
The Effects of Ascorbate, N-Acetylcysteine, and Resveratrol on Fibroblasts from Patients with Mitochondrial Disorders


Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Tissue Culture
2.3. Assays in Microtiter Wells
2.4. Respiratory Chain Enzymes
2.5. Statistical Analysis
3. Results
3.1. Mitochondrial Respiratory Chain Enzymes
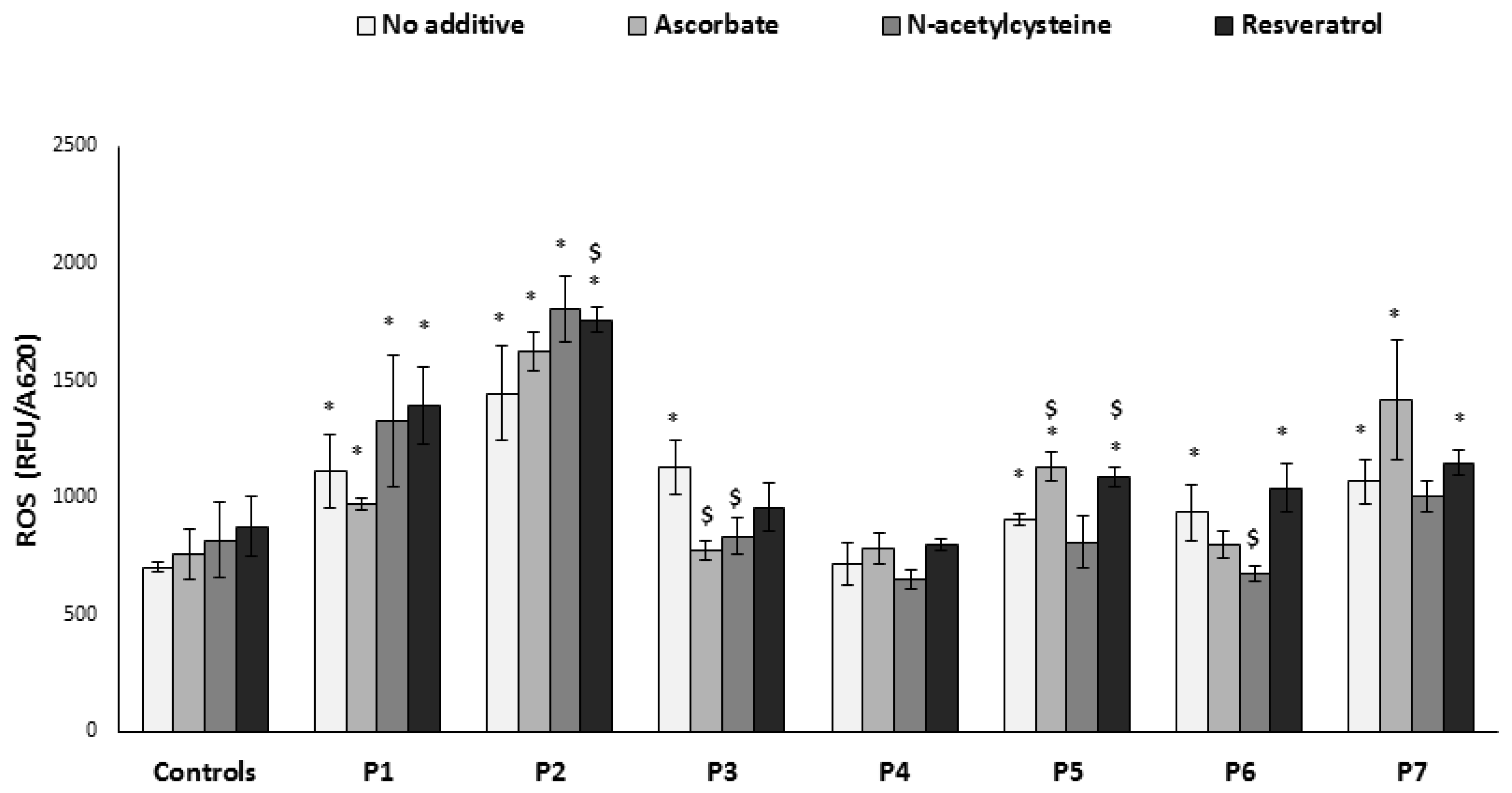
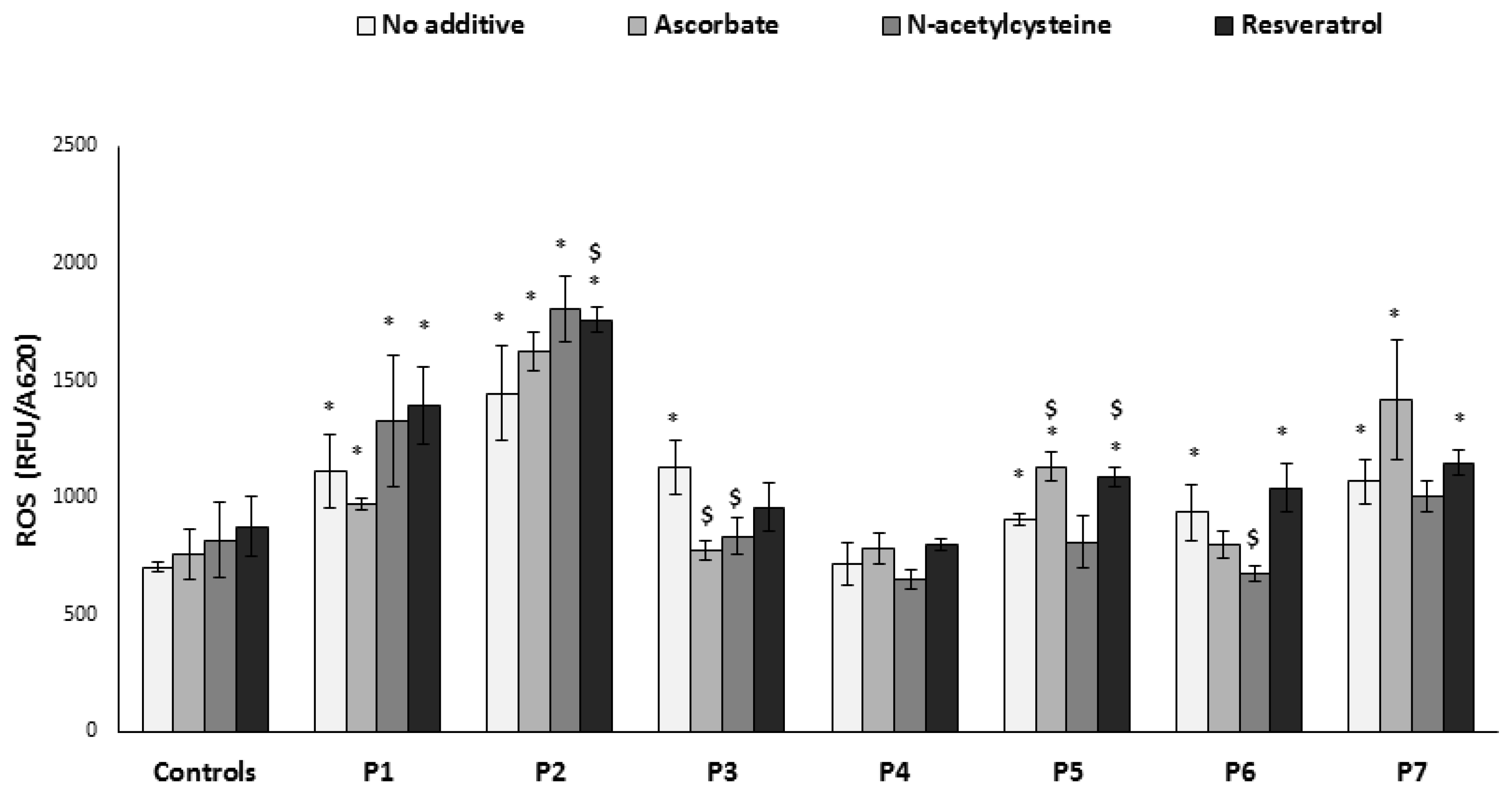
3.2. ROS Production
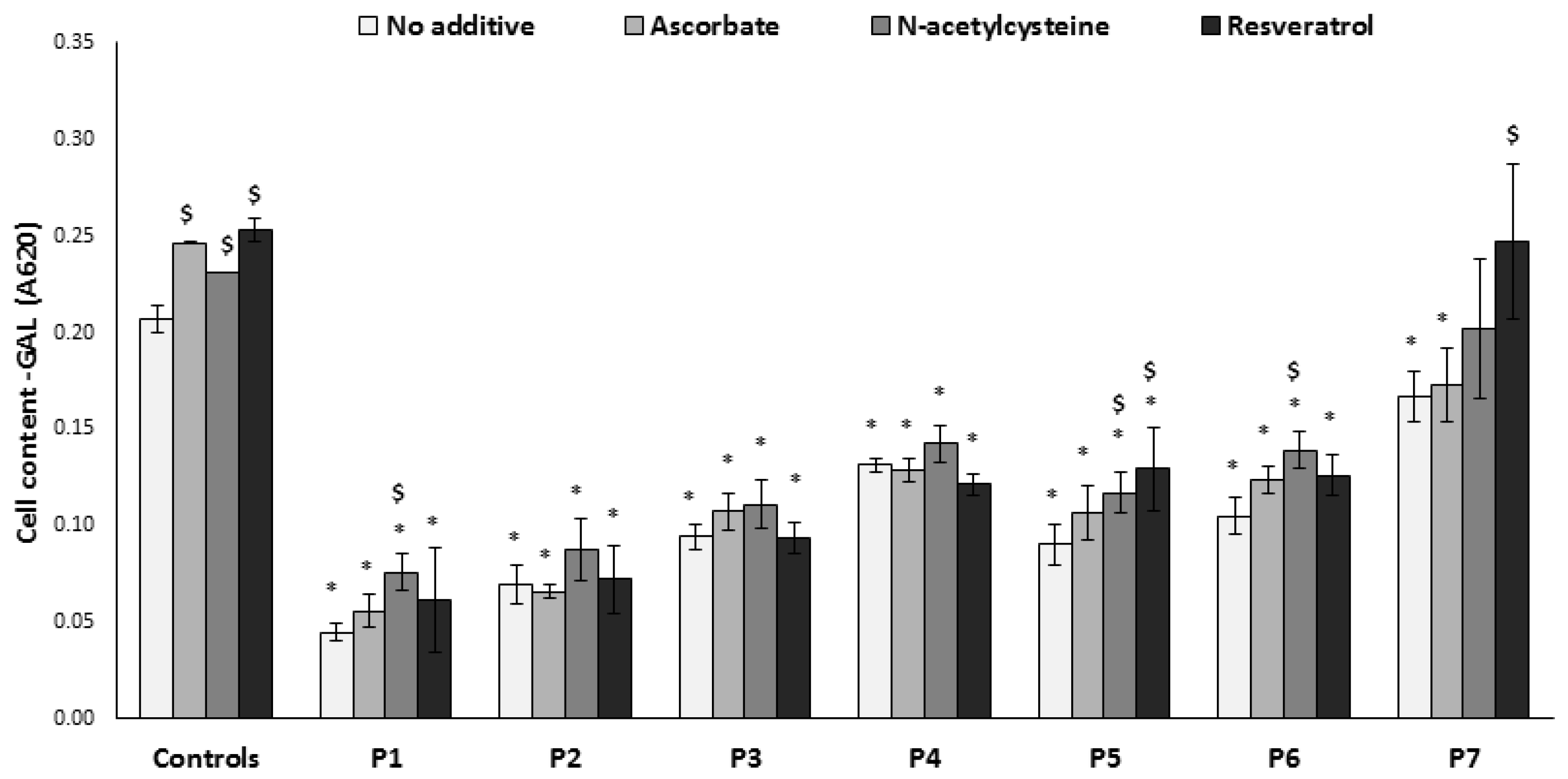
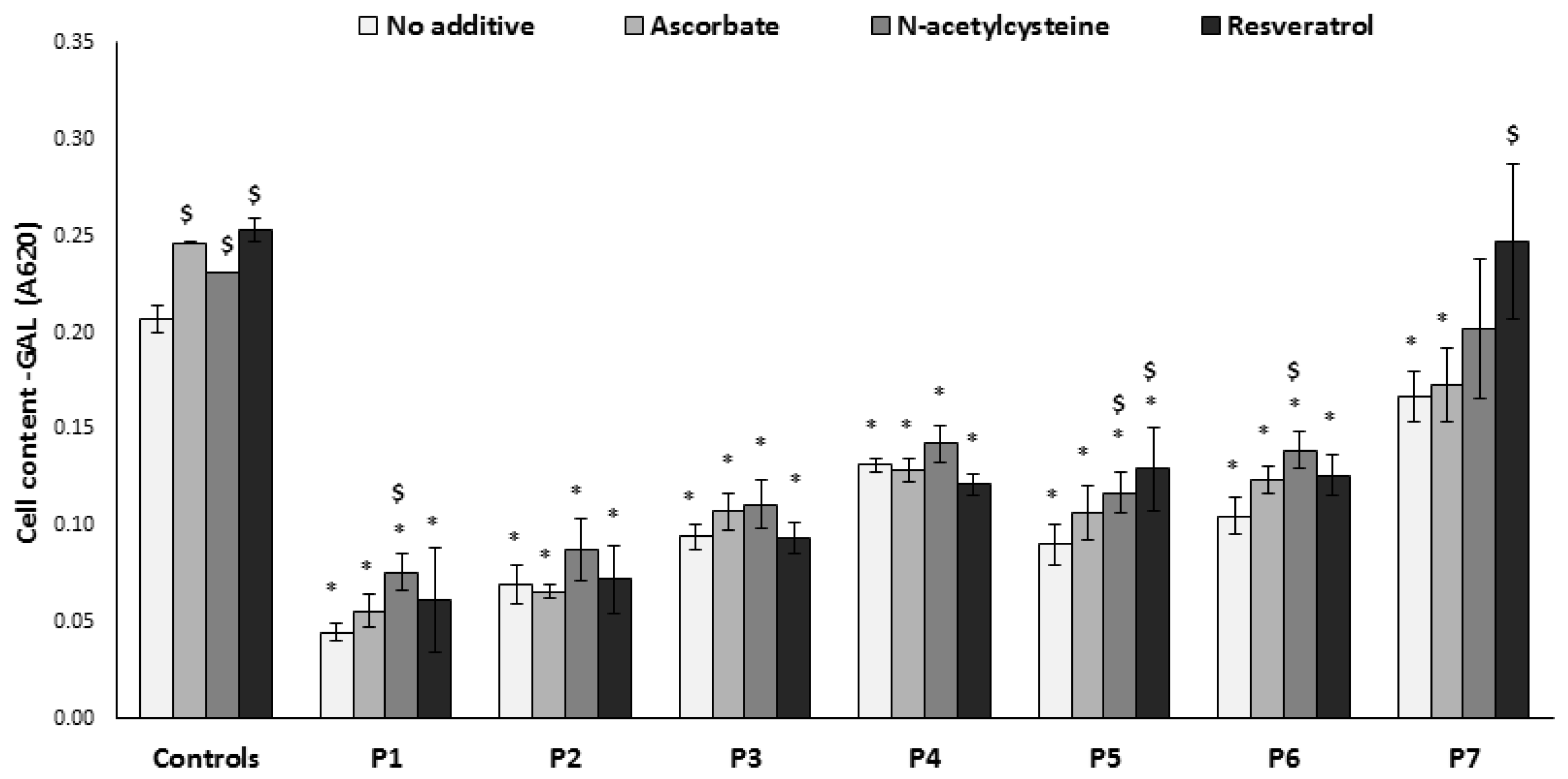
3.3. Growth on Glucose-Free Medium
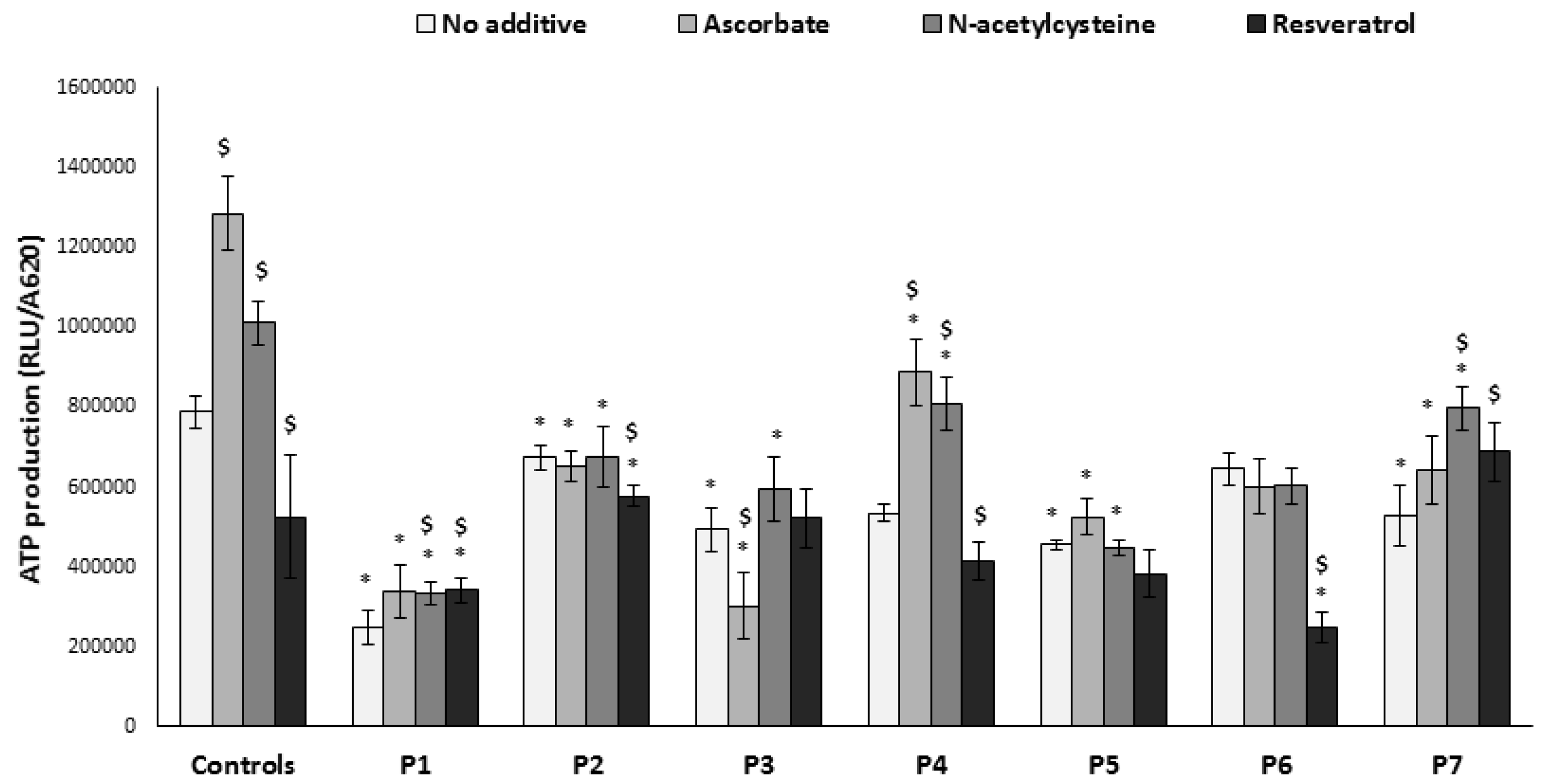
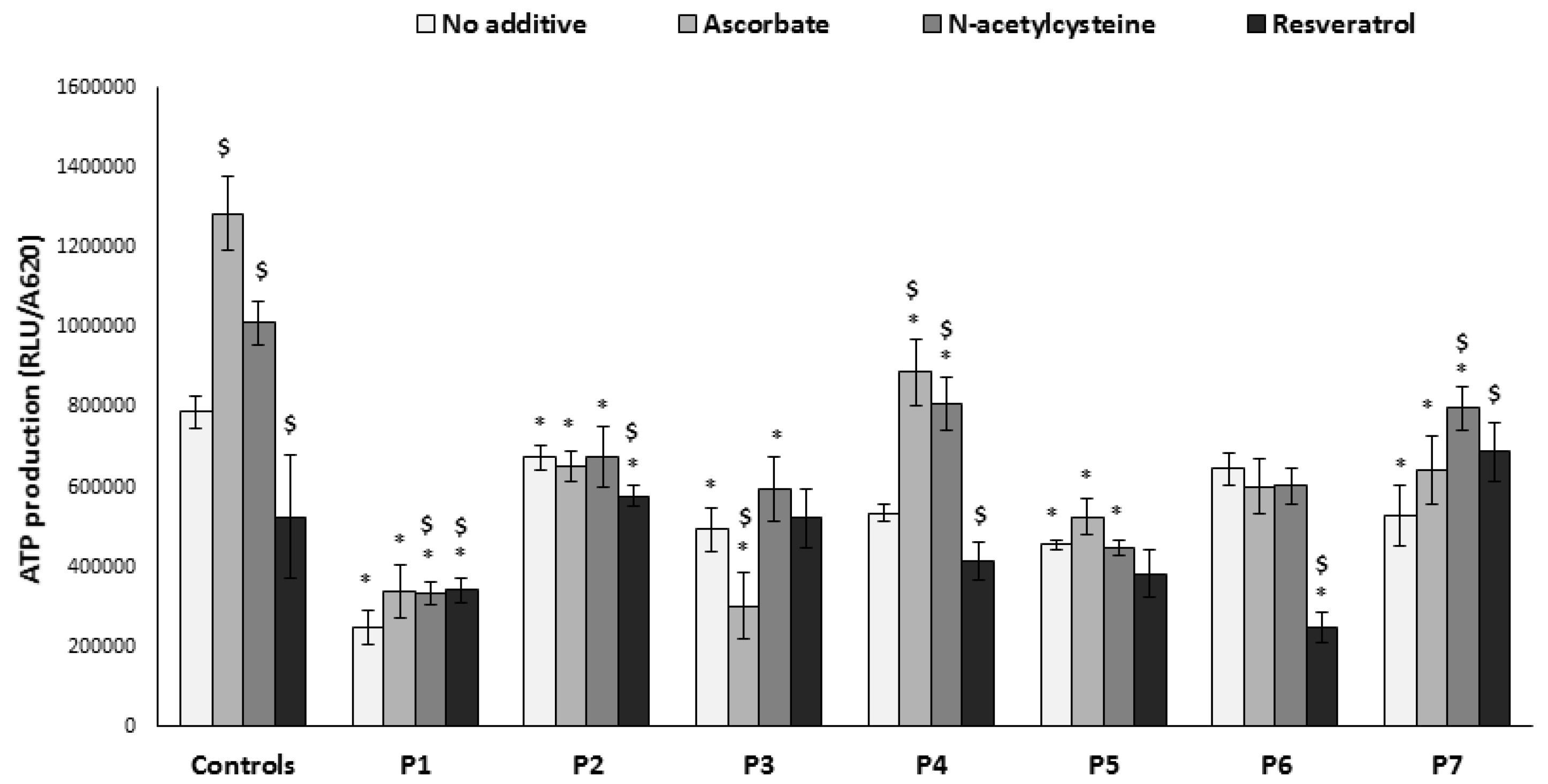
3.4. ATP Production
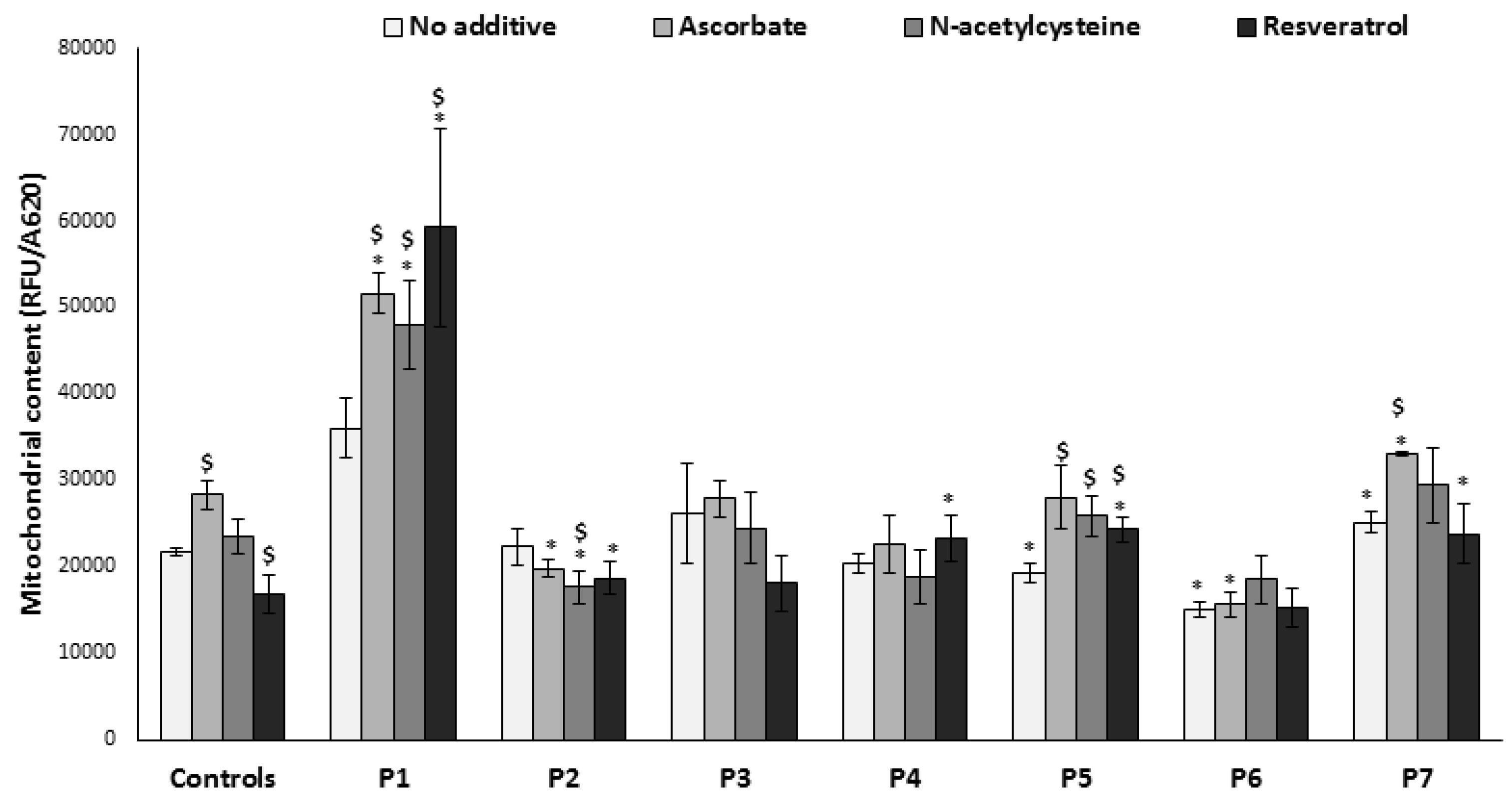
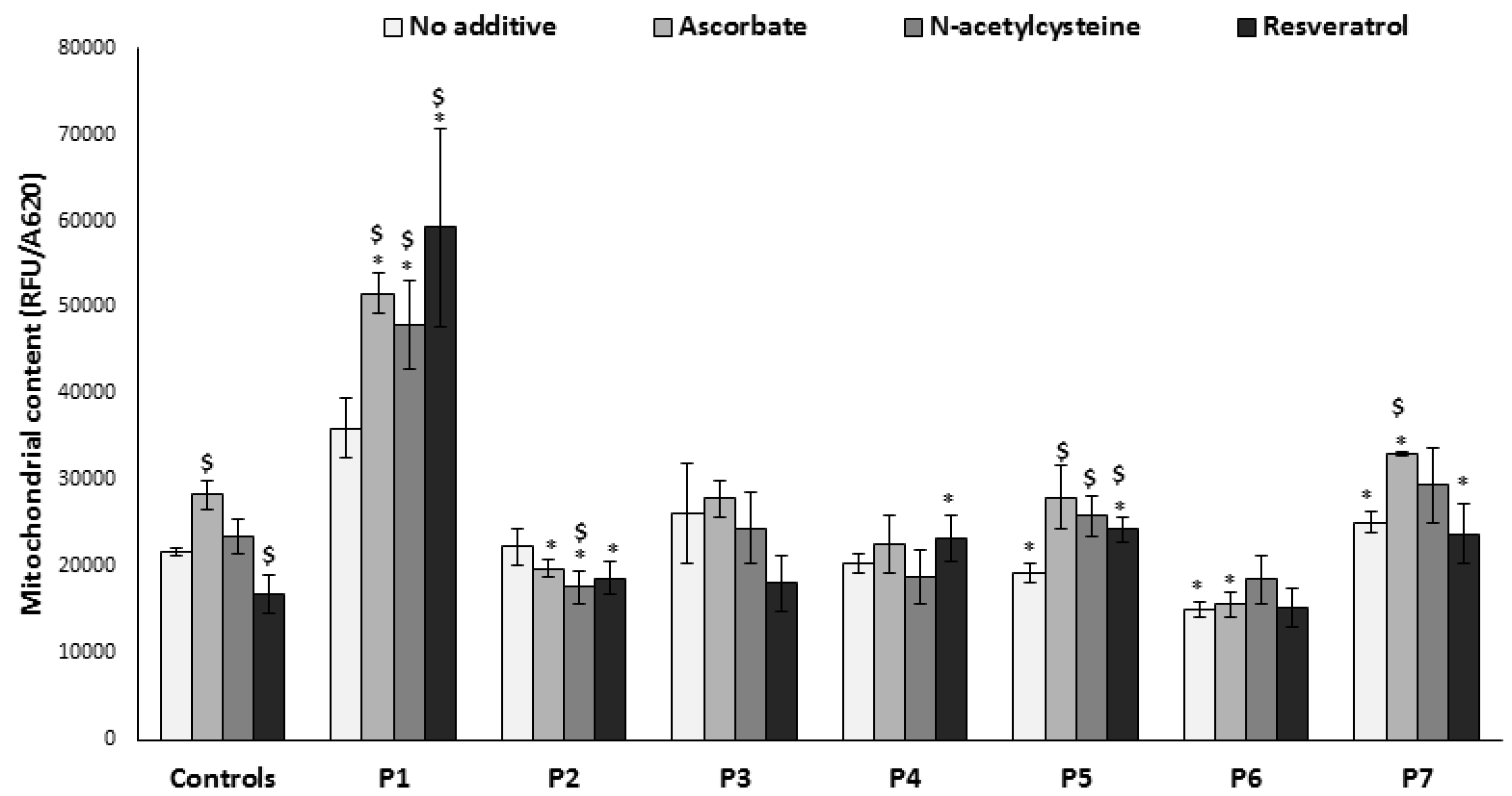
3.5. Mitochondrial Content
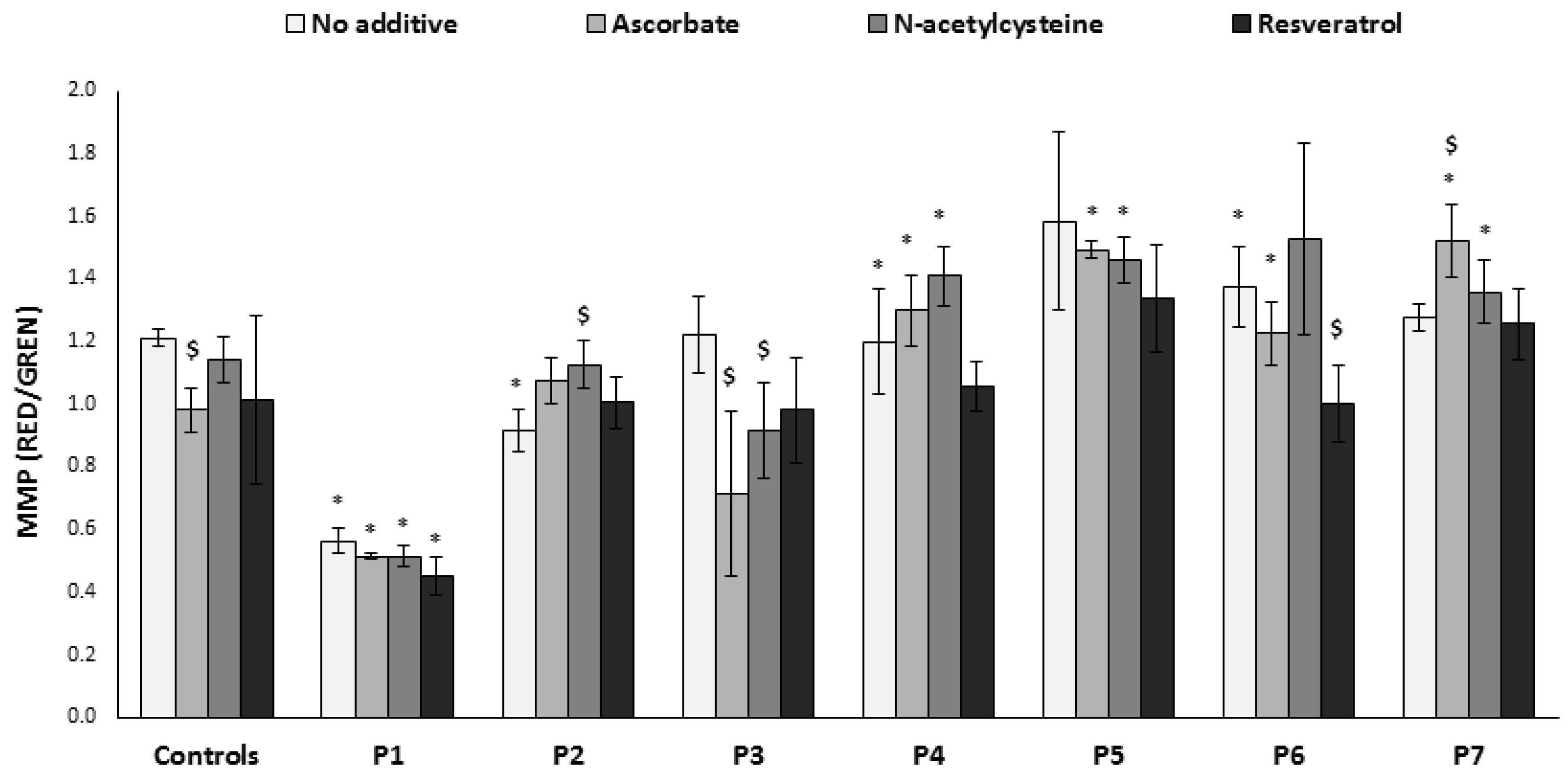
3.6. Mitochondrial Membrane Potential
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Skladal, D.; Halliday, J.; Thorburn, D.R. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 2003, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Rötig, A. Human diseases with impaired mitochondrial protein synthesis. Biochim. Biophys. Acta 2011, 1807, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Saada, A. The use of individual patient’s fibroblasts in the search for personalized treatment of nuclear encoded OXPHOS diseases. Mol. Genet. Metab. 