
Hypoxia, Epithelial-Mesenchymal Transition, and TET-Mediated Epigenetic Changes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
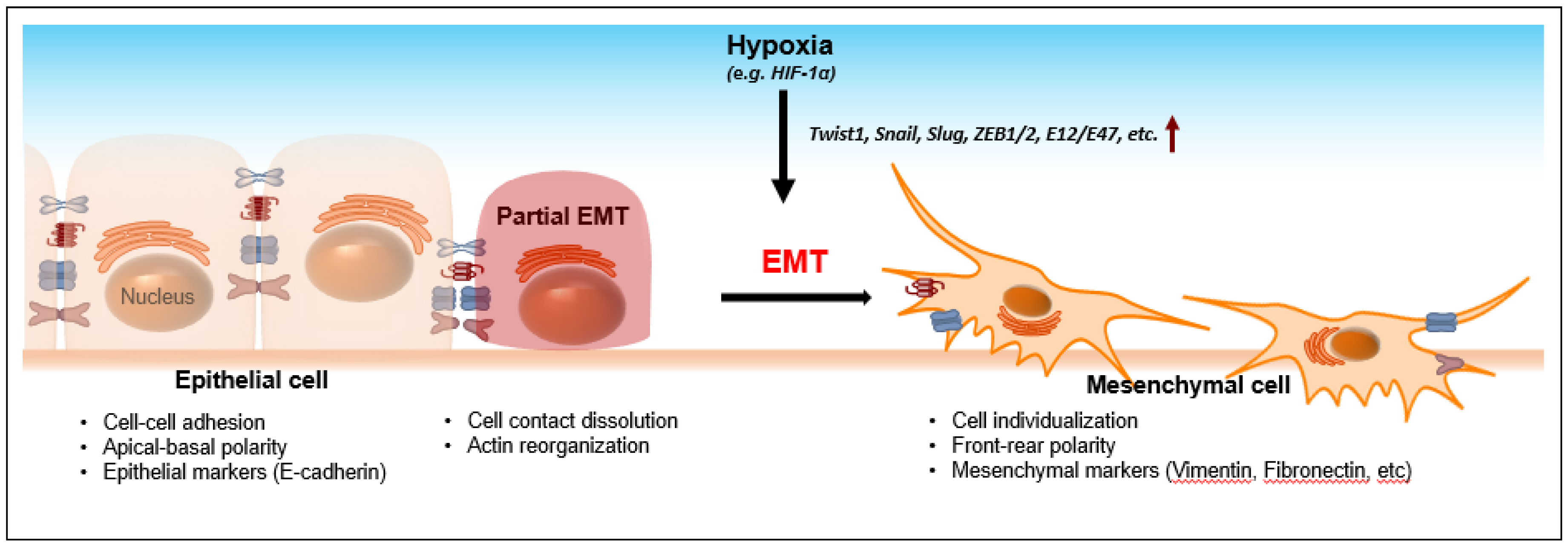
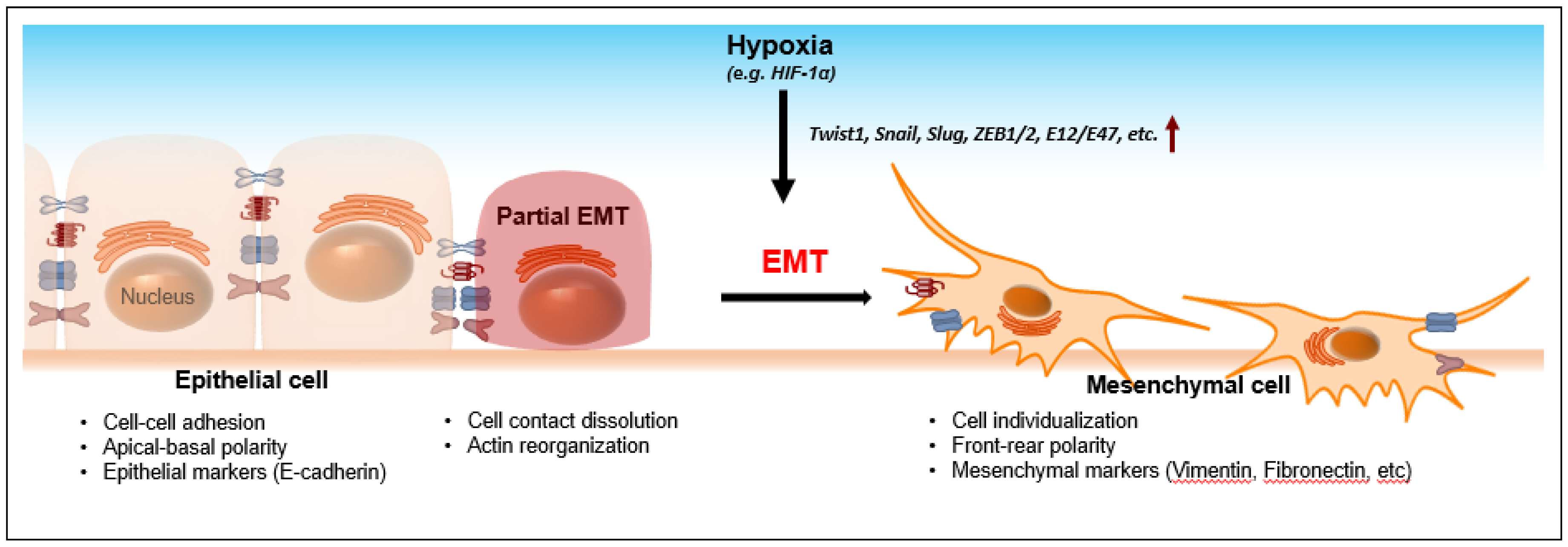
2. Hypoxia-Induced EMT
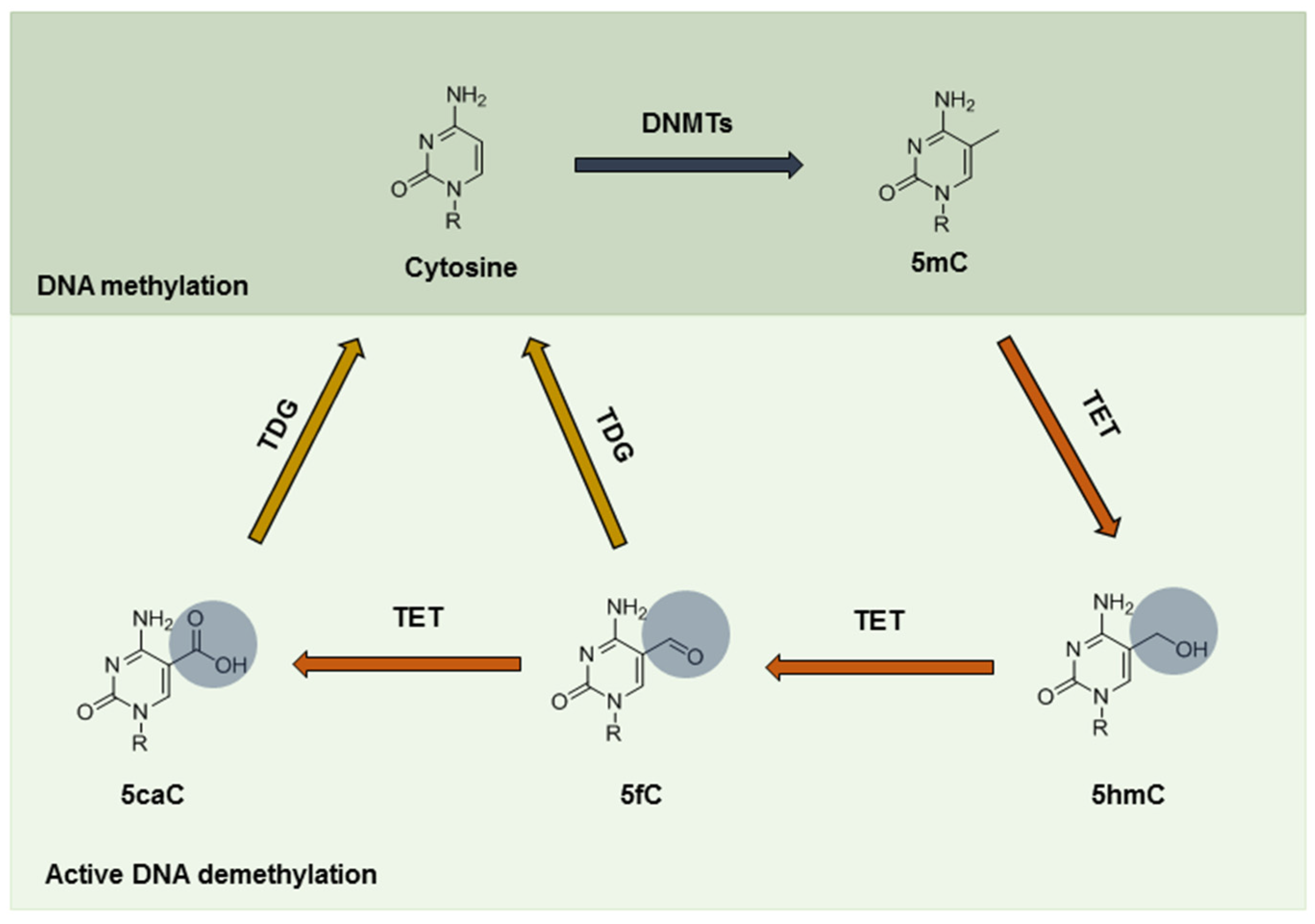
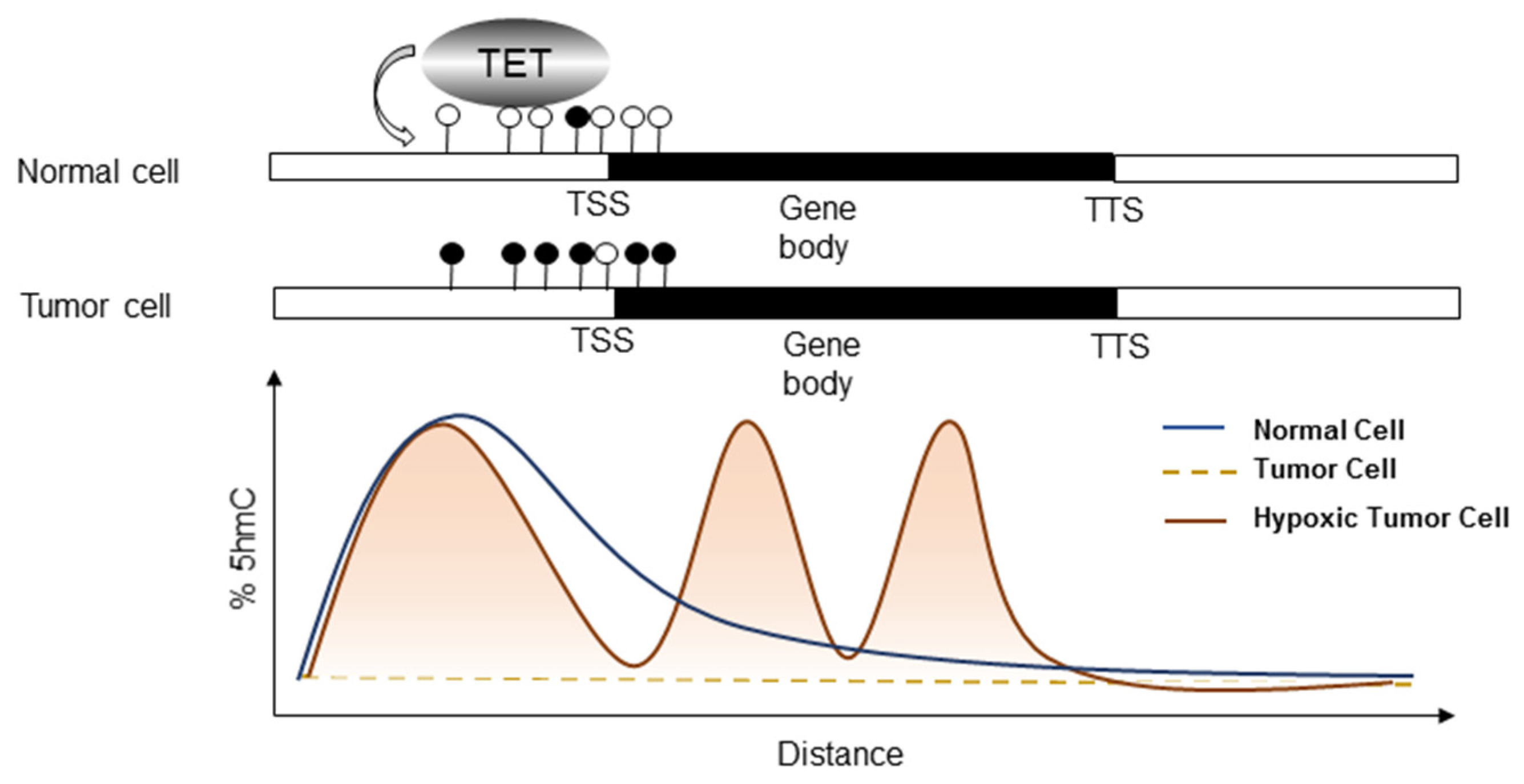
3. Epigenetic Alterations under Hypoxia
4. Detection of DNA Methylation Status under Hypoxia
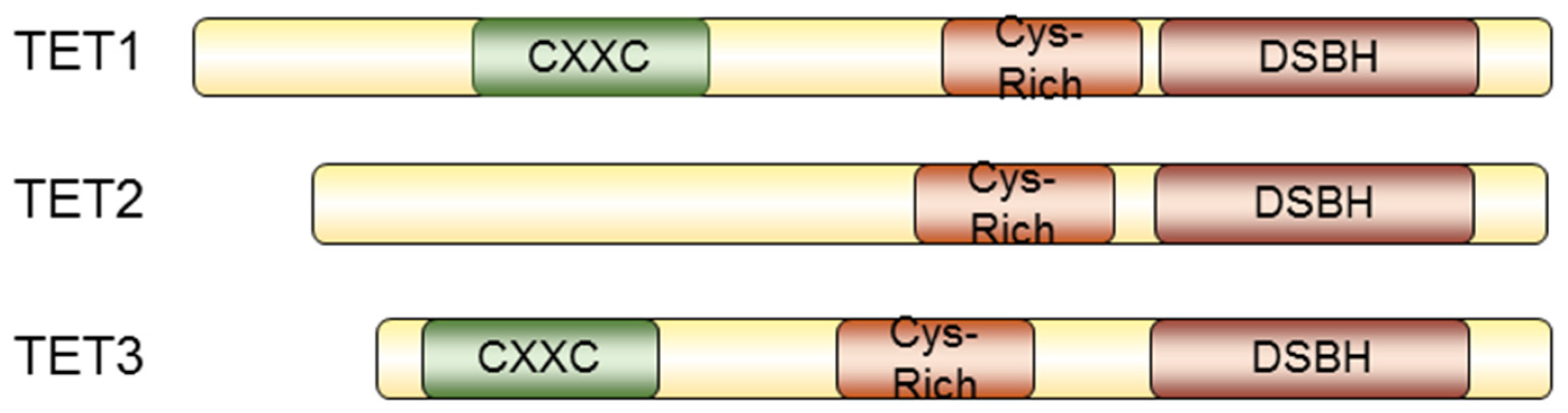
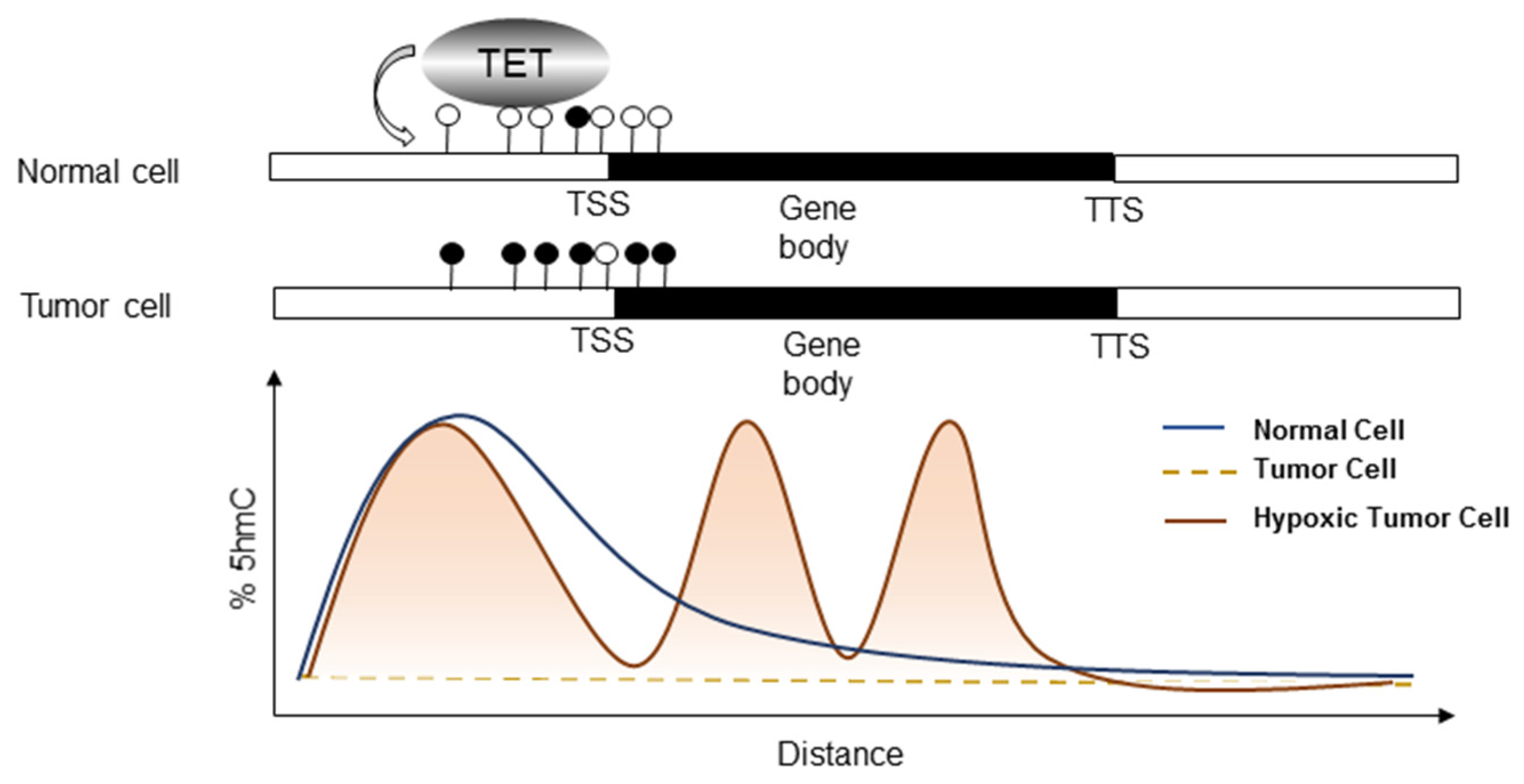
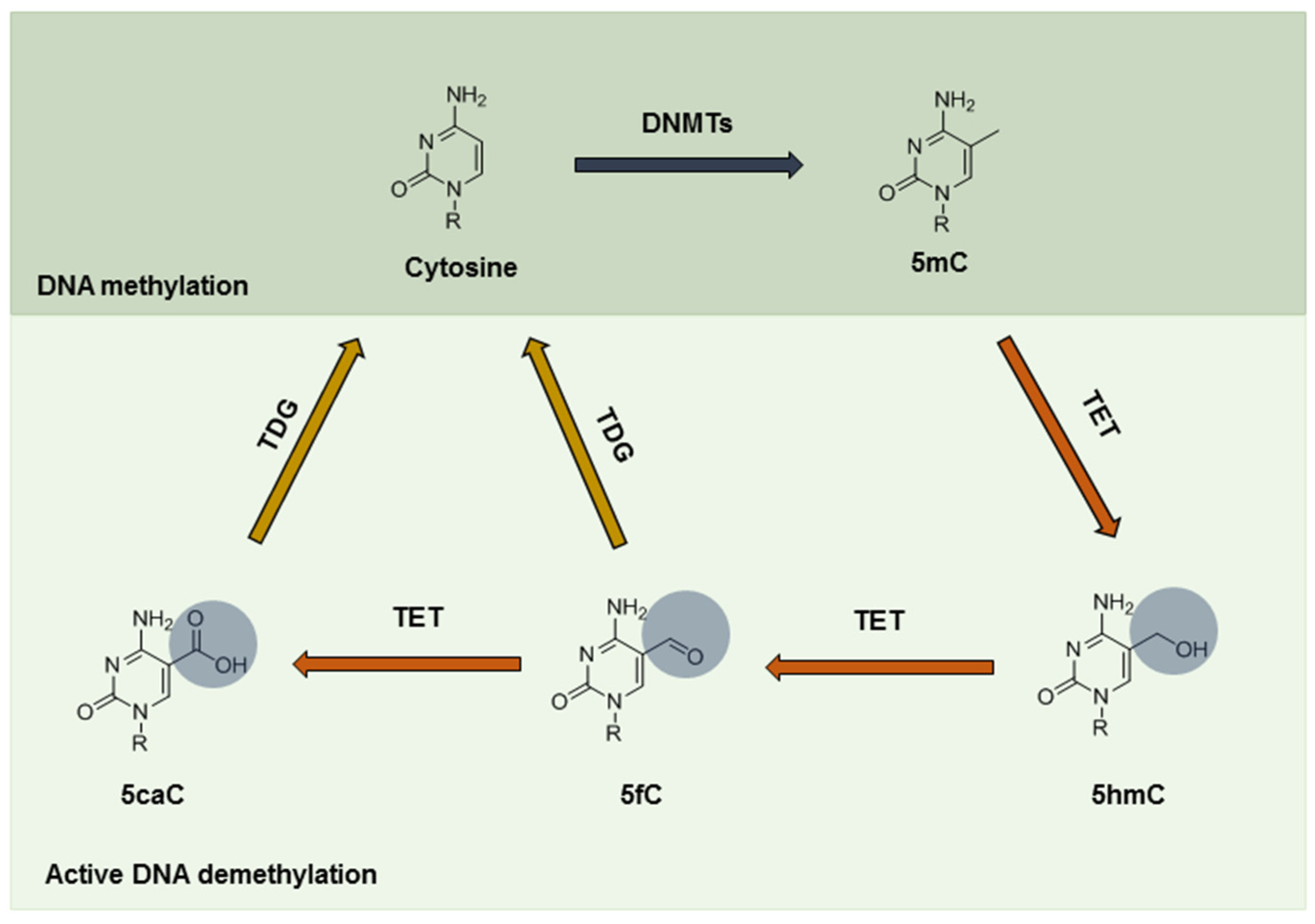
5. TET-Mediated Demethylation in Cancer

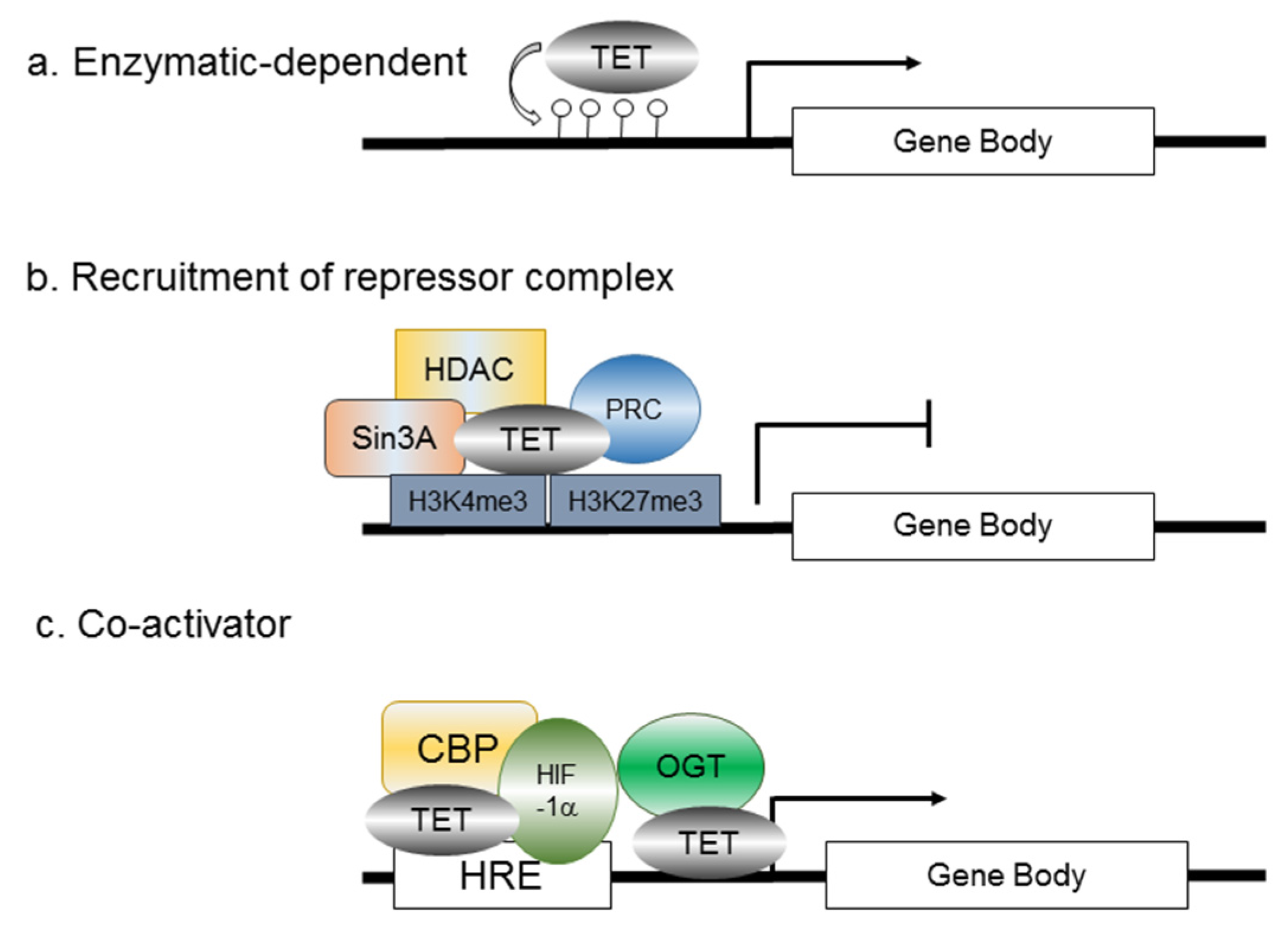
6. Bridging Hypoxia-Induced EMT to TET
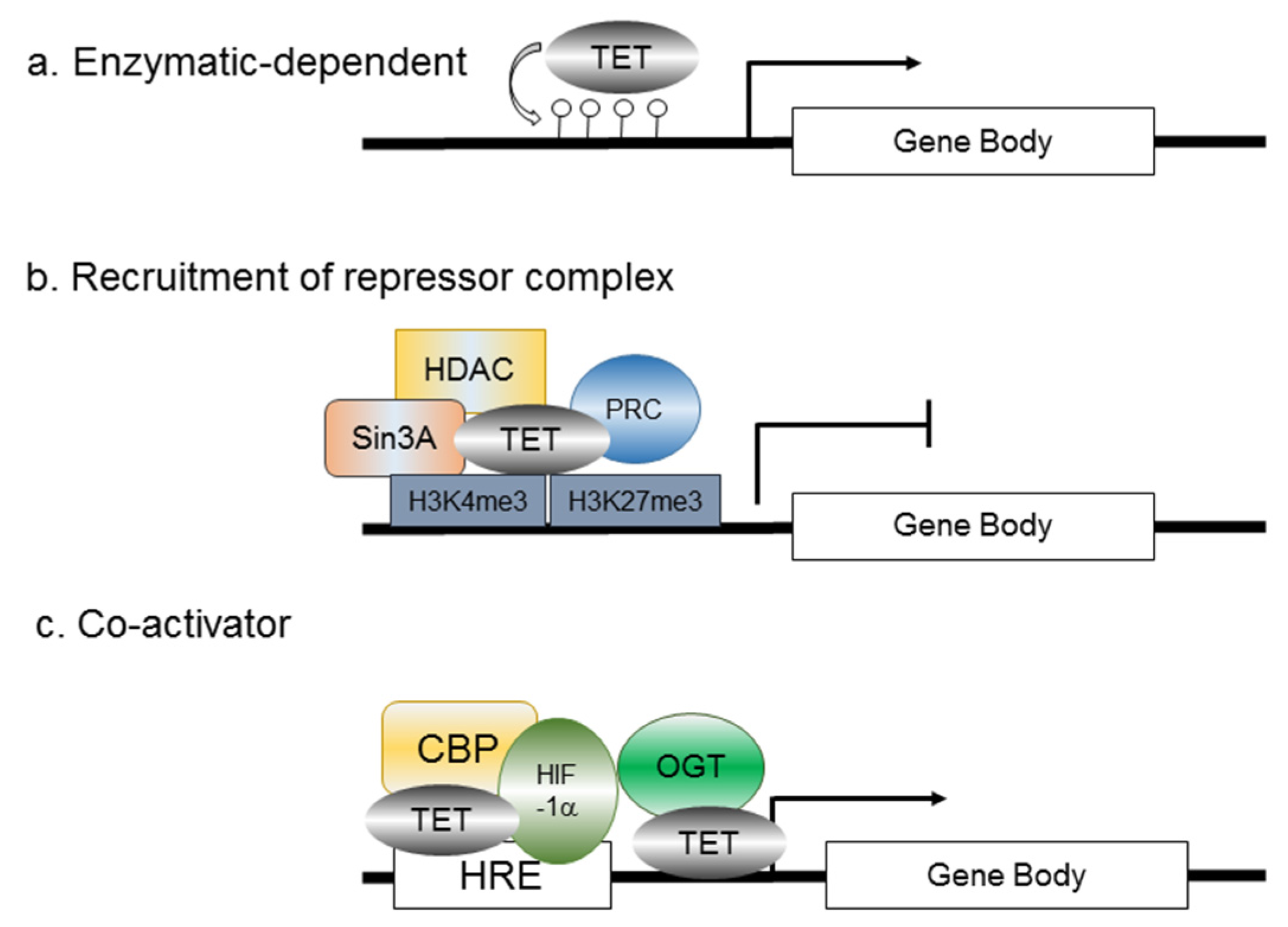
7. Hypoxia-Activated Prodrugs (HAPs) as a Potential Therapeutic Approach
8. Perspectives
Acknowledgments
Conflicts of Interest
References
- Pouyssegur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Rosmorduc, O.; Housset, C. Hypoxia: A link between fibrogenesis, angiogenesis, and carcinogenesis in liver disease. Semin. Liver Dis. 2010, 30, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, H.; Wakabayashi, M.; Hattori, N.; Yamashita, S.; Ushijima, T. Identification of coexistence of DNA methylation and h3k27me3 specifically in cancer cells as a promising target for epigenetic therapy. Carcinogenesis 2015, 36, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; Bestor, T.H. The colorful history of active DNA demethylation. Cell 2008, 133, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.C.; Zhang, Y. Active DNA demethylation: Many roads lead to rome. Nat. Rev. Mol. Cell Biol. 2010, 11, 607–620. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by tdg in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by mll partner tet1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.M.; Zhang, Y. Tet enzymes, tdg and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Pectasides, E.; Rampias, T.; Sasaki, C.; Perisanidis, C.; Kouloulias, V.; Burtness, B.; Zaramboukas, T.; Rimm, D.; Fountzilas, G.; Psyrri, A. Markers of epithelial to mesenchymal transition in association with survival in head and neck squamous cell carcinoma (hnscc). PLoS ONE 2014, 9, e94273. [Google Scholar] [CrossRef] [PubMed]
