, 41279–41287. [Google Scholar] [CrossRef] [PubMed]
- Pullar, J.M.; Vissers, M.C.; Winterbourn, C.C. Glutathione oxidation by hypochlorous acid in endothelial cells produces glutathione sulfonamide as a major product but not glutathione disulfide. J. Biol. Chem. 2001, 276, 22120–22125. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L. The role of hypothiocyanous acid (HOSCN) in biological systems. Free Radic. Res. 2009, 43, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.E.; Tan, J.T.; Hawkins, C.L.; Heather, A.K.; Davies, M.J. The myeloperoxidase-derived oxidant HOSCN inhibits protein tyrosine phosphatases and modulates cell signalling via the mitogen-activated protein kinase (MAPK) pathway in macrophages. Biochem. J. 2010, 430, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Podrez, E.A.; Schmitt, D.; Hoff, H.F.; Hazen, S.L. Myeloperoxidase-generated reactive nitrogen species convert LDL into an atherogenic form in vitro. J. Clin. Investing. 1999, 103, 1547–1560. [Google Scholar] [CrossRef] [PubMed]
- Bergt, C.; Pennathur, S.; Fu, X.; Byun, J.; O’Brien, K.; McDonald, T.O.; Singh, P.; Anantharamaiah, G.M.; Chait, A.; Brunzell, J.; et al. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc. Natl. Acad. Sci. USA 2004, 101, 13032–13037. [Google Scholar] [CrossRef] [PubMed]
- Exner, M.; Hermann, M.; Hofbauer, R.; Hartmann, B.; Kapiotis, S.; Gmeiner, B. Thiocyanate catalyzes myeloperoxidase-initiated lipid oxidation in LDL. Free Radic. Biol. Med. 2004, 37, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Baldus, S.; Heitzer, T.; Eiserich, J.P.; Lau, D.; Mollnau, H.; Ortak, M.; Petri, S.; Goldmann, B.; Duchstein, H.J.; Berger, J.; et al. Myeloperoxidase enhances nitric oxide catabolism during myocardial ischemia and reperfusion. Free Radic. Biol. Med. 2004, 37, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Nicholls, S.J.; Rodriguez, E.R.; Kummu, O.; Horkko, S.; Barnard, J.; Reynolds, W.F.; Topol, E.J.; DiDonato, J.A.; Hazen, S.L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Vozarova, B.; Weyer, C.; Lindsay, R.S.; Pratley, R.E.; Bogardus, C.; Tataranni, P.A. High white blood cell count is associated with a worsening of insulin sensitivity and predicts the development of type 2 diabetes. Diabetes 2002, 51, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Orio, F., Jr.; Palomba, S.; Cascella, T.; Di Biase, S.; Manguso, F.; Tauchmanova, L.; Nardo, L.G.; Labella, D.; Savastano, S.; Russo, T.; et al. The increase of leukocytes as a new putative marker of low-grade chronic inflammation and early cardiovascular risk in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2005, 90, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Rovira-Llopis, S.; Rocha, M.; Falcon, R.; de Pablo, C.; Alvarez, A.; Jover, A.; Hernandez-Mijares, A.; Victor, V.M. Is myeloperoxidase a key component in the ROS-induced vascular damage related to nephropathy in type 2 diabetes? Antioxid. Redox Signal. 2013, 19, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Victor, V.M.; Rovira-Llopis, S.; Banuls, C.; Diaz-Morales, N.; Martinez de Maranon, A.; Rios-Navarro, C.; Alvarez, A.; Gomez, M.; Rocha, M.; Hernandez-Mijares, A. Insulin resistance in PCOS patients enhances oxidative stress and leukocyte adhesion: Role of myeloperoxidase. PLoS ONE 2016, 11, e0151960. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.L.; Scapinelli, A.; Tamanaha, S.; Oliveira, R.M.; Kowastch, I.; Mathias, W., Jr.; Aoki, T.; Aldrighi, J.M. Myeloperoxidases and polycystic ovary syndrome. Gynecol. Endocrinol. 2012, 28, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Shishehbor, M.H.; Brennan, M.L.; Aviles, R.J.; Fu, X.; Penn, M.S.; Sprecher, D.L.; Hazen, S.L. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation 2003, 108, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Laursen, J.B.; Rajagopalan, S.; Galis, Z.; Tarpey, M.; Freeman, B.A.; Harrison, D.G. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation 1997, 95, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Ono, H.; Minatoguchi, S.; Watanabe, K.; Yamada, Y.; Mizukusa, T.; Kawasaki, H.; Takahashi, H.; Uno, T.; Tsukamoto, T.; Hiei, K.; et al. Candesartan decreases carotid intima-media thickness by enhancing nitric oxide and decreasing oxidative stress in patients with hypertension. Hypertens. Res. 2008, 31, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, R.H.; Armas-Hernandez, M.J.; Velasco, M.; Israili, Z.H.; Armas-Padilla, M.C. Calcium antagonists and atherosclerosis protection in hypertension. Am. J. Ther. 2003, 10, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, P.; Perrone-Filardi, P.; Petretta, M.; Marciano, C.; Vassallo, E.; Gargiulo, P.; Paolillo, S.; Petretta, A.; Chiariello, M. Calcium channel blockers and cardiovascular outcomes: A meta-analysis of 175,634 patients. J. Hypertens. 2009, 27, 1136–1151. [Google Scholar] [CrossRef]