2011, 104, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Reddy, P.H.; Iijima, M.; Sesaki, H. Mitochondrial division and fusion in metabolism. Curr. Opin. Cell Biol. 2015, 33, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Scandalios, J.G. Oxidative stress: Molecular perception and transduction of signals triggering antioxidant gene defenses. Braz. J. Med. Biol. Res. 2005, 38, 995–1014. [Google Scholar] [CrossRef] [PubMed]
- Pitkanen, S.; Robinson, B.H. Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J. Clin. Investig. 1996, 98, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.; Beyrath, J.; Fung, C.-W.; Koene, S.; Rodenburg, R.J.; Willems, P.H.; Smeitink, J.A. Mitochondrial disorders in children: Toward development of small-molecule treatment strategies. EMBO Mol. Med. 2016, 8, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Golubitzky, A.; Dan, P.; Weissman, S.; Link, G.; Wikstrom, J.D.; Saada, A. Screening for active small molecules in mitochondrial complex I deficient patient’s fibroblasts, reveals AICAR as the most beneficial compound. PLoS ONE 2011, 6, e26883. [Google Scholar] [CrossRef] [PubMed]
- Linster, C.L.; Van Schaftingen, E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007, 274, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Vitamin C: Antioxidant or pro-oxidant in vivo? Free Radic. Res. 1996, 25, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Malhi, G.S.; Gray, L.J.; Dean, O.M. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol. Sci. 2013, 34, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 4117–4129. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.H.W.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in Translational Medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef] [PubMed]