- Singhai, R.; Patil, V.W.; Jaiswal, S.R.; Patil, S.D.; Tayade, M.B.; Patil, A.V. E-cadherin as a diagnostic biomarker in breast cancer. N. Am. J. Med. Sci. 2011, 3, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.M.; Li, A.; Olino, K.; Wolfgang, C.L.; Herman, J.M.; Schulick, R.D.; Iacobuzio-Donahue, C.; Hruban, R.H.; Goggins, M. Loss of e-cadherin expression and outcome among patients with resectable pancreatic adenocarcinomas. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. 2011, 24, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.C. Coming up for air: Hif-1 and mitochondrial oxygen consumption. Cell Metab. 2006, 3, 150–151. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. Hif-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. Hif and c-myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of o2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of twist by hif-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Wu, K.J. Twist activation by hypoxia inducible factor-1 (hif-1): Implications in metastasis and development. Cell Cycle 2008, 7, 2090–2096. [Google Scholar] [CrossRef] [PubMed]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by hif-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Takahashi, K.; Okita, K.; Ichisaka, T.; Yamanaka, S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 2009, 5, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.P.; Wu, K.J. Hypoxia-regulated target genes implicated in tumor metastasis. J. Biomed. Sci. 2012, 19, 102. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, zeb and bhlh factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Gan, H.; Cai, Z.; Li, N.; Yang, Z.; Lu, G.; Chen, J. Aberrant expression of hypoxia-inducible factor 1alpha, twist and e-cadherin is associated with aggressive tumor phenotypes in endometrioid endometrial carcinoma. Jpn. J. Clin. Oncol. 2013, 43, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cheng, Q.; Zhou, Y.; Wang, Y.; Chen, X. Slug is a key mediator of hypoxia induced cadherin switch in hnscc: Correlations with poor prognosis. Oral Oncol. 