
Signal Transduction Pathways of EMT Induced by TGF-β, SHH, and WNT and Their Crosstalks


{kind=link}
{kind=link}
{kind=link}
Abstract
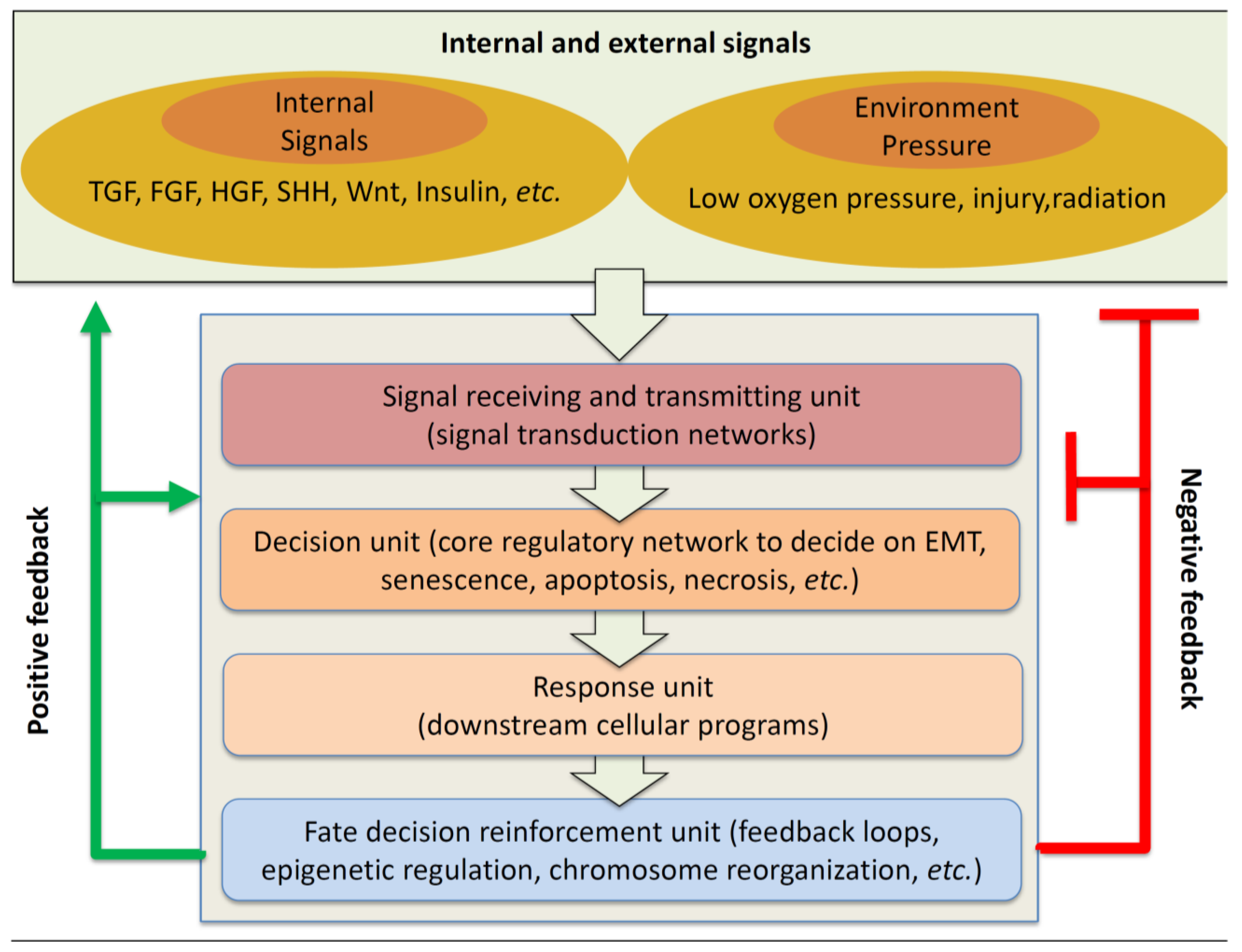
:1. Biomedical Significance of EMT
2. Multiple Signal Transduction Pathway and Their Crosstalks
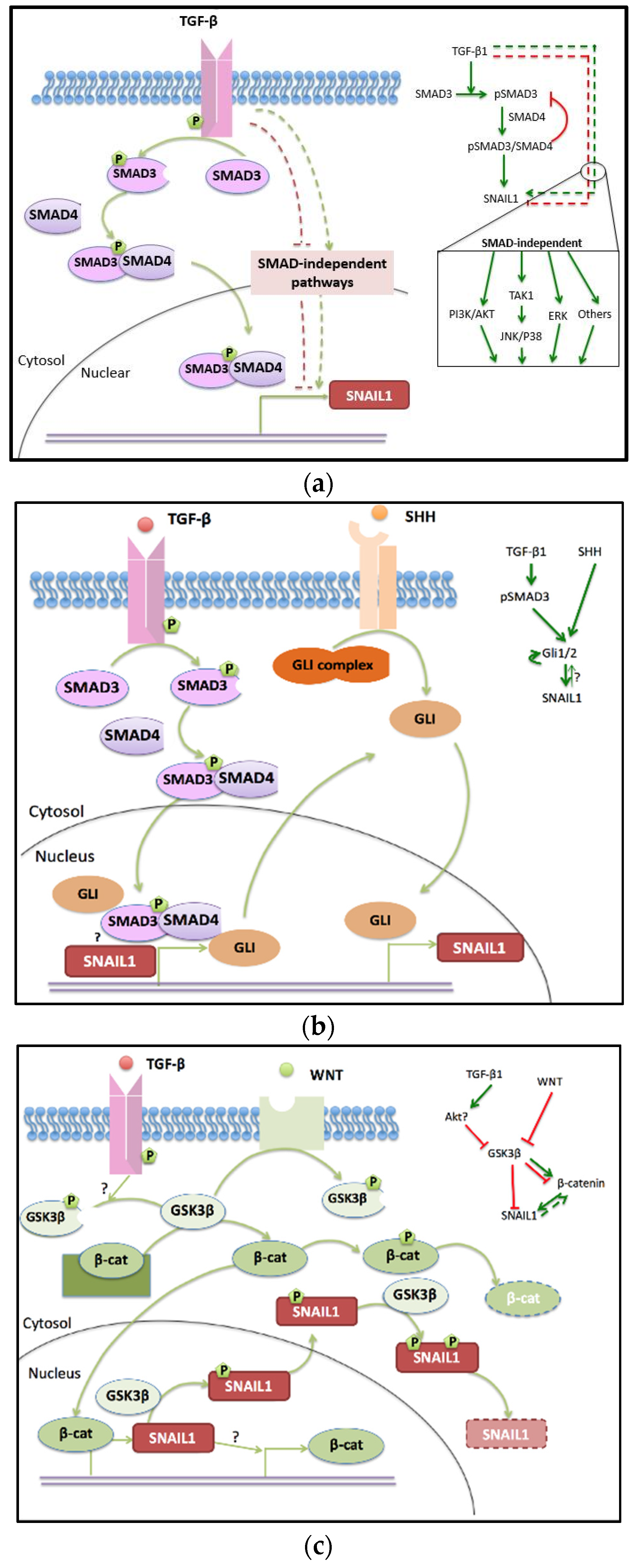
2.1. TGF-β Pathway
2.1.1. SMAD-Dependent TGF-β Pathway
2.1.2. SMAD Independent Pathway
2.1.3. Drugs Targeted to TGF-β Pathway
2.2. SHH Pathway Engages in EMT and Crosstalks to TGF-β
2.2.1. The off and on States of the SHH Pathway
2.2.2. Regulation of GLI Proteins and Crosstalk to the TGF-β Pathway
2.2.3. Clinical Observation and Interventions of SHH Signaling Pathway in Cancer
2.3. WNT Pathway in Cancer Progress and EMT
Clinical Observation and Interventions of the WNT Signaling Pathway in Cancer
3. Systems Biology in Signaling Crosstalk and Drug Discovery
- Which of the proteins/regulators to target for clinic and commercial consideration?
- How to select chemicals that are suitable for therapy from the gigantic data pool?
- How to design treatments that target to specific population of tumors in patients?
Acknowledgments
Conflicts of Interest
References
- Debnath, J.; Brugge, J.S. Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer 2005, 5, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Mather, J.P.; Roberts, P.E. Introduction to Cell and Tissue Culture: Theory and Technique; Springer Science & Business Media: New York, NY, USA, 1998; Volume 1. [Google Scholar]
- Mackenzie, T.C.; Flake, A.W. Human mesenchymal stem cells persist, demonstrate site-specific multipotential differentiation, and are present in sites of wound healing and tissue regeneration after transplantation into fetal sheep. Blood Cells Mol. Dis. 2001, 27, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.; Spaeth, E.; Dembinski, J.L.; Dietrich, M.; Watson, K.; Klopp, A.; Battula, V.L.; Weil, M.; Andreeff, M.; Marini, F.C. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells 2009, 27, 2614–2623. [Google Scholar] [CrossRef] [PubMed]
- Hoogduijn, M.J.; Dor, F.J. Mesenchymal stem cells in transplantation and tissue regeneration. Front. Immunol. 2011, 2, 84. [Google Scholar] [CrossRef] [PubMed]
- Hematti, P.; Keating, A. Mesenchymal Stromal Cells: Biology and Clinical Applications; Springer Science & Business Media: New York, NY, USA, 2013. [Google Scholar]
- Hay, E.D. Organization and fine structure of epithelium and mesenchyme in the developing chick embryo. Epithel. Mesenchymal Interact. 1968, 2, 31–35. [Google Scholar]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Ten Berge, D.; Koole, W.; Fuerer, C.; Fish, M.; Eroglu, E.; Nusse, R. Wnt signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell 2008, 3, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Gill, J.G.; Kyba, M.; Murphy, T.L.; Murphy, K.M. Canonical wnt signaling is required for development of embryonic stem cell-derived mesoderm. Development 2006, 133, 3787–3796. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, R.T.; Niehrs, C. Fibroblast growth factor signaling during early vertebrate development. Endocr. Rev. 2005, 26, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Eastham, A.M.; Spencer, H.; Soncin, F.; Ritson, S.; Merry, C.L.; Stern, P.L.; Ward, C.M. Epithelial-mesenchymal transition events during human embryonic stem cell differentiation. Cancer Res. 2007, 67, 11254–11262. [Google Scholar] [CrossRef] [PubMed]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-β1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.S.; Ayerinskas, I.I.; Vincent, E.B.; McKinney, L.A.; Weeks, D.L.; Runyan, R.B. TGFβ2 and TGFβ3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev. Biol. 1999, 208, 530–545. [Google Scholar] [CrossRef] [PubMed]