- Leonard, S.S.; Xia, C.; Jiang, B.-H.; Stinefelt, B.; Klandorf, H.; Harris, G.K.; Shi, X. Resveratrol scavenges reactive oxygen species and effects radical-induced cellular responses. Biochem. Biophys. Res. Commun. 2003, 309, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Udenigwe, C.C.; Ramprasath, V.R.; Aluko, R.E.; Jones, P.J.H. Potential of resveratrol in anticancer and anti-inflammatory therapy. Nutr. Rev. 2008, 66, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Negari, S.B.-H.; Aouizerat, T.; Tenenbaum, A.; Cohen-Cymberknoh, M.; Shoseyov, D.; Kerem, E.; Saada, A. Mitochondrial OXPHOS function is unaffected by chronic azithromycin treatment. J. Cyst. Fibros. 2013, 12, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Saada, A.; Shaag, A.; Elpeleg, O. mtDNA depletion myopathy: Elucidation of the tissue specificity in the mitochondrial thymidine kinase (TK2) deficiency. Mol. Genet. Metab. 2003, 79, 1–5. [Google Scholar] [CrossRef]
- Saada, A.; Bar-Meir, M.; Belaiche, C.; Miller, C.; Elpeleg, O. Evaluation of enzymatic assays and compounds affecting ATP production in mitochondrial respiratory chain complex I deficiency. Anal. Biochem. 2004, 335, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Smeitink, J.A.M.; Elpeleg, O.; Antonicka, H.; Diepstra, H.; Saada, A.; Smits, P.; Sasarman, F.; Vriend, G.; Jacob-Hirsch, J.; Shaag, A.; et al. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am. J. Hum. Genet. 2006, 79, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Saada, A.; Shaag, A.; Arnon, S.; Dolfin, T.; Miller, C.; Fuchs-Telem, D.; Lombes, A.; Elpeleg, O. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J. Med. Genet. 2007, 44, 784–786. [Google Scholar] [CrossRef] [PubMed]
- Abdulhag, U.N.; Soiferman, D.; Schueler-Furman, O.; Miller, C.; Shaag, A.; Elpeleg, O.; Edvardson, S.; Saada, A. Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur. J. Hum. Genet. 2014, 23, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Shany, E.; Saada, A.; Landau, D.; Shaag, A.; Hershkovitz, E.; Elpeleg, O.N. Lipoamide dehydrogenase deficiency due to a novel mutation in the interface domain. Biochem. Biophys. Res. Commun. 1999, 262, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Sheffer, R.; Douiev, L.; Edvardson, S.; Shaag, A.; Tamimi, K.; Soiferman, D.; Meiner, V.; Saada, A. Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. Am. J. Med. Genet. A 2016, 170, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M. Treatment of mitochondrial disorders: Antioxidants and beyond. J. Child Neurol. 2014, 29, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, J.; Duchen, M.R.; Hothersall, J.; Clark, J.B.; Heales, S.J.R. Induction of mitochondrial oxidative stress in astrocytes by nitric oxide precedes disruption of energy metabolism. J. Neurochem. 