2013, 49, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yan, Y.; Davidson, T.L.; Shinkai, Y.; Costa, M. Hypoxic stress induces dimethylated histone h3 lysine 9 through histone methyltransferase g9a in mammalian cells. Cancer Res. 2006, 66, 9009–9016. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Lemieux, M.E.; Li, W.; Carroll, J.S.; Brown, M.; Liu, X.S.; Kung, A.L. Integrative analysis of hif binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. USA 2009, 106, 4260–4265. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Chen, L.; Yang, J.; Ye, T.; Chen, Y.; Fang, J. Hif-1alpha-induced histone demethylase jmjd2b contributes to the malignant phenotype of colorectal cancer cells via an epigenetic mechanism. Carcinogenesis 2012, 33, 1664–1673. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Chang, R.; Zhong, J.; Pandey, A.; Semenza, G.L. Histone demethylase jmjd2c is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc. Natl. Acad. Sci. USA 2012, 109, E3367–E3376. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Z.; Tsai, Y.P.; Yang, M.H.; Huang, C.H.; Chang, S.Y.; Chang, C.C.; Teng, S.C.; Wu, K.J. Interplay between hdac3 and wdr5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol. Cell 2011, 43, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Tsai, Y.P.; Wu, M.Z.; Teng, S.C.; Wu, K.J. Epigenetic reprogramming and post-transcriptional regulation during the epithelial-mesenchymal transition. Trends Genet. TIG 2012, 28, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Tamamizu-Kato, S.; Shibasaki, F. Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J. Biol. Chem. 2004, 279, 41966–41974. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.W.; Kim, E.J.; Na, H.; Lee, M.O. Transcriptional activation of hypoxia-inducible factor-1alpha by hdac4 and hdac5 involves differential recruitment of p300 and fih-1. FEBS Lett. 2009, 583, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Kenneth, N.S.; Mudie, S.; van Uden, P.; Rocha, S. Swi/snf regulates the cellular response to hypoxia. J. Biol. Chem. 2009, 284, 4123–4131. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Shahrzad, S.; Bertrand, K.; Minhas, K.; Coomber, B.L. Induction of DNA hypomethylation by tumor hypoxia. Epigenetics 2007, 2, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Srivastava, T.; Sharma, M.K.; Mehndiratta, M.; Das, P.; Sinha, S.; Chattopadhyay, P. Aberrant methylation and associated transcriptional mobilization of alu elements contributes to genomic instability in hypoxia. J. Cell. Mol. Med. 2010, 14, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, L.; Zhao, Y.; Zhang, J.; Wang, D.; Chen, J.; He, Y.; Wu, J.; Zhang, Z.; Liu, Z. Hypoxia induces genomic DNA demethylation through the activation of hif-1alpha and transcriptional upregulation of mat2a in hepatoma cells. Mol. Cancer Ther. 2011, 10, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Collier, P.; Tea, I.; Neary, R.; Watson, J.A.; Robinson, C.; Phelan, D.; Ledwidge, M.T.; McDonald, K.M.; McCann, A.; et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum. Mol. Genet. 2014, 23, 2176–2188. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.A.; Watson, C.J.; McCrohan, A.M.; Woodfine, K.; Tosetto, M.; McDaid, J.; Gallagher, E.; Betts, D.; Baugh, J.; O’Sullivan, J.; et al. Generation of an epigenetic signature by chronic hypoxia in prostate cells. Hum. Mol. Genet. 2009, 18, 3594–3604. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wu, X.; Shen, L.; Zhang, Y. Single-base resolution analysis of active DNA demethylation using methylase-assisted bisulfite sequencing. Nat. Biotechnol. 2014, 32, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, N.; Tamura, G.; Honda, T.; Endoh, M.; Nishizuka, S.; Motoyama, T. Demethylation of the synuclein gamma gene cpg island in primary gastric cancers and gastric cancer cell lines. Clin. Cancer Res. 2004, 10, 2447–2451. [Google Scholar] [CrossRef] [PubMed]
- Koslowski, M.; Luxemburger, U.; Tureci, O.; Sahin, U. Tumor-associated cpg demethylation augments hypoxia-induced effects by positive autoregulation of hif-1alpha. Oncogene 2011, 30, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Hatzimichael, E.; Dasoula, A.; Shah, R.; Syed, N.; Papoudou-Bai, A.; Coley, H.M.; Dranitsaris, G.; Bourantas, K.L.; Stebbing, J.; Crook, T. The prolyl-hydroxylase egln3 and not egln1 is inactivated by methylation in plasma cell neoplasia. Eur. J. Haematol. 