- Arnoux, V.; Nassour, M.; L’Helgoualc’h, A.; Hipskind, R.A.; Savagner, P. Erk5 controls slug expression and keratinocyte activation during wound healing. Mol. Biol. Cell 2008, 19, 4738–4749. [Google Scholar] [CrossRef] [PubMed]
- Carretero, M.; Escámez, M.J.; García, M.; Duarte, B.; Holguín, A.; Retamosa, L.; Jorcano, J.L.; del Río, M.; Larcher, F. In vitro and in vivo wound healing-promoting activities of human cathelicidin ll-37. J. Investig. Dermatol. 2008, 128, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Grimm, W.A.; Garner, W.L.; Qin, L.; Travis, T.; Tan, N.; Han, Y.-P. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-α through bone morphogenic protein-2. Am. J. Pathol. 2010, 176, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R. The Biology of Cancer; Garland Science: New York, NY, USA, 2013. [Google Scholar]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Cajal, S.R.Y. Manual de Anatomia Patologica General; Casa Provincial Caridad: Barcelona, Spain, 1890. [Google Scholar]
- Nieto, M.A. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Yingling, J.M.; Blanchard, K.L.; Sawyer, J.S. Development of TGF-β signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 2004, 3, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.D.; Klaus, A.; Garratt, A.N.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Curr. Opin. Cell Biol. 2013, 25, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.; Sasser, A.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.; Hall, B. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Louise Jones, J. Jekyll and hyde: The role of the microenvironment on the progression of cancer. J. Pathol. 2011, 223, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Parri, M.; Chiarugi, P. Emt and oxidative stress: A bidirectional interplay affecting tumor malignancy. Antioxid. Redox Signal. 2012, 16, 1248–1263. [Google Scholar] [CrossRef] [PubMed]
- Montserrat, N.; Mozos, A.; Llobet, D.; Dolcet, X.; Pons, C.; de Herreros, A.G.; Matias-Guiu, X.; Prat, J. Epithelial to mesenchymal transition in early stage endometrioid endometrial carcinoma. Hum. Pathol. 2012, 43, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Lee, S.-J.; Jung, Y.S.; Xu, Y.; Kang, H.S.; Ha, N.-C.; Park, B.-J. Blocking of p53-SNAIL binding, promoted by oncogenic K-Ras, recovers p53 expression and function. Neoplasia 2009, 11, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Hill, C.S. TGF-β superfamily signaling in embryonic development and homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Zi, Z.; Chapnick, D.A.; Liu, X. Dynamics of TGF-β/SMAD signaling. FEBS Lett. 2012, 586, 1921–1928. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H.; Miyazono, K.; Ten Dijke, P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-W.; Fairman, R.; Penry, J.; Shi, Y. Formation of a stable heterodimer between SMAD2 and SMAD4. J. Biol. Chem. 2001, 276, 20688–20694. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.M.; Qin, B.; Correia, J.J.; Lam, S.S.; de Caestecker, M.P.; Lin, K. The L3 loop and C-terminal phosphorylation jointly define SMAD protein trimerization. Nat. Struct. Mol. Biol. 2001, 8, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.M.; Qin, B.Y.; Tiwari, A.; Shi, G.; Lam, S.; Hayward, L.J.; De Caestecker, M.; Lin, K. Structural basis of heteromeric SMAD protein assembly in TGF-β signaling. Mol. Cell 2004, 15, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Thuault, S.; Tan, E.-J.; Peinado, H.; Cano, A.; Heldin, C.-H.; Moustakas, A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J. Biol. Chem. 2008, 283, 33437–33446. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Neve, E.P.A.