- Dewey, C.M.; Spitler, K.M.; Ponce, J.M.; Hall, D.D.; Grueter, C.E. Cardiac-secreted factors as peripheral metabolic regulators and potential disease biomarkers. J. Am. Heart Assoc. 2016. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Walker, A.E.; Magerko, K.A.; Bramwell, R.C.; Black, A.D.; Henson, G.D.; Lawson, B.R.; Lesniewski, L.A.; Seals, D.R. Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell 2013, 12, 772–783. [Google Scholar] [CrossRef]
- Gano, L.B.; Donato, A.J.; Pasha, H.M.; Hearon, C.M., Jr.; Sindler, A.L.; Seals, D.R. The sirt1 activator srt1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1754–H1763. [Google Scholar] [CrossRef] [PubMed]
- Koentges, C.; Bode, C.; Bugger, H. SIRT3 in Cardiac Physiology and Disease. Front. Cardiovasc. Med. 2016, 13, 3–38. [Google Scholar] [CrossRef] [PubMed]
- De Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.; Seals, D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Hearon, C.M., Jr.; Henson, G.D.; Seals, D.R. Mitochondrial quality control and age-associated arterial stiffening. Exp. Gerontol. 2014, 58, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, R.E.; Hill, S.D.; Bispham, N.Z.; Santos-Parker, J.R.; Nowlan, M.J.; Snyder, L.L.; Chonchol, M.; LaRocca, T.J.; McQueen, M.B.; Seals, D.R. Oral trehalose supplementation improves resistance artery endothelial function in healthy middle-aged and older adults. Aging 2016, 8, 1167–1183. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Villar, L.; Perez, B.; Ugarte, M.; Desviat, L.R.; Richard, E. Antioxidants successfully reduce ROS production in propionic acidemia fibroblasts. Biochem. Biophys. Res. Commun. 2014, 452, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Lopez, I.; Diaz-Morales, N.; Rovira-Llopis, S.; de Marañon, A.M.; Orden, S.; Alvarez, A.; Bañuls, C.; Rocha, M.; Murphy, M.P.; Hernandez-Mijares, A.; et al. The mitochondria-targeted antioxidant mitoq modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biol. 2016, 10, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Hassinen, I.; Hartikainen, J.; Hedman, A.; Kivelä, A.; Saraste, A.; Knuuti, J.; Husso, M.; Mussalo, H.; Hedman, M.; Toivanen, P.; et al. Abstract 11987: Adenoviral intramyocardial VEGF-D gene transfer increases myocardial perfusion in refractory angina patients. Circulation 2015, 132, A11987. [Google Scholar]
- Van Wanrooij, E.J.; de Vos, P.; Bixel, M.G.; Vestweber, D.; van Berkel, T.J.; Kuiper, J. Vaccination against CD99 inhibits atherogenesis in low-density lipoprotein receptor-deficient mice. Cardiovasc. Res. 2008, 78, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, D.; Kethireddy, S.; Janero, D.R.; Amiji, M.M. Therapeutic efficacy of an ω-3-fatty acid-containing 17-β estradiol nano-delivery system against experimental atherosclerosis. PLoS ONE 2016, 11, e0147337. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Hasan, A.; Kindi, H.A.; Gaharwar, A.K.; Rao, V.T.; Nikkhah, M.; Shin, S.R.; Krafft, D.; Dokmeci, M.R.; Shum-Tim, D.; et al. Injectable graphene oxide/hydrogel-based angiogenic gene delivery system for vasculogenesis and cardiac repair. ACS Nano 2014, 8, 8050–8062. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Qian, H.; Yang, H.; Xu, L.; Xu, W.; Yan, J. Regression of atherosclerosis plaques in apolipoprotein e−/− mice after lentivirus-mediated RNA interference of CD40. Int. J. Cardiol. 2013, 163, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.S.; Lintermans, L.L.; Morselt, H.W.; Leus, N.G.; Ruiters, M.H.; Molema, G.; Kamps, J.A. Anti-VCAM-1 and Anti-E-selectin SAINT-O-Somes for selective delivery of siRNA into inflammation-activated primary endothelial cells. Mol. Pharm. 2013, 10, 3033–3044. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.G.; Crosby, J.; Baker, B.F.; Graham, M.J.; Crooke, R.M. Antisense technology: An emerging platform for cardiovascular disease therapeutics. J. Cardiovasc. Transl. Res. 2013, 6, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Philippen, L.E.; Dirkx, E.; Wit, J.B.; Burggraaf, K.; de Windt, L.J.; da Costa Martins, P.A. Antisense microrna therapeutics in cardiovascular disease: Quo vadis? Mol. Ther. 2015, 23, 1810–1818. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yang, Y.; Hong, H.; Zhang, Y.; Cai, W.; Fang, D. Aptamers as therapeutics in cardiovascular diseases. Curr. Med. Chem. 2011, 18, 4169–4174. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Li, L.; Bennett, D.; Guo, Y.; Key, T.J.; Bian, Z.; Sherliker, P.; Gao, H.; Chen, Y.; Yang, L.; et al. Fresh fruit consumption and major cardiovascular disease in china. N. Engl. J. Med. 2016, 374, 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Toh, J.Y.; Tan, V.M.; Lim, P.C.; Lim, S.T.; Chong, M.F. Flavonoids from fruit and vegetables: A focus on cardiovascular risk factors. Curr. Atheroscler. Rep. 2013, 15, 368. [Google Scholar] [CrossRef] [PubMed]
- Noratto, G.; Martino, H.S.; Simbo, S.; Byrne, D.; Mertens-Talcott, S.U. Consumption of polyphenol-rich peach and plum juice prevents risk factors for obesity-related metabolic disorders and cardiovascular disease in zucker rats. J. Nutr. Biochem. 2015, 26, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.L.; Arruda, F.C.; Reis, P.P.; Felix, T.F.; Santos, P.P.; Rafacho, B.P.; Goncalves, A.F.; Claro, R.T.; Azevedo, P.S.; Polegato, B.F.; et al. Tomato (Lycopersicon esculentum) supplementation induces changes in cardiac mirna expression, reduces oxidative stress and left ventricular mass, and improves diastolic function. Nutrients 2015, 7, 9640–9649. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.; Kay, C.; Abdelhamid, A.; Kroon, P.A.; Cohn, J.S.; Rimm, E.B.; Cassidy, A. Effects of chocolate, cocoa, and flavan-3-ols on cardiovascular health: A systematic review and meta-analysis of randomized trials. Am. J. Clin. Nutr. 2012, 95, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Ghayur, M.N.; Gilani, A.H. Ginger lowers blood pressure through blockade of voltage-dependent calcium channels. J. Cardiovasc. Pharmacol. 2005, 45, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, B.; Saedisomeolia, A.; Allman-Farinelli, M. Association between antioxidant intake/status and obesity: A systematic review of observational studies. Biol. Trace Elem. Res. 2017, 175, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Sudheer, A.R.; Menon, V.P. Ferulic acid: Therapeutic potential through its antioxidant property. J. Clin. Biochem. Nutr. 2007, 40, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Liu, Y.H.; Chen, C.M.; Chang, W.H.; Chen, C.Y. The effect of almonds on inflammation and oxidative stress in Chinese patients with type 2 diabetes mellitus: A randomized crossover controlled feeding trial. Eur. J. Nutr. 2012, 52, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Uriarte, P.; Nogues, R.; Saez, G.; Bullo, M.; Romeu, M.; Masana, L. Effect of nut consumption on oxidative stress and the endothelial function in metabolic syndrome. Clin. Nutr. 2010, 29, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Chirumbolo, S. Role of oxidative stress and antioxidants in daily nutrition and human health. Nutrition 2017, 33, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Montague, C.T.; Farooqi, I.S.; Whitehead, J.P.; Soos, M.A.; Rau, H.; Wareham, N.J. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 1997, 387, 903–908. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cardiokines | Expression Status | Condition |
|---|---|---|
| Atrial and B-Type Natriuretic Peptides | Highly expressed | In rats with pressure overload |
| Low level of expression | In mice under fasting conditions | |
| GDF-8 | Highly expressed | When skeletal muscle mass is increased |
| GDF-15 | Highly expressed | In patients with cardiac hypertrophy and chronic HF |
| CTRP9 | Low level of expression | In diabetic animals and humans. In subjects that have suffered an acute myocardial infarction |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and Oxidative Stress. J. Clin. Med. 2017, 6, 22. https://doi.org/10.3390/jcm6020022
Cervantes Gracia K, Llanas-Cornejo D, Husi H. CVD and Oxidative Stress. Journal of Clinical Medicine. 2017; 6(2):22. https://doi.org/10.3390/jcm6020022
Chicago/Turabian StyleCervantes Gracia, Karla, Daniel Llanas-Cornejo, and Holger Husi. 2017. "CVD and Oxidative Stress" Journal of Clinical Medicine 6, no. 2: 22. https://doi.org/10.3390/jcm6020022
APA StyleCervantes Gracia, K., Llanas-Cornejo, D., & Husi, H. (2017). CVD and Oxidative Stress. Journal of Clinical Medicine, 6(2), 22. https://doi.org/10.3390/jcm6020022