2010, 84, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Skowronski, K.; Dubey, S.; Rodenhiser, D.; Coomber, B. Ischemia dysregulates DNA methyltransferases and p16ink4a methylation in human colorectal cancer cells. Epigenetics 2010, 5, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.D.; Grummitt, C.G.; Hilcenko, C.; Min, S.Y.; Tonkin, L.M.; Johnson, C.M.; Freund, S.M.; Bycroft, M.; Warren, A.J. Solution structure of the nonmethyl-cpg-binding cxxc domain of the leukaemia-associated mll histone methyltransferase. EMBO J. 2006, 25, 4503–4512. [Google Scholar] [CrossRef] [PubMed]
- Hajkova, P.; Jeffries, S.J.; Lee, C.; Miller, N.; Jackson, S.P.; Surani, M.A. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science 2010, 329, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef] [PubMed]
- Wossidlo, M.; Nakamura, T.; Lepikhov, K.; Marques, C.J.; Zakhartchenko, V.; Boiani, M.; Arand, J.; Nakano, T.; Reik, W.; Walter, J. 5-hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun. 2011, 2, 241. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of tet proteins in 5mc to 5hmc conversion, es-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.P.; Yabuuchi, A.; Rao, S.; Huang, Y.; Cunniff, K.; Nardone, J.; Laiho, A.; Tahiliani, M.; Sommer, C.A.; Mostoslavsky, G.; et al. Tet1 and tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 2011, 8, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; D’Alessio, A.C.; Ito, S.; Xia, K.; Wang, Z.; Cui, K.; Zhao, K.; Sun, Y.E.; Zhang, Y. Dual functions of tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 2011, 473, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Christensen, J.; Pedersen, M.T.; Johansen, J.V.; Cloos, P.A.; Rappsilber, J.; Helin, K. Tet1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 2011, 473, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Ganz, K.; Powell, B.E.; Hu, Y.C.; Markoulaki, S.; Cheng, A.W.; Gao, Q.; Kim, J.; Choi, S.W.; Page, D.C.; et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 2011, 9, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Ficz, G.; Branco, M.R.; Seisenberger, S.; Santos, F.; Krueger, F.; Hore, T.A.; Marques, C.J.; Andrews, S.; Reik, W. Dynamic regulation of 5-hydroxymethylcytosine in