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Brandl, M.; Seidler, B.; Haller, F.; Adamski, J.; Schmid, R.M.; Saur, D.; Schneider, G. IKKα controls canonical TGFβ–SMAD signaling to regulate genes expressing SNAIL and SLUG during EMT in Panc1 cells. J. Cell Sci. 2010, 123, 4231–4239. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Park, S.Y.; Joo, C.-K. Transforming growth factor-β1 represses E-cadherin production via slug expression in lens epithelial cells. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2708–2718. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Afrakhte, M.; Morn, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.-E.; Heldin, C.-H. Identification of SMAD7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 1997, 389, 631–635. [Google Scholar] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and SMAD-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Srinivasan, R.; Wig, J.D.; Radotra, B.D. A study of Smad4, Smad6 and Smad7 in surgically resected samples of pancreatic ductal adenocarcinoma and their correlation with clinicopathological parameters and patient survival. BMC Res. Notes 2011, 4, 560. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, K.; Krebs, T.L.; Yang, J.; Danielpour, D. Smad7 is inactivated through a direct physical interaction with the lim protein Hic-5/ARA55. Oncogene 2008, 27, 6791–6805. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Kamaraju, A.K.; Roberts, A.B. Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-β-mediated SMAD-dependent growth inhibition of human breast carcinoma cells in vivo. J. Biol. Chem. 2005, 280, 1024–1036. [Google Scholar] [CrossRef] [PubMed]
- Kanamaru, C.; Yasuda, H.; Fujita, T. Involvement of SMAD proteins in TGF-β and activin A-induced apoptosis and growth inhibition of liver cells. Hepatol. Res. 2002, 23, 211–219. [Google Scholar] [CrossRef]
- Liu, X.; Sun, Y.; Constantinescu, S.N.; Karam, E.; Weinberg, R.A.; Lodish, H.F. Transforming growth factor β-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10669–10674. [Google Scholar] [CrossRef] [PubMed]
- Fleming, N.I.; Jorissen, R.N.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013, 73, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Groth, S.; Sebens, S.; Lehnert, H.; Gieseler, F.; Fändrich, F. Differential roles of Smad2 and Smad3 in the regulation of TGF-β1-mediated growth inhibition and cell migration in pancreatic ductal adenocarcinoma cells: Control by Rac1. Mol. Cancer 2011, 10, 67. [Google Scholar] [CrossRef] [PubMed]
- Yakicier, M.; Irmak, M.; Romano, A.; Kew, M.; Ozturk, M. Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene 1999, 18, 4879–4883. [Google Scholar] [CrossRef] [PubMed]
- Riggins, G.J.; Kinzler, K.W.; Vogelstein, B.; Thiagalingam, S. Frequency of Smad gene mutations in human cancers. Cancer Res. 1997, 57, 2578–2580. [Google Scholar] [PubMed]
- Miyaki, M.; Kuroki, T. Role of Smad4 (DPC4) inactivation in human cancer. Biochem. Biophys. Res. Commun. 2003, 306, 799–804. [Google Scholar] [CrossRef]
- Chen, H.-S.; Bai, M.-H.; Zhang, T.; Li, G.-D.; Liu, M. Ellagic acid induces cell cycle arrest and apoptosis through TGF-β/Smad3 signaling pathway in human breast cancer MCF-7 cells. Int. J. Oncol. 2015, 46, 1730–1738. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.-C.; Auersperg, N.; Leung, P.C. TGF-β induces serous borderline ovarian tumor cell invasion by activating EMTEMT but triggers apoptosis in low-grade serous ovarian carcinoma cells. PLoS ONE 2012, 7, e42436. [Google Scholar]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; de Guise, C.; Kim, C.; Lemay, S.; Wang, X.-F.; Lebrun, J.-J. Activin induces hepatocyte cell growth arrest through induction of the cyclin-dependent kinase Inhibitor p15 INK4b and Sp1. Cell. Signal. 