Is Exaggerated Release of Arginine Vasopressin an Endocrine Disorder? Pathophysiology and Treatment

Abstract
:1. Introduction
2. Clinical Causes of Inappropriate Secretion of AVP
3. Pathogenesis of Exaggerated AVP Release
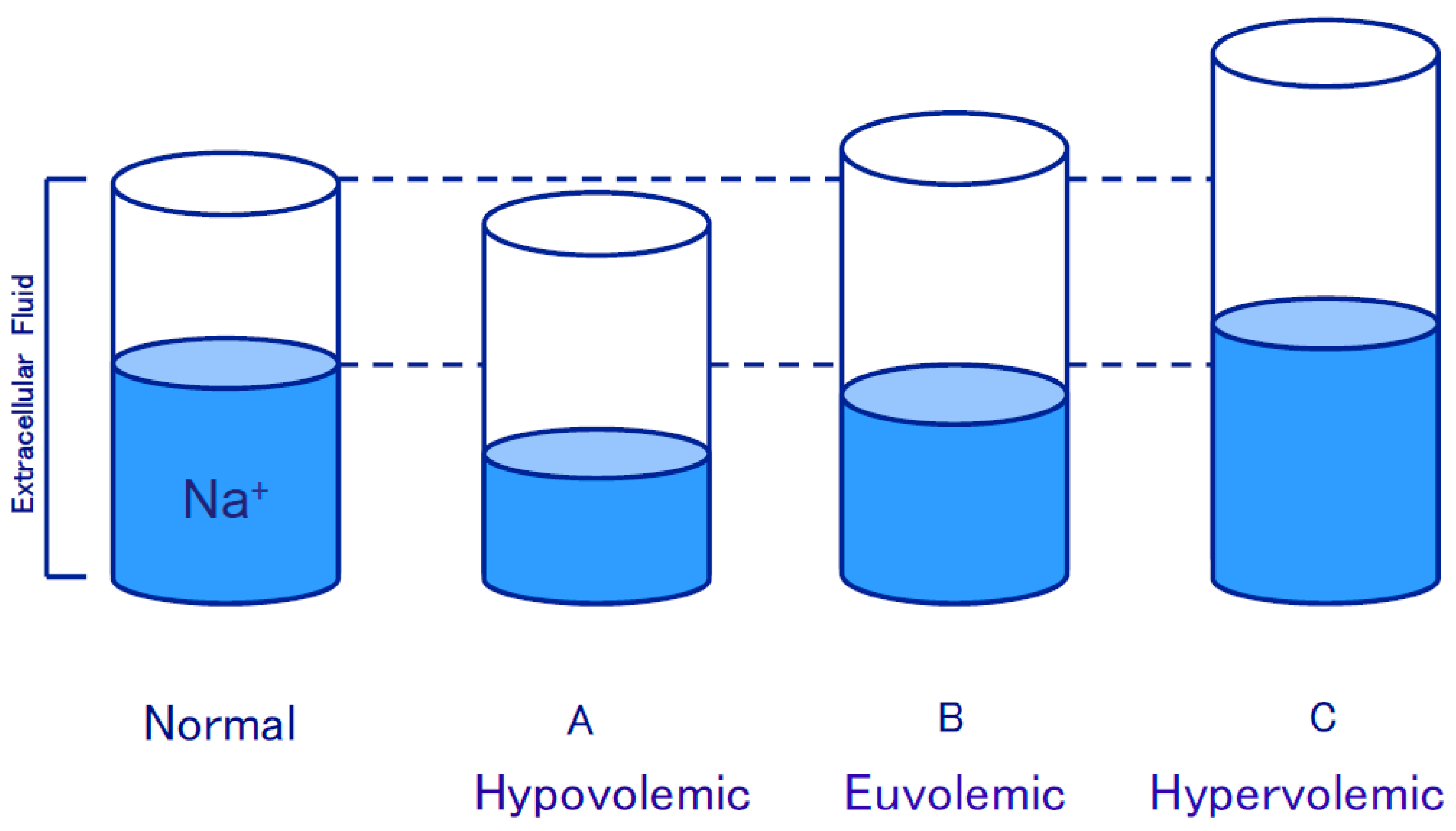
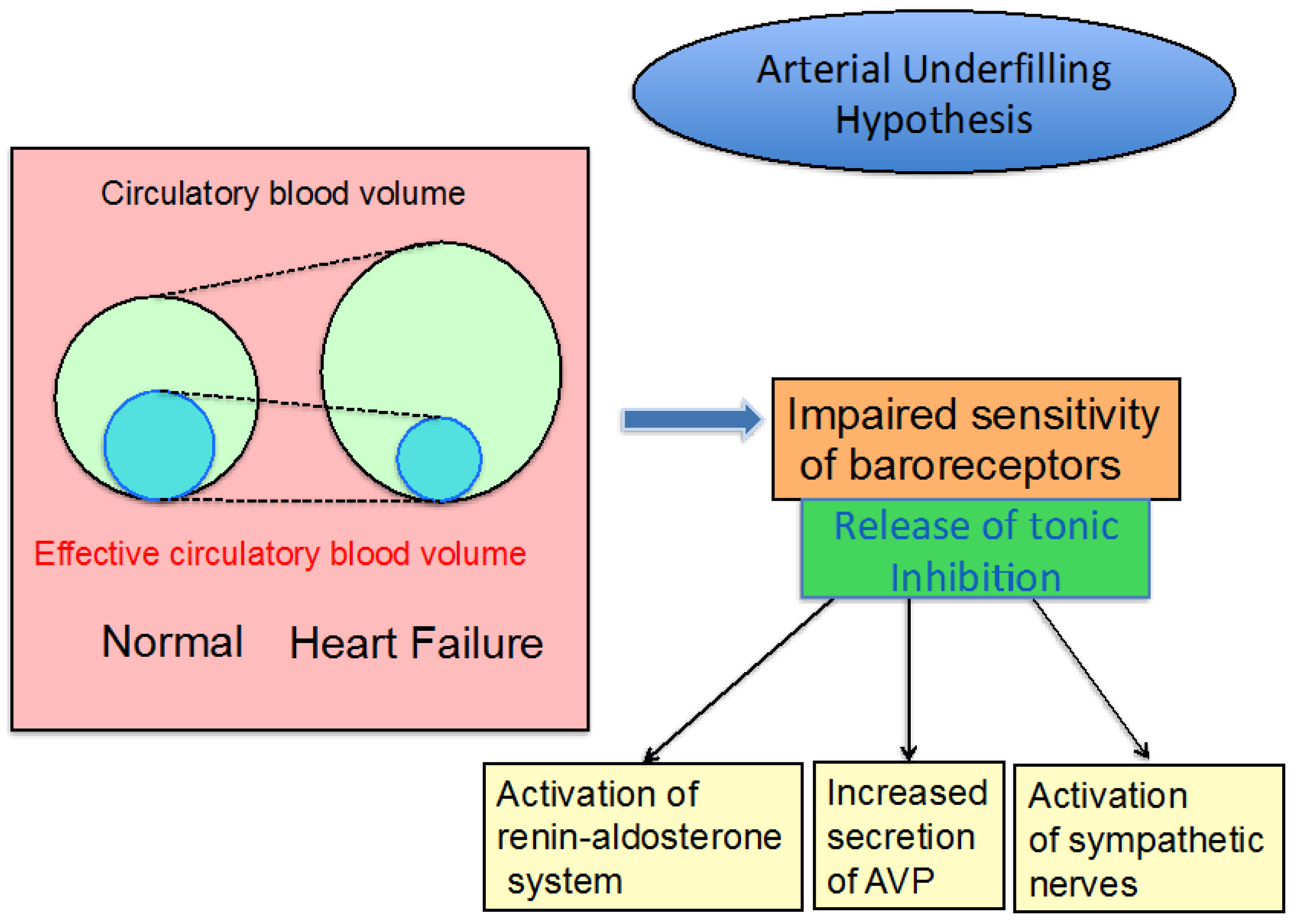
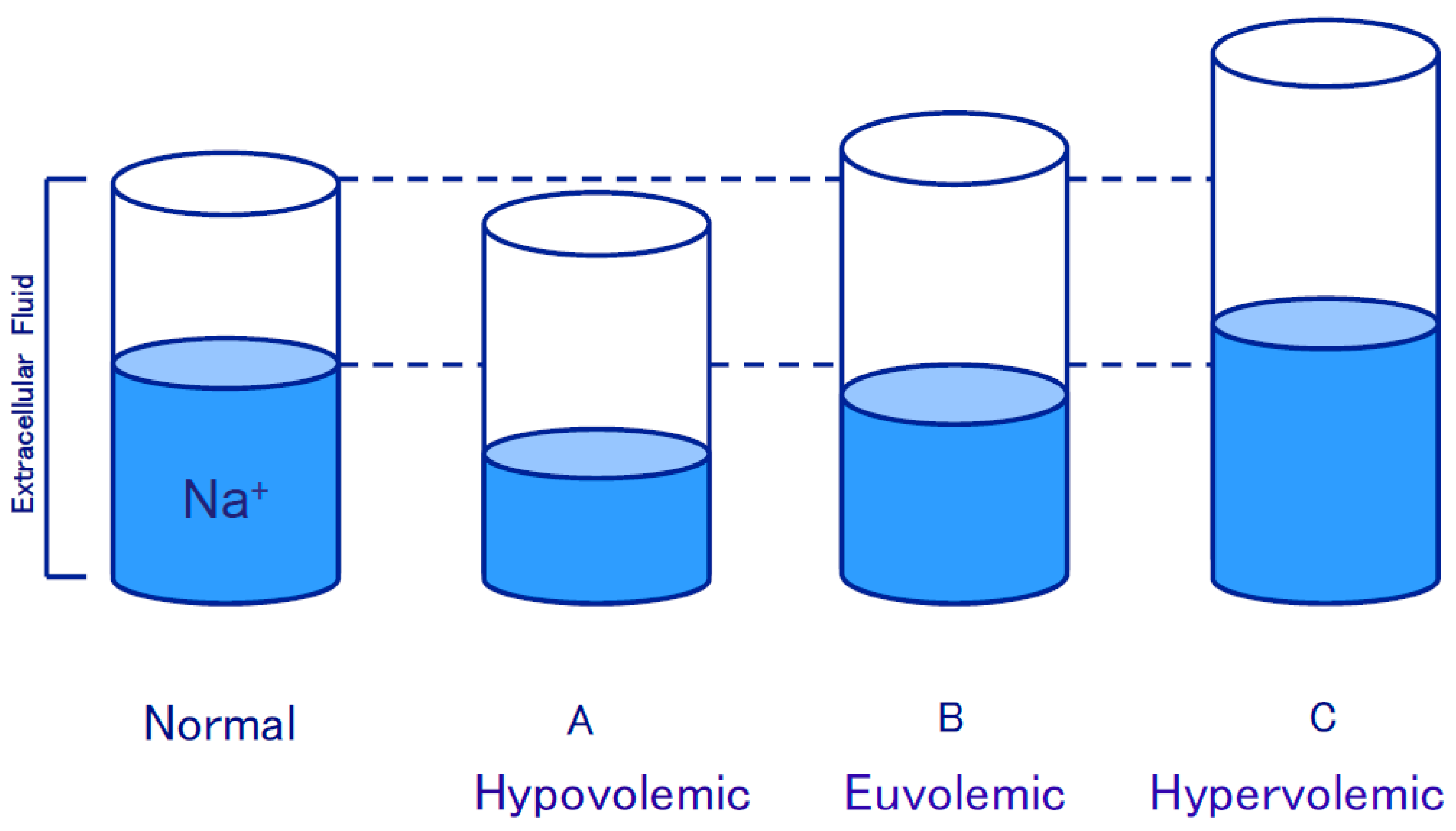
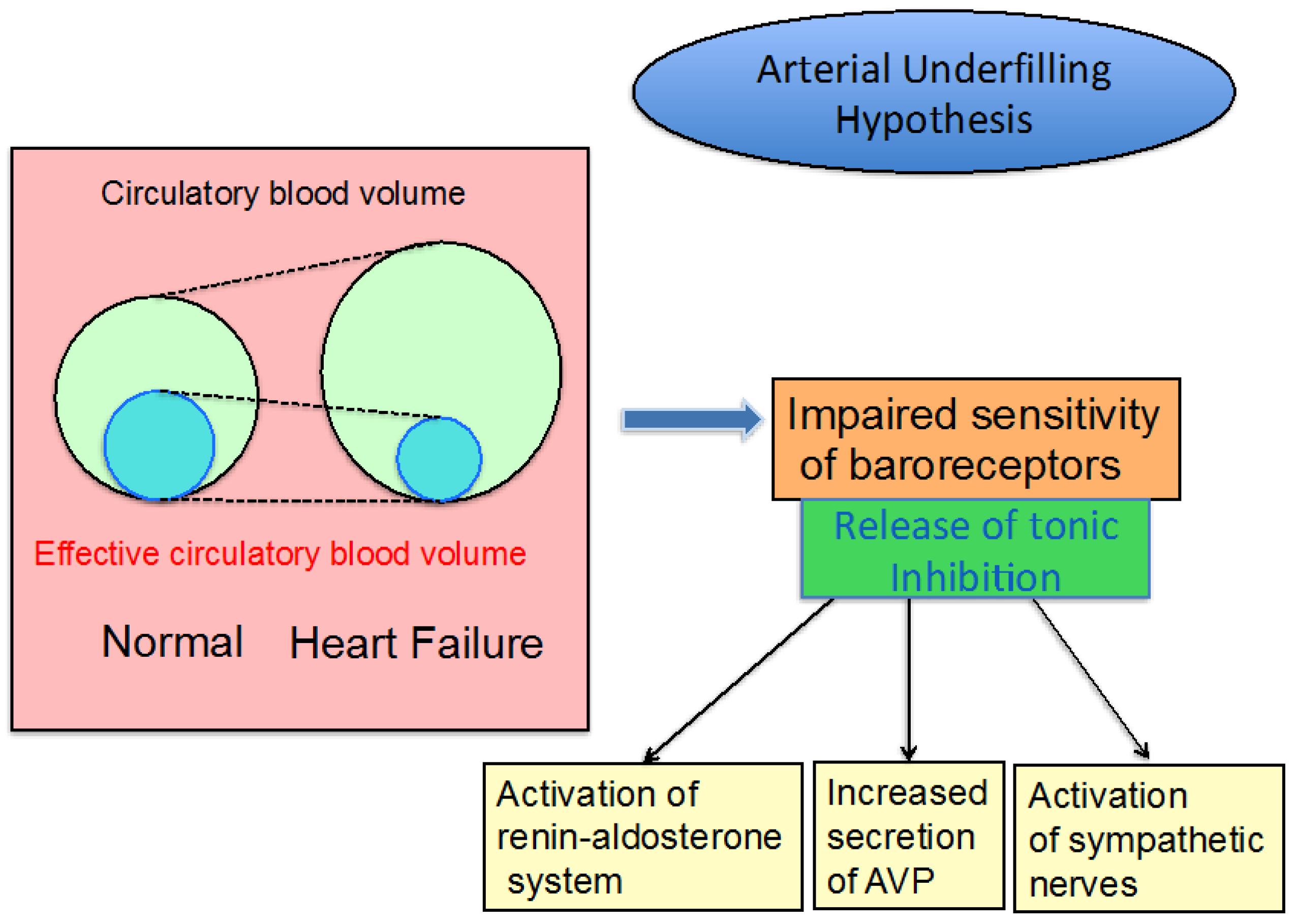
3.1. Edematous Diseases
3.2. SIADH
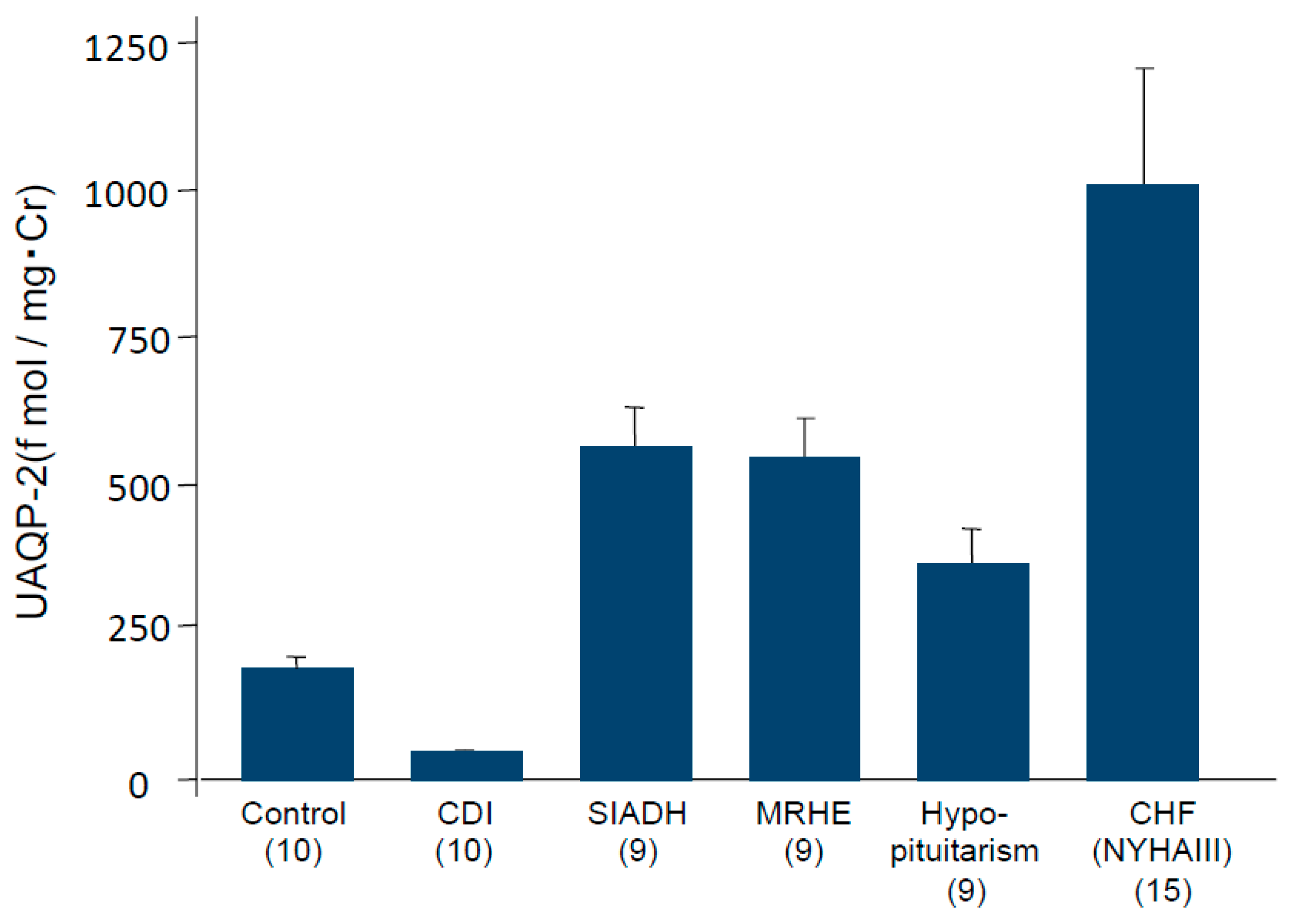
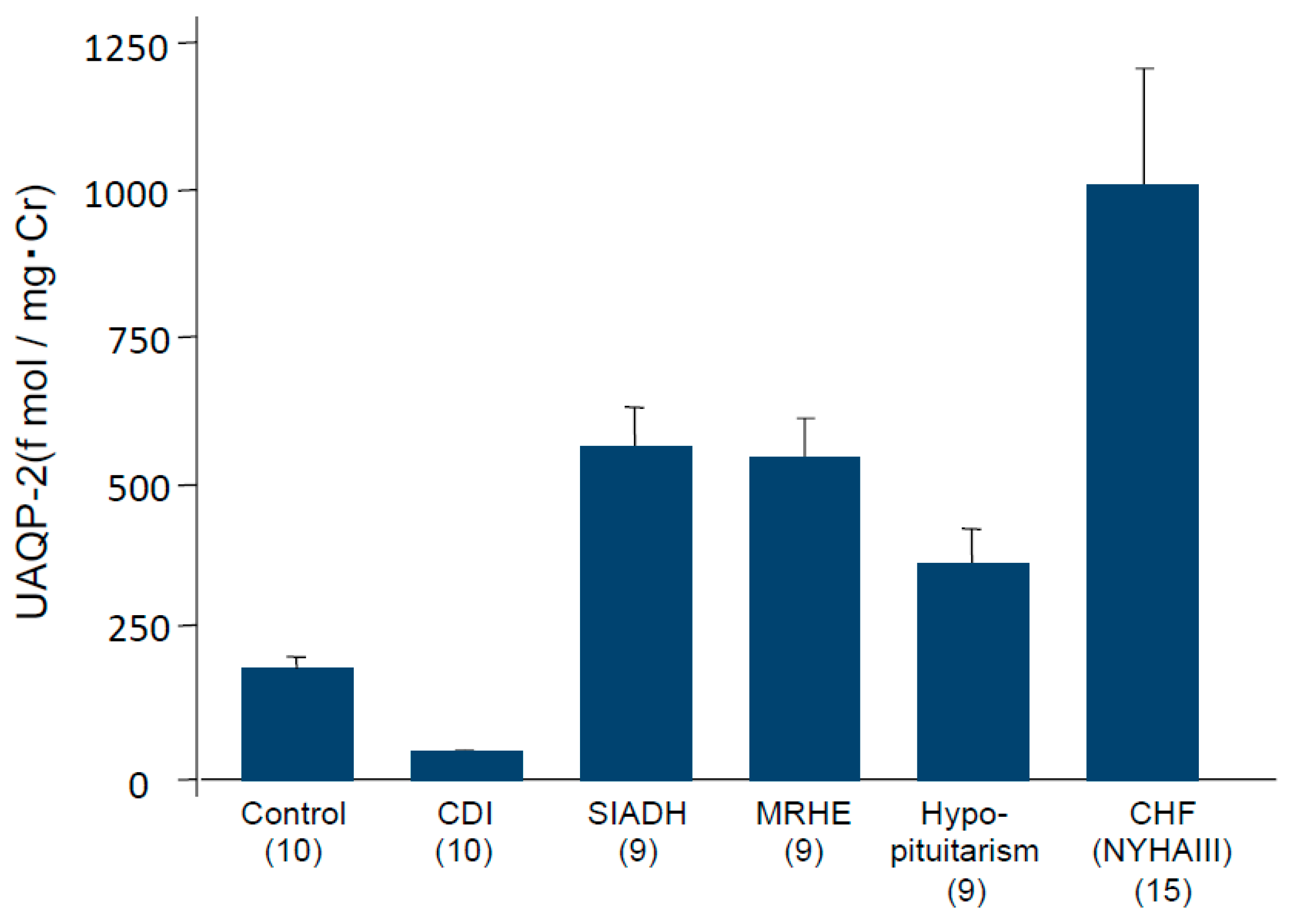
4. Impaired Water Excretion
5. Hyponatremia Predicts Poor Prognosis
6. AVP V2 Receptor Antagonists and Impaired Water Excretion
7. Conclusions
Conflicts of Interest
References
- Schrier, R.W. Pathogenesis of sodium and water retention in high-output and low-output cardiac failure, nephrotic syndrome, cirrhosis and pregnancy. N. Engl. J. Med. 1988, 319, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W. Pathogenesis of sodium and water retention in high-output and low-output cardiac failure, nephrotic syndrome, cirrhosis and pregnancy (2). N. Engl. J. Med. 1988, 319, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W.; Gross, P.; Gheroghiade, M.; Berl, T.; Verbalis, J.G.; Czerwiec, F.S.; Orlandi, C.; For the SALT Investigators. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N. Engl. J. Med. 2006, 355, 2099–2112. [Google Scholar] [CrossRef] [PubMed]
- Gheroghiade, M.; Gattis, W.A.; O’Connor, C.M.; Adams, K.F.; Elkayam, U.; Barbagelata, A.; Ghali, K.; Benka, R.I.; McGrew, F.A.; Klapholz, M.; et al. Effect of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure: A randomized controlled trial. JAMA 2004, 291, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Konstam, M.A.; Gheroghiade, M.; Burnett, J.C., Jr.; Grinfeld, L.; Maggioni, A.P.; Swedberg, K.; Udelson, J.E.; Zannad, F.; Cook, T.; Ouyang, J.; et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: The EVEREST Trial. JAMA 2007, 297, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Schrier, R.W. Pathophysiological roles of arginine vasopressin and aquaporin-2 in impaired water excretion. Clin. Endocrinol. 2003, 58, 1–17. [Google Scholar] [CrossRef]
- Schrier, R.W.; Abraham, W. Hormone and hemodynamics in heart failure. N. Engl. J. Med. 1999, 341, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Szatalovitz, V.L.; Arnold, P.E.; Chaimovitz, C.; Bichet, D.G.; Berl, T.; Schrier, R.W. Radioimmunoassay of plasma arginine vasopressin in hyponatremic patients with congestive heart failure. N. Engl. J. Med. 1981, 305, 263–266. [Google Scholar]
- Goldsmith, S.R.; Frances, C.G.; Cowley, A.W.; Levine, B.; Cohn, J.N. Increased plasma arginine vasopressin levels in patients with congestive heart failure. J. Am. Coll. Cardiol. 1983, 1, 1385–1390. [Google Scholar] [CrossRef]
- Nakamura, T.; Funayama, H.; Yoshimura, A.; Tsuruya, Y.; Saito, M.; Kawakami, M.; Ishikawa, S. Possible vascular role of increased plasma arginine vasopressin in congestive heart failure. Int. J. Cardiol. 2006, 106, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Kinugawa, K.; Hatano, M.; Fujino, T.; Inaba, T.; Maki, H.; Kinoshita, O.; Nawata, K.; Kyo, S.; Ono, M.; et al. Low cardiac output stimulates vasopressin release in patients with stage D heart failure. Circ. J. 2014, 78, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Gines, P.; Arroyo, V.; Rodes, J. Disorders of renal function in cirrhosis: Pathophysiology and clinical aspects. In Hepatology, 3rd ed.; Zakim, D., Boyer, T.D., Eds.; Saunders: Philadelphia, PA, USA, 1996; pp. 650–675. [Google Scholar]
- Bichet, D.G.; Szatalowicz, V.; Chaimovitz, C.; Schrier, R.W. Role of vasopressin in abnormal water excretion in cirrhotic patients. Ann. Intern. Med. 1982, 96, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Summer, S.; Howard, R.L.; Schrier, R.W. Vasopressin gene expression in rats with experimental cirrhosis. Hepatology 1993, 17, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Bichet, B.G.; van Puttan, V.; Schrier, R.W. Potential role of increased sympathetic activity in impaired sodium and water excretion in cirrhosis. N. Engl. J. Med. 1982, 307, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W.; Berl, T.; Anderson, R.J. Osmotic and non-osmotic control of vasopressin release. Am. J. Physiol. 1979, 236, F321–F332. [Google Scholar] [PubMed]
- Yamashita, T.; Yoshida, M.; Yamada, H.; Asano, T.; Aoki, A.; Ikoma, A.; Kusaka, I.; Kakei, M.; Ishikawa, S. Prompt efficacy of tolvaptan in treating hyponatremia of syndrome of inappropriate secretion of antidiuretic hormone (SIADH) closely associated with rupture of a gastric artery aneurysm. Intern. Med. 2014, 53, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Verbalis, J.G.; Drutarosky, M.D. Adaptation to chronic hypoosmolality in rats. Kidney Int. 1988, 34, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Fushimi, K.; Uchida, S.; Hara, Y.; Hirata, Y.; Marumo, F.; Sasaki, S. Cloning and expression of apical membrane water channel of rat kidney collecting tubule. Nature 1993, 361, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Ishikawa, S.; Sasaki, S.; Fujisawa, G.; Fushimi, K.; Marumo, F.; Saito, T. Role of water channel AQP-CD in water retention in SIADH and cirrhotic rats. Am. J. Physiol. 1995, 269, F926–F931. [Google Scholar] [PubMed]
- Saito, T.; Higashiyama, M.; Nagasaka, S.; Sasaki, S.; Saito, T.; Ishikawa, S. Role of aquaporin-2 gene expression in hyponatremic rats with chronic vasopressin-induced antidiuresis. Kidney Int. 2001, 60, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.L.; Martin, P.Y.; Ohara, M.; StJohn, J.; Pattison, T.; Meng, X.; Morris, K.; Kim, J.K.; Schrier, R.W. Upregulation of aquaporin-2 water channel expression in chronic heart failure rat. J. Clin. Investig. 1997, 99, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Terris, J.; Anderson, D.; Ecelbarger, C.; Froklaer, J.; Jonassen, T.; Marples, D.; Knepper, M.A.; Petersen, J.S. Congestive heart failure in rats is associated with increased expression and targeting of aquaporin-2 water channel in collecting duct. Proc. Natl. Acad. Sci. USA 1997, 94, 5450–5455. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Saito, T.; Kasono, K.; Tamemoto, H.; Kawakami, M.; Sasaki, S.; Ishikawa, S. Hypotonicity reduces the activity of murine aquaporin-2 promoter induced by dibutyryl cAMP. Exp. Physiol. 2008, 93, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Kanno, K.; Sasaki, S.; Hirata, Y.; Ishikawa, S.; Fushimi, K.; Nakanishi, S.; Bichet, D.G.; Marumo, F. Urinary excretion of aquaporin-2 in patients with diabetes insipidus. N. Engl. J. Med. 1995, 332, 1540–1545. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ishikawa, S.; Sasaki, S.; Nakamura, T.; Rokkaku, K.; Kawakami, A.; Honda, K.; Saito, T. Urinary excretion of aquaporin-2 in the diagnosis of central diabetes insipidus. J. Clin. Endocrinol. Metab. 1997, 82, 1823–1827. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Ohmoto, Y.; Mori, T.; Iwata, F.; Muraguchi, M. Daily variance of urinary excretion of AQP2 determined by sandwich ELISA method. Clin. Exp. Nephrol. 2012, 16, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Rai, T.; Sekine, K.; Kanno, K.; Hata, K.; Miura, M.; Mizushima, A.; Marumo, F.; Sasaki, S. Urinary excretion of aquaporin-2 water channel in human and rat. J. Am. Soc. Nephrol. 1997, 8, 1357–1362. [Google Scholar] [PubMed]
- Saito, T.; Ishikawa, S.; Ando, F.; Okada, N.; Nakamura, T.; Kusaka, I.; Higashiyama, M.; Nagasaka, S.; Saito, T. Exaggerated urinary excretion of aquaporin-2 in the pathological state of impaired water excretion dependent upon arginine vasopressin. J. Clin. Endocrinol. Metab. 1998, 83, 4034–4040. [Google Scholar] [CrossRef] [PubMed]
- Funayama, H.; Nakamura, T.; Saito, T.; Yoshimura, A.; Saito, M.; Kawakami, M.; Ishikawa, S. Urinary excretion of aquaporin-2 water channel exaggerated dependent upon vasopressin in congestive heart failure. Kidney Int. 2004, 66, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Gheroghiade, M.; Abraham, W.T.; Albert, N.M.; Gattis-Stough, W.; Greenberg, B.H.; O’Connor, C.M.; She, L.; Yancy, C.W.; Young, J.; Fonarow, G.C.; et al. Relationship between admission serum sodium concentration and clinical outcomes in patients hospitalized for heart failure: An analysis from the OPTIMIZE-HF registry. Eur. Heart J. 2007, 28, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Arao, K.; Fujiwara, T.; Sakakura, K.; Wada, H.; Sugawara, Y.; Suga, C.; Ako, J.; Ishikawa, S.; Momomura, S. Hyponatremia for worsening heart failure in patients receiving cardiac resynchronization therapy. Circ. J. 2013, 77, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.; O’Connor, C.M.; Leimberger, J.O.; Gattis-Stough, W.; Pina, I.L.; Felker, G.M.; Adams, K.F.; Califf, R.M.; Gheroghiade, M.; OPTIMIZE-CHF Investigators. Low serum sodium is associated with increased short-term mortality in hospitalized patients with worsening heart failure. Results from the Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIMIZE-CHF) study. Circulation 2005, 111, 2454–2460. [Google Scholar] [PubMed]
- Gheroghiade, M.; Rossi, J.S.; Cotts, W.; Shin, D.O.; Hellkamp, A.S.; Pina, I.L.; Fonarow, G.C.; DeMarco, T.; Pauly, D.F.; Rogers, J. Characterization and prognostic value of persistent hyponatremia in patients with severe heart failure in the ESCAPE Trial. Arch. Intern. Med. 2007, 167, 1998–2005. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S. Arginine vasopressin in heart failure. Circ. J. 2014, 78, 2159–2161. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, A.; Gines, P. Predicting mortality in cirrhosis: Serum sodium helps. N. Engl. J. Med. 2008, 359, 1060–1062. [Google Scholar] [CrossRef] [PubMed]
- Guevara, M.; Baccaro, M.E.; Torre, A.; Gomez-Anson, B.; Rios, J.; Torres, F.; Rami, L.; Monte-Rubio, G.C.; Martin-Llahi, M.; Arroyo, V.; et al. Hyponatremia is a risk factor of hepatic encephalopathy in patients with cirrhosis: A prospective study with time-dependent analysis. Am. J. Gastroenterol. 2009, 104, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Jenq, C.C.; Tsai, M.H.; Tian, Y.C.; Chang, M.Y.; Lin, C.Y.; Lien, C.M.; Chen, Y.C.; Fang, J.T.; Chen, P.C.; Yang, C.W. Serum sodium predicts prognosis in critically ill cirrhotic patients. J. Clin. Gastroenterol. 2010, 44, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, Y.; Ogawa, H.; Yamashita, H.; Chihara, T.; Miyamoto, H.; Nakamura, S.; Onogawa, T.; Yamashita, T.; Hosokawa, T.; Mori, T.; et al. Characterization of a novel aquaretic agent, OPC-31260, as an orally effective, nonpeptide vasopressin V2 receptor antagonist. Br. J. Pharmacol. 1992, 105, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, Y.; Nakamura, T.; Itoh, J.; Hirano, T.; Onogawa, T.; Yamashita, T.; Yamada, Y.; Tsujimae, K.; Aoyama, M.; Kotosai, K.; et al. OPC-41061, a highly potent human vasopressin V2-receptor antagonist: Pharmacological profile and aquaretic effect by single and multiple oral dosing in rats. J. Pharmacol. Exp. Ther. 1998, 287, 860–866. [Google Scholar] [PubMed]
- Fujisawa, G.; Ishikawa, S.; Tsuboi, Y.; Okada, K.; Saito, T. Therapeutic efficacy of non-peptide ADH antagonist OPC-31260 in SIADH rats. Kidney Int. 1993, 44, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, Y.; Ishikawa, S.; Fujisawa, G.; Okada, K.; Saito, T. Therapeutic efficacy of the non-peptide AVP antagonist OPC-31260 in cirrhotic rats. Kidney Int. 1994, 46, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ishikawa, S.; Abe, K.; Kamoi, K.; Yamada, K.; Shimizu, K.; Saruta, T.; Yoshida, S. Acute aquaresis by the nonpeptide arginine vasopressin (AVP) antagonist OPC-31260 improves hyponatremia in patients with syndrome of inappropriate secretion of antidiuretic hormone (SIADH). J. Clin. Endocrinol. Metab. 1997, 82, 1054–1057. [Google Scholar] [CrossRef] [PubMed]
- Berl, T.; Quittant-Pelletier, F.; Verbalis, J.G.; Schrier, R.W.; Bichet, D.G.; Ouyang, J.; Czerwiec, F.S. Oral tolvaptan is safe and effective in chronic hyponatremia. J. Am. Soc. Nephrol. 2010, 21, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Gheroghiade, M.; Niazi, I.; Ouyang, J.; Czerwiec, F.; Kambayashi, J.; Zampino, M.; Otlandi, C. Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: Results from a double-blind, randomized trial. Circulation 2003, 107, 2690–2696. [Google Scholar] [CrossRef] [PubMed]
- Gheroghiade, M.; Konstam, M.A.; Burnett, J.C.; Grinfeld, L.; Maggioni, A.P.; Swedberg, K.; Udelson, J.E.; Zannad, F.; Cook, T.; Ouyang, J.; et al. Short-term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure. The EVEREST clinical status trial. JAMA 2007, 297, 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Kinugawa, K.; Sato, N.; Inomata, T.; Shimokawa, Y.; Iwatake, N.; Mizuguchi, K. Efficacy and safety of tolvaptan in heart failure patients with volume overload: An interim result of post-marketing surveillance in Japan. Circ. J. 2014, 78, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Kinugawa, K.; Shiga, T.; Kato, N.; Muraoka, H.; Minatsuki, S.; Inaba, T.; Maki, H.; Hatano, M.; Yao, A.; et al. Novel criteria of urinary osmolality effectively predict response to tolvaptan in decompensated heart failure patients: Association between non-responders and chronic kidney disease. Circ. J. 2013, 77, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Toda, H.; Nakamura, K.; Nakahama, M.; Wada, T.; Watanabe, A.; Hashimoto, K.; Terasaka, R.; Tokioka, K.; Nishii, N.; Miyoshi, T.; et al. Clinical characteristics of responders to treatment with tolvaptan in patients with acute decompensated heart failure: Importance of preserved kidney size. J. Cardiol. 2016, 67, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Uemura, Y.; Shibata, R.; Takemoto, K.; Uchikawa, T.; Koyasu, M.; Ishikawa, S.; Imai, R.; Ozaki, Y.; Watanabe, S.; Teraoka, T.; et al. Safety and efficacy of long-term use of tolvaptan in patients with heart failure and chronic kidney disease. Circ. J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Matsue, Y.; Suzuki, M.; Torii, S.; Yamaguchi, S.; Fukamizu, S.; Ono, Y.; Fujii, H.; Kitai, T.; Nishioka, T.; Sugi, K.; et al. Prognostic impact of early treatment with tolvaptan in patients with acute heart failure and renal dysfunction. Int. J. Cardiol. 2016, 221, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Gaglio, P.; Marfo, K.; Chiodo, J. Hyponatremia in cirrhosis and end-stage liver disease: Treatment with the vasopressin V2-receptor antagonist tolvaptan. Dig. Dis. Sci. 2012, 57, 2774–2785. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, A.; Gines, P.; Marotta, P.; Czewiec, F.; Ouyang, J.; Guevara, M.; Afdhal, N.H. Tolvaptan, an oral vasopressin antagonist, in the treatment of hyponatremia in cirrhosis. J. Hepatol. 2012, 56, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Sakaida, I.; Kawazoe, S.; Kajimura, K.; Saito, T.; Okuse, C.; Takaguchi, K.; Okada, M.; Okita, K.; The ASCITES-DOUBLEBLIND Study Group. Tolvaptan for improvement of hepatic edema: A phase 3, multicenter, randomized, double-blind, placebo-controlled trial. Hepatol. Res. 2014, 44, 73–82. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Cancer | Diseases of Central Nervous System | Intrathoracic Diseases | Drugs |
|---|---|---|---|
| Lung cancer | Encephalils | Pneumonia | Vincristine |
| Duodenal cancer | Meningitis | Tuberculosis | Cyclophosphamide |
| Pancreatic cancer | Cerebrovascular diseases (Cerebral infarction and hemorrhage) | Pulmonary abscess | Clofiblate |
| Malignant lymphoma | Subarachnoid hemorrhage | Pulmonary fungus diseases | Carbamazepine |
| Prostatic cancer | Subdural hematoma | Lung cancer (non-ectopic production) | Nicotine |
| Ewing sarcoma | Head injury | ||
| Acute porphyria | |||
| Pituitary tumor |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishikawa, S.-e. Is Exaggerated Release of Arginine Vasopressin an Endocrine Disorder? Pathophysiology and Treatment. J. Clin. Med. 2017, 6, 102. https://doi.org/10.3390/jcm6110102
Ishikawa S-e. Is Exaggerated Release of Arginine Vasopressin an Endocrine Disorder? Pathophysiology and Treatment. Journal of Clinical Medicine. 2017; 6(11):102. https://doi.org/10.3390/jcm6110102
Chicago/Turabian StyleIshikawa, San-e. 2017. "Is Exaggerated Release of Arginine Vasopressin an Endocrine Disorder? Pathophysiology and Treatment" Journal of Clinical Medicine 6, no. 11: 102. https://doi.org/10.3390/jcm6110102
APA StyleIshikawa, S.-e. (2017). Is Exaggerated Release of Arginine Vasopressin an Endocrine Disorder? Pathophysiology and Treatment. Journal of Clinical Medicine, 6(11), 102. https://doi.org/10.3390/jcm6110102