mouse es cells and during differentiation. Nature 2011, 473, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Breiling, A.; Le, T.; Raddatz, G.; Barrasa, M.I.; Cheng, A.W.; Gao, Q.; Powell, B.E.; Li, Z.; Xu, M.; et al. Combined deficiency of tet1 and tet2 causes epigenetic abnormalities but is compatible with postnatal development. Developmental cell 2013, 24, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Costa, Y.; Ding, J.; Theunissen, T.W.; Faiola, F.; Hore, T.A.; Shliaha, P.V.; Fidalgo, M.; Saunders, A.; Lawrence, M.; Dietmann, S.; et al. Nanog-dependent function of tet1 and tet2 in establishment of pluripotency. Nature 2013, 495, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Bartolomei, M.S.; Ferguson-Smith, A.C. Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Shen, L.; Liu, Y.; Sendler, D.; Zhang, Y. Role of tet1 in erasure of genomic imprinting. Nature 2013, 504, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Hong, K.; Liu, R.; Shen, L.; Inoue, A.; Diep, D.; Zhang, K.; Zhang, Y. Tet1 controls meiosis by regulating meiotic gene expression. Nature 2012, 492, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.P.; Guo, F.; Yang, H.; Wu, H.P.; Xu, G.F.; Liu, W.; Xie, Z.G.; Shi, L.; He, X.; Jin, S.G.; et al. The role of tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 2011, 477, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Song, C.X.; Szulwach, K.E.; Fu, Y.; Dai, Q.; Yi, C.; Li, X.; Li, Y.; Chen, C.H.; Zhang, W.; Jian, X.; et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2011, 29, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Chaux, A.; Meeker, A.K.; Esopi, D.M.; Gerber, J.; Pellakuru, L.G.; Toubaji, A.; Argani, P.; Iacobuzio-Donahue, C.; Nelson, W.G.; et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2011, 2, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Nestor, C.E.; Ottaviano, R.; Reddington, J.; Sproul, D.; Reinhardt, D.; Dunican, D.; Katz, E.; Dixon, J.M.; Harrison, D.J.; Meehan, R.R. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012, 22, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.Y.; Ma, S.H.; Liu, J.; Xu, Z.D.; Zhu, H.G.; Ling, Z.Q.; Ye, D.; et al. Tumor development is associated with decrease of tet gene expression and 5-methylcytosine hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Dettori, D.; Incarnato, D.; Krepelova, A.; Rapelli, S.; Maldotti, M.; Parlato, C.; Paliogiannis, P.; Oliviero, S. Tet1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the wnt pathway. Oncogene 2015, 34, 4168–4176. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Peng, K.L.; Kang, M.L.; Chen, Y.R.; Yang, Y.C.; Tsai, C.H.; Chu, C.S.; Jeng, Y.M.; Chen, Y.T.; Lin, F.M.; et al. Tet1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep. 2012, 2, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yu, S.J.; Hong, Q.; Yang, Y.; Shao, Z.M. Reduced expression of tet1, tet2, tet3 and tdg mrnas are associated with poor prognosis of patients with early breast cancer. PLoS ONE 2015, 10, e0133896. [Google Scholar] [CrossRef] [PubMed]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in tet2 in myeloid cancers. N. Engl. J. med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Langemeijer, S.M.; Kuiper, R.P.; Berends, M.; Knops, R.; Aslanyan, M.G.; Massop, M.; Stevens-Linders, E.; van Hoogen, P.; van Kessel, A.G.; Raymakers, R.A.; et al. Acquired mutations in tet2 are common in myelodysplastic syndromes. Nat. Genet. 2009, 41, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of tet1, tet2, and tet3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Levine, R.L.; Lim, K.H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Finke, C.M.; Mullally, A.; Li, C.Y.; Pardanani, A.; et al. Frequent tet2 mutations in systemic mastocytosis: Clinical, kitd816v and fip1l1-pdgfra correlates. Leukemia 2009, 23, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Pronier, E.; Almire, C.; Mokrani, H.; Vasanthakumar, A.; Simon, A.; da Costa Reis Monte Mor, B.; Masse, A.; Le Couedic, J.P.; Pendino, F.; Carbonne, B.; et al. Inhibition of tet2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood 2011, 118, 2551–2555. [Google Scholar] [CrossRef] [PubMed]
- Asmar, F.; Punj, V.; Christensen, J.; Pedersen, M.T.; Pedersen, A.; Nielsen, A.B.; Hother, C.; Ralfkiaer, U.; Brown, P.; Ralfkiaer, E.; et al. Genome-wide profiling identifies a DNA methylation signature that associates with tet2 mutations in diffuse large b-cell lymphoma. Haematologica 2013, 98, 1912–1920. [Google Scholar] [CrossRef] [PubMed]