2004, 16, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Bae, G.-U.; Kang, J.K.; Park, J.W.; Lee, E.K.; Lee, H.Y.; Choi, W.S.; Lee, H.W.; Han, J.-W. Cooperation of H2O2-mediated ERK activation with Smad pathway in TGF-β1 induction of p21 WAF1/Cip1. Cell. Signal. 2006, 18, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Fuxe, J.; Vincent, T.; Garcia de Herreros, A. Transcriptional crosstalk between TGFβ and stem cell pathways in tumor cell invasion: Role of EMT promoting SMAD complexes. Cell Cycle 2010, 9, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-F.; Zhang, H.-J.; Wang, H.-B.; Zhu, J.; Zhou, W.-Y.; Zhang, H.; Zhao, M.-C.; Su, J.-M.; Gao, W.; Zhang, L. Transforming growth factor-β1 induces epithelial-to-mesenchymal transition in human lung cancer cells via PI3K/Akt and MEK/Erk1/2 signaling pathways. Mol. Biol. Rep. 2012, 39, 3549–3556. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-SMAD pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Cell size and invasion in TGF-β-induced epithelial to mesenchymal transition is regulated by activation of the mtor pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.; Bakin, A.V.; Rodeck, U.; Brunet, A.; Arteaga, C.L. Transforming growth factor β enhances epithelial cell survival via Akt-dependent regulation of fkhrl1. Mol. Biol. Cell 2001, 12, 3328–3339. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Wang, H.; Krebs, T.L.; Danielpour, D. Novel roles of Akt and mTOR in suppressing TGF-β/Alk5-mediated Smad3 activation. EMBO J. 2006, 25, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Masszi, A.; Di Ciano, C.; Sirokmany, G.; Arthur, W.T.; Rotstein, O.D.; Wang, J.; McCulloch, C.A.; Rosivall, L.; Mucsi, I.; Kapus, A. Central role for rho in TGF-β1-induced α-smooth muscle actin expression during epithelial-mesenchymal transition. Am. J. Physiol.-Ren. Physiol. 2003, 284, F911–F924. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gudey, S.K.; Landström, M. Non-Smad signaling pathways. Cell Tissue Res. 2012, 347, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, Y.; Hua, X.; Bergelson, S.; Lodish, H.F. Critical role of SMADs and AP-1 complex in transforming growth factor-β-dependent apoptosis. J. Biol. Chem. 2000, 275, 36295–36302. [Google Scholar] [CrossRef] [PubMed]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.-F. Transforming growth factor β induces the cyclin-dependent kinase Inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Akhurst, R.J. Transforming growth factor-β in breast cancer: Too much, too late. Breast Cancer Res 2009, 11, 202. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L. Inhibition of TGFβ signaling in cancer therapy. Curr. Opin. Genet. Dev. 2006, 16, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Ambrosino, E.; Takaku, S.; O’Konek, J.J.; Venzon, D.; Lonning, S.; McPherson, J.M.; Berzofsky, J.A. Synergistic enhancement of CD8+ T cell-mediated tumor vaccine efficacy by an anti–transforming growth factor-β monoclonal antibody. Clin. Cancer Res. 2009, 15, 6560–6569. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.; Shapiro, G.; Tan, A.; Lawrence, D.; Olencki, T.; Dezube, B.; Hsu, F.; Reiss, M.; Berzofsky, J. Phase I/II study of GC1008: A human anti-transforming growth factor-β (TGF β) monoclonal antibody (MAb) in patients with advanced malignant melanoma (MM) or renal cell carcinoma (RCC). J. Clin. Oncol. 2008, 26, 9028. [Google Scholar]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-β (TGF-β) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.-E.; Yu, J. TGF-β signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955. [Google Scholar] [PubMed]
- Schlingensiepen, K.H.; Jaschinski, F.; Lang, S.A.; Moser, C.; Geissler, E.K.; Schlitt, H.J.; Kielmanowicz, M.; Schneider, A. Transforming growth factor-β 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011, 102, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Shinto, O.; Yashiro, M.; Yamazoe, S.; Iwauchi, T.; Muguruma, K.; Kubo, N.; Ohira, M.; Hirakawa, K. Transforming growth factor β signaling inhibitor, SB-431542, induces maturation of dendritic cells and enhances anti-tumor activity. Oncol. Rep. 2010, 24, 1637–1643. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Smith, J.A.; Schiemann, W.P. Transforming growth factor-β-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene 2010, 29, 6485–6498. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a novel transforming growth factor β receptor type i and type ii dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Carroll, K.D.; Policarpio, D.; Osborn, C.; Gregory, M.; Bassi, R.; Jimenez, X.; Prewett, M.; Liebisch, G.; Persaud, K. Anti-transforming growth factor β receptor II antibody has therapeutic efficacy against primary tumor growth and metastasis through multieffects on cancer, stroma, and immune cells. Clin. Cancer Res. 2010, 16, 1191–1205. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.M.; Hoffmann, F.M. Inhibition of transforming growth factor-β1-induced signaling and epithelial-to-mesenchymal transition by the Smad-binding peptide aptamer Trx-SARA. Mol. Biol. Cell 2006, 17, 3819–3831. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.A.; Kang, M.H.; Lee, H.J.; Kim, B.-H.; Park, J.K.; Kim, H.K.; Kim, J.S.; Oh, S.C. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. 2011, 71, 7061–7070. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhou, Y.; Xie, C.; Wei, S.; Gan, H.; He, S.; Wang, F.; Xu, L.; Lu, J.; Dai, W. Genome-wide screening reveals an EMT molecular network mediated by Sonic hedgehog-Gli1 signaling in pancreatic cancer cells. PLoS ONE 2012, 7, e43119. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Mokhtari, R.; Noman, A.; Uddin, M.; Rahman, M.; Azadi, M.; Zlotta, A.; van der Kwast, T.; Yeger, H.; Farhat, W. Sonic hedgehog (SHH) signaling promotes tumorigenicity and stemness via activation of epithelial-to-mesenchymal transition (EMT) in bladder cancer. Mol. Carcinog. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-H.; Li, Y.-J.; Kawakami, T.; Xu, S.-M.; Chuang, P.-T. Palmitoylation is required for the production of a soluble multimeric hedgehog protein complex and long-range signaling in vertebrates. Genes Dev. 2004, 18, 641–659. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hui, C.-C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat.Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.M.; Hynes, M.; Armanini, M.; Swanson, T.A.; Gu, Q.; Johnson, R.L.; Scott, M.P.; Pennica, D.; Goddard, A.; Phillips, H. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 1996, 384, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Anderson, K.V. Signaling from Smo to Ci/Gli: Conservation and divergence of hedgehog pathways from Drosophila to vertebrates. Development 2006, 133, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Murone, M.; Rosenthal, A.; de Sauvage, F.J. Sonic hedgehog signaling by the patched-smoothened receptor complex. Curr. Biol. 1999, 9, 76–84. [Google Scholar] [CrossRef]
- Roessler, E.; Ermilov, A.N.; Grange, D.K.; Wang, A.; Grachtchouk, M.; Dlugosz, A.A.; Muenke, M. A previously unidentified amino-terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum. Mol. Genet. 2005, 14, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Nishizaki, Y.; Hui, C.-C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar] [PubMed]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed]
- Regl, G.; Neill, G.W.; Eichberger, T.; Kasper, M.; Ikram, M.S.; Koller, J.; Hintner, H.; Quinn, A.G.; Frischauf, A.-M.; Aberger, F. Human Gli2 and Gli1 are part of a positive feedback mechanism in basal cell carcinoma. Oncogene 2002, 21, 5529–5539. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Venugopal, C.; Manoranjan, B.; McFarlane, N.; O’Farrell, E.; Nolte, S.; Gunnarsson, T.; Hollenberg, R.; Kwiecien, J.; Northcott, P. Sonic hedgehog regulates Bmi1 in human medulloblastoma brain tumor-initiating cells. Oncogene 2012, 31, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.N.; Borges, I.; i Altaba, A.R. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with Gli1 and p53. EMBO J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, W.; Nail, C.D.; Bailey, S.K.; Kraus, M.H.; Ruppert, J.M.; Lobo-Ruppert, S.M. Snail induction is an early response to Gli1 that determines the efficiency of epithelial transformation. Oncogene 2006, 25, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, W.; Lobo-Ruppert, S.M.; Ruppert, J.M. Gli1 acts through Snail and E-cadherin to promote nuclear signaling by β-catenin. Oncogene 2007, 26, 4489–4498. [Google Scholar] [CrossRef] [PubMed]
- Villavicencio, E.H.; Yoon, J.W.; Frank, D.J.; Füchtbauer, E.M.; Walterhouse, D.O.; Iannaccone, P.M. Cooperative E-box regulation of human GLI1 by TWIST and USF. Genesis 2002, 32, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; André, J.; Alexaki, I.; Li, A.; Magnaldo, T.; Ten Dijke, P.; Wang, X.-J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-β: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; André, J.; Verrecchia, F.; Mauviel, A. Cloning of the human GLI2 promoter transcriptional activation by transforming growth factor-β via Smad3/β-catenin cooperation. J. Biol. Chem. 2009, 284, 31523–31531. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Alexaki, V.I.; Dennler, S.; Mohammad, K.S.; Guise, T.A.; Mauviel, A. TGF-β/SMAD/GLI2 signaling axis in cancer progression and metastasis. Cancer Res. 2011, 71, 5606–5610. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, G.; Fan, Q.; Xie, J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 2010, 29, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.-H.; Sun, H.; Xue, H.; Zhang, G.; Zhang, C.-H.; Liu, X.-L.; Su, J.; Li, S.-J. Expression and clinical significance of Shh/Gli-1 in papillary thyroid carcinoma. Tumor Biol. 2014, 35, 10523–10528. [Google Scholar] [CrossRef] [PubMed]
- Yue, D.; Li, H.; Che, J.; Zhang, Y.; Tseng, H.-H.K.; Jin, J.Q.; Luh, T.M.; Giroux-Leprieur, E.; Mo, M.; Zheng, Q. Hedgehog/Gli promotes epithelial-mesenchymal transition in lung squamous cell carcinomas. J. Exp. Clin. Cancer Res. 2014, 33, 34. [Google Scholar] [CrossRef] [PubMed]
- Behnsawy, H.M.; Shigemura, K.; Meligy, F.Y.; Yamamichi, F.; Yamashita, M.; Haung, W.-C.; Li, X.; Miyake, H.; Tanaka, K.; Kawabata, M. Possible role of sonic hedgehog and epithelial-mesenchymal transition in renal cell cancer progression. Korean J. Urol. 2013, 54, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, O.; Hennen, E.; Koch, I.; Lindner, M.; Eickelberg, O. Gli1 mediates lung cancer cell proliferation and sonic hedgehog-dependent mesenchymal cell activation. PLoS ONE 2013, 8, e63226. [Google Scholar] [CrossRef] [PubMed]
- Ruiz i Altaba, A.; Sánchez, P.; Dahmane, N. Gli and hedgehog in cancer: Tumours, embryos and stem cells. Nat. Rev. Cancer 2002, 2, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Dhara, S.; Fendrich, V.; Bedja, D.; Beaty, R.; Mullendore, M.; Karikari, C.; Alvarez, H.; Iacobuzio-Donahue, C.; Jimeno, A. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: A new paradigm for combination therapy in solid cancers. Cancer Res. 2007, 67, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B.; Fehrenbacher, L.; Holloway, R.; Amit, A.; Karlan, B.; Slomovitz, B.; Sabbatini, P.; Fu, L.; Yauch, R.L.; Chang, I. A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin. Cancer Res. 2012, 18, 6509–6518. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Fu, J.; Srivastava, R.K.; Shankar, S. Hedgehog signaling antagonist GDC-0449 (vismodegib) inhibits pancreatic cancer stem cell characteristics: Molecular mechanisms. PLoS ONE 2011, 6, e27306. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergström, Å.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Polakis, P. Wnt signaling in oncogenesis and embryogenesis—A look outside the nucleus. Science 2000, 287, 1606–1609. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Hung, M.-C. Wnt, hedgehog, and SNAIL: Sister pathways that control by GSK-3β and β-Trcp in the regulation of metastasis. Cell Cycle 2005, 4, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Fodde, R.; Brabletz, T. Wnt/β-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol. 2007, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, V.; Pishvaian, M.; Byers, S. Cross-regulation of β-catenin-LEF/TCF and retinoid signaling pathways. Curr. Biol. 1999, 9, 1415–1419. [Google Scholar] [CrossRef]
- Stemmer, V.; De Craene, B.; Berx, G.; Behrens, J. Snail promotes Wnt target gene expression and interacts with β-catenin. Oncogene 2008, 27, 5075–5080. [Google Scholar] [CrossRef] [PubMed]
- Orsulic, S.; Huber, O.; Aberle, H.; Arnold, S.; Kemler, R. E-cadherin binding prevents β-catenin nuclear localization and β-catenin/LEF-1-mediated transactivation. J. Cell Sci. 1999, 112, 1237–1245. [Google Scholar] [PubMed]
- Yook, J.I.; Li, X.-Y.; Ota, I.; Hu, C.; Kim, H.S.; Kim, N.H.; Cha, S.Y.; Ryu, J.K.; Choi, Y.J.; Kim, J. A Wnt-Axin2-GSK3β cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol. 2006, 8, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Mizuarai, S.; Kawagishi, A.; Kotani, H. Inhibition of p70S6K2 down-regulates Hedgehog/GLI pathway in non-small cell lung cancer cell lines. Mol. Cancer 2009, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, K.; Kise, Y.; Miki, H. GSK3β positively regulates Hedgehog signaling through Sufu in mammalian cells. Biochem. Biophys. Res. Commun. 2007, 353, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Noubissi, F.K.; Goswami, S.; Sanek, N.A.; Kawakami, K.; Minamoto, T.; Moser, A.; Grinblat, Y.; Spiegelman, V.S. Wnt signaling stimulates transcriptional outcome of the Hedgehog pathway by stabilizing GLI1 mRNA. Cancer Res. 2009, 69, 8572–8578. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Gili, E.; Calafiore, M.; Failla, M.; La Rosa, C.; Crimi, N.; Sortino, M.A.; Nicoletti, F.; Copani, A.; Vancheri, C. TGF-β1 targets the GSK-3β/β-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol. Res. 2008, 57, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Ramirez, A.; Waddell, D.S.; Li, Z.; Liu, X.; Wang, X.-F. Axin and GSK3-β control Smad3 protein stability and modulate TGF-β signaling. Genes Dev. 2008, 22, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Yanaga, F.; Kahn, M. Targeting wnt signaling: Can we safely eradicate cancer stem cells? Clin. Cancer Res. 2010, 16, 3153–3162. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. Can we safely target the Wnt pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Gala, M.K.; Chan, A.T. Molecular pathways: Aspirin and Wnt signaling—A molecularly targeted approach to cancer prevention and treatment. Clin. Cancer Res. 2015, 21, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Revenu, C.; Gilmour, D. EMT 2.0: Shaping epithelia through collective migration. Curr. Opin. Genet. Dev. 2009, 19, 338–342. [Google Scholar] [CrossRef] [PubMed]
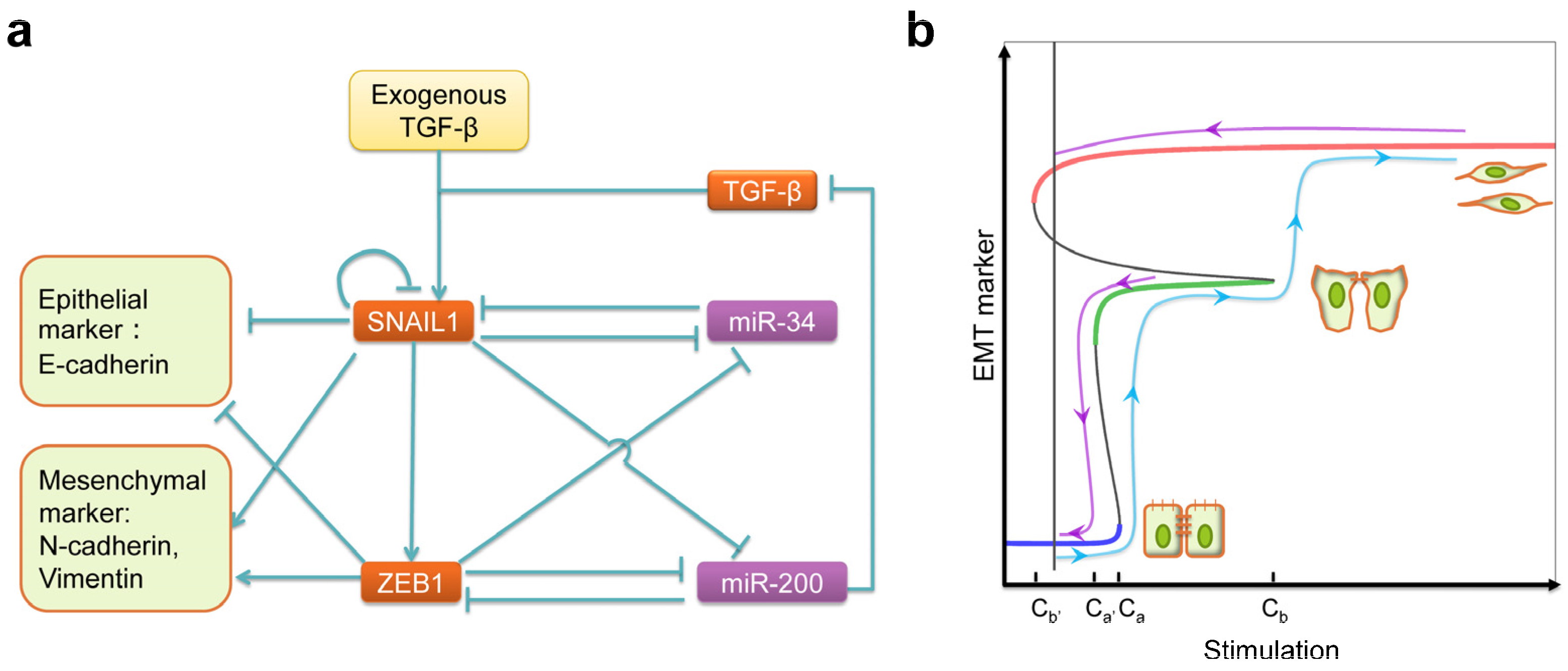
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.-J.; Zhang, H.; Xing, J. Coupled reversible and irreversible bistable switches underlying TGFβ-induced epithelial to mesenchymal transition. Biophys. J. 2013, 105, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Siemens, H.; Jackstadt, R.; Hünten, S.; Kaller, M.; Menssen, A.; Götz, U.; Hermeking, H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.-J.; Zhang, H.; Teng, Y.; Li, R.; Bai, F.; Elankumaran, S.; Xing, J. TGF-β-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci. Signal. 2014, 7, ra91. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Watanabe, K.; Ta, C.H.; Villarreal-Ponce, A.; Nie, Q.; Dai, X. An Ovol2-Zeb1 mutual inhibitory circuit governs bidirectional and multi-step transition between epithelial and mesenchymal states. PLoS Comput. Biol. 2015, 11, e1004569. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Wong, M.; Tan, T.; Kuay, K.; Ng, A.; Chung, V.; Chu, Y.; Matsumura, N.; Lai, H.; Lee, Y. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to E-cadherin restoration by a src-kinase Inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef] [PubMed]
- Steinway, S.N.; Zañudo, J.G.; Ding, W.; Rountree, C.B.; Feith, D.J.; Loughran, T.P.; Albert, R. Network modeling of TGF-β signaling in hepatocellular carcinoma epithelial-to-mesenchymal transition reveals joint sonic hedgehog and wnt pathway activation. Cancer Res. 2014, 74, 5963–5977. [Google Scholar] [CrossRef] [PubMed]
- Steinway, S.N.; Zañudo, J.G.T.; Michel, P.J.; Feith, D.J.; Loughran, T.P.; Albert, R. Combinatorial interventions inhibit TGF-β-driven epithelial-to-mesenchymal transition and support hybrid cellular phenotypes. NPJ Syst. Biol. Appl. 2015, 1, 15014. [Google Scholar] [CrossRef]
- Nam, S.; Chang, H.; Kim, K.; Kook, M.; Hong, D.; Kwon, C.; Jung, H.; Park, H.; Powis, G.; Liang, H. Pathome: An algorithm for accurately detecting differentially expressed subpathways. Oncogene 2014, 33, 4941–4951. [Google Scholar] [CrossRef] [PubMed]
- Behar, M.; Barken, D.; Werner, S.L.; Hoffmann, A. The dynamics of signaling as a pharmacological target. Cell 2013, 155, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Albert, S.Y.; Gardino, A.K.; Heijink, A.M.; Sorger, P.K.; MacBeath, G.; Yaffe, M.B. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 2012, 149, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Kreeger, P.K.; Lauffenburger, D.A. Cancer systems biology: A network modeling perspective. Carcinogenesis 2010, 31, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Bakan, A.; Kapralov, A.A.; Bayir, H.; Hu, F.; Kagan, V.E.; Bahar, I. Inhibition of peroxidase activity of cytochrome c: De novo compound discovery and validation. Mol. Pharmacol. 2015, 88, 421–427. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Tian, X.-J.; Xing, J. Signal Transduction Pathways of EMT Induced by TGF-β, SHH, and WNT and Their Crosstalks. J. Clin. Med. 2016, 5, 41. https://doi.org/10.3390/jcm5040041
Zhang J, Tian X-J, Xing J. Signal Transduction Pathways of EMT Induced by TGF-β, SHH, and WNT and Their Crosstalks. Journal of Clinical Medicine. 2016; 5(4):41. https://doi.org/10.3390/jcm5040041
Chicago/Turabian StyleZhang, Jingyu, Xiao-Jun Tian, and Jianhua Xing. 2016. "Signal Transduction Pathways of EMT Induced by TGF-β, SHH, and WNT and Their Crosstalks" Journal of Clinical Medicine 5, no. 4: 41. https://doi.org/10.3390/jcm5040041
APA StyleZhang, J., Tian, X.-J., & Xing, J. (2016). Signal Transduction Pathways of EMT Induced by TGF-β, SHH, and WNT and Their Crosstalks. Journal of Clinical Medicine, 5(4), 41. https://doi.org/10.3390/jcm5040041