2005, 95, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, H.; Pfeffer, G.; Bargiela, D.; Horvath, R.; Chinnery, P.F. Emerging therapies for mitochondrial disorders. Brain 2016, 139, 1633–1648. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.E.; Lodi, R.; Rajagopalan, B.; Bradley, J.L.; Crilley, J.G.; Turner, C.; Blamire, A.M.; Manners, D.; Styles, P.; Schapira, A.H.V.; et al. Antioxidant treatment of patients with Friedreich ataxia: Four-year follow-up. Arch. Neurol. 2005, 62, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, D.; Catteruccia, M.; Piemonte, F.; Pastore, A.; Tozzi, G.; Dionisi-Vici, C.; Pontrelli, G.; Corsetti, T.; Livadiotti, S.; Kheifets, V.; et al. EPI-743 reverses the progression of the pediatric mitochondrial disease-Genetically defined Leigh Syndrome. Mol. Genet. Metab. 2012, 107, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Finichiu, P.G.; Larsen, D.S.; Evans, C.; Larsen, L.; Bright, T.P.; Robb, E.L.; Trnka, J.; Prime, T.A.; James, A.M.; Smith, R.A.J.; et al. A mitochondria-targeted derivative of ascorbate: MitoC. Free Radic. Biol. Med. 2015, 89, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Salmi, H.; Leonard, J.V.; Rahman, S.; Lapatto, R. Plasma thiol status is altered in children with mitochondrial diseases. Scand. J. Clin. Lab. Investig. 2012, 72, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Miquel, J.; Ferrándiz, M.L.; De Juan, E.; Sevila, I.; Martínez, M. N-acetylcysteine protects against age-related decline of oxidative phosphorylation in liver mitochondria. Eur. J. Pharmacol. 1995, 292, 333–335. [Google Scholar] [CrossRef]
- Cocco, T.; Sgobbo, P.; Clemente, M.; Lopriore, B.; Grattagliano, I.; Di Paola, M.; Villani, G. Tissue-specific changes of mitochondrial functions in aged rats: Effect of a long-term dietary treatment with N-acetylcysteine. Free Radic. Biol. Med. 2005, 38, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Kamboj, S.S.; Sandhir, R. Protective effect of N-acetylcysteine supplementation on mitochondrial oxidative stress and mitochondrial enzymes in cerebral cortex of streptozotocin-treated diabetic rats. Mitochondrion 2011, 11, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Soiferman, D.; Ayalon, O.; Weissman, S.; Saada, A. The effect of small molecules on nuclear-encoded translation diseases. Biochimie 2014, 100, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Mongan, P.D.; Morgan, P.D. Ascorbate reduces superoxide production and improves mitochondrial respiratory chain function in human fibroblasts with electron transport chain deficiencies. Mitochondrion 2001, 1, 191–198. [Google Scholar] [CrossRef]
- Ghneim, H.K.; Al-Sheikh, Y.A. The effect of aging and increasing ascorbate concentrations on respiratory chain activity in cultured human fibroblasts. Cell Biochem. Funct. 2010, 28, 283–292. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, M.R.; Nabavi, S.F.; Manayi, A.; Daglia, M.; Hajheydari, Z.; Nabavi, S.M. Resveratrol and the mitochondria: From triggering the intrinsic apoptotic pathway to inducing mitochondrial biogenesis, a mechanistic view. Biochim. Biophys. Acta 2016, 1860, 727–745. [Google Scholar] [CrossRef] [PubMed]
- Lopes Costa, A.; Le Bachelier, C.; Mathieu, L.; Rotig, A.; Boneh, A.; De Lonlay, P.; Tarnopolsky, M.A.; Thorburn, D.R.; Bastin, J.; Djouadi, F. Beneficial effects of resveratrol on respiratory chain defects in patients’ fibroblasts involve estrogen receptor and estrogen-related receptor alpha signaling. Hum. Mol. Genet. 2014, 23, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiecki, A.; Roomi, M.W.; Kalinovsky, T.; Rath, M. Anticancer Efficacy of Polyphenols and Their Combinations. Nutrients 2016. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, B.; Vandemeulebroecke, K.; Smet, J.; Vanlander, A.; Seneca, S.; Lissens, W.; Van Hove, J.L.; Deschepper, E.; Briones, P.; Van Coster, R. Effect of resveratrol on cultured skin fibroblasts from patients with oxidative phosphorylation defects. Phytother. Res. 2014, 28, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Gottschalk, B.; Parichatikanond, W.; Eroglu, E.; Klec, C.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Resveratrol Specifically Kills Cancer Cells by a Devastating Increase in the Ca2+ Coupling Between the Greatly Tethered Endoplasmic Reticulum and Mitochondria. Cell. Physiol. Biochem. 2016, 39, 1404–1420. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fibroblast | Defect | Activity * | Mutation | |
|---|---|---|---|---|
| Fibroblasts | Muscle | |||
| P1 | Combined defect | CI 44% CIV 25% | CI 12% | EFTsR333W/R333W |
| CII + III 17% | ||||
| CIV 14% | ||||
| P2 | CI 51% CIV 65% | CI 42% | MRPS22R170H/R170H | |
| CII + III 12% | ||||
| CIV 8% | ||||
| P3 | Single defect | CI 44% | - | undefined |
| P4 | CIV 43% | - | undefined | |
| P5 | CIV Undetectable | CIV undetectable | COX6B1R20C/R20C | |
| P6 | Indirect defect | Lipoamide dehydrogenase 36% | - | LADD479V/D479V |
| P7 | CI 58% | CIV 47% | DNM1LG362S/+ | |
| CIV 29% | ||||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Douiev, L.; Soiferman, D.; Alban, C.; Saada, A. The Effects of Ascorbate, N-Acetylcysteine, and Resveratrol on Fibroblasts from Patients with Mitochondrial Disorders. J. Clin. Med. 2017, 6, 1. https://doi.org/10.3390/jcm6010001
Douiev L, Soiferman D, Alban C, Saada A. The Effects of Ascorbate, N-Acetylcysteine, and Resveratrol on Fibroblasts from Patients with Mitochondrial Disorders. Journal of Clinical Medicine. 2017; 6(1):1. https://doi.org/10.3390/jcm6010001
Chicago/Turabian StyleDouiev, Liza, Devorah Soiferman, Corinne Alban, and Ann Saada. 2017. "The Effects of Ascorbate, N-Acetylcysteine, and Resveratrol on Fibroblasts from Patients with Mitochondrial Disorders" Journal of Clinical Medicine 6, no. 1: 1. https://doi.org/10.3390/jcm6010001
APA StyleDouiev, L., Soiferman, D., Alban, C., & Saada, A. (2017). The Effects of Ascorbate, N-Acetylcysteine, and Resveratrol on Fibroblasts from Patients with Mitochondrial Disorders. Journal of Clinical Medicine, 6(1), 1. https://doi.org/10.3390/jcm6010001