- Muto, H.; Sakata-Yanagimoto, M.; Nagae, G.; Shiozawa, Y.; Miyake, Y.; Yoshida, K.; Enami, T.; Kamada, Y.; Kato, T.; Uchida, K.; et al. Reduced tet2 function leads to t-cell lymphoma with follicular helper t-cell-like features in mice. Blood Cancer J. 2014, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, M.; Chen, X.; Chen, L.; Xu, Y.; Lv, L.; Wang, P.; Yang, H.; Ma, S.; Lin, H.; et al. Wt1 recruits tet2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol. Cell 2015, 57, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant tet2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Dawlaty, M.M.; Ndiaye-Lobry, D.; Yap, Y.S.; Bakogianni, S.; Yu, Y.; Bhattacharyya, S.; Shaknovich, R.; Geng, H.; Lobry, C.; et al. Tet1 is a tumor suppressor of hematopoietic malignancy. Nat. Immunol. 2015, 16, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Mariani, C.J.; Vasanthakumar, A.; Madzo, J.; Yesilkanal, A.; Bhagat, T.; Yu, Y.; Bhattacharyya, S.; Wenger, R.H.; Cohn, S.L.; Nanduri, J.; et al. Tet1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep. 2014, 7, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chen, Y.; Bian, C.; Fujiki, R.; Yu, X. Tet2 promotes histone o-glcnacylation during gene transcription. Nature 2013, 493, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Deplus, R.; Delatte, B.; Schwinn, M.K.; Defrance, M.; Mendez, J.; Murphy, N.; Dawson, M.A.; Volkmar, M.; Putmans, P.; Calonne, E.; et al. Tet2 and tet3 regulate glcnacylation and h3k4 methylation through ogt and set1/compass. EMBO J. 2013, 32, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.P.; Chen, H.F.; Chen, S.Y.; Cheng, W.C.; Wang, H.W.; Shen, Z.J.; Song, C.; Teng, S.C.; He, C.; Wu, K.J. Tet1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 2014, 15, 513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting hdac2 to specifically repress il-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Z.; Chen, S.F.; Nieh, S.; Benner, C.; Ger, L.P.; Jan, C.I.; Ma, L.; Chen, C.H.; Hishida, T.; Chang, H.T.; et al. Hypoxia drives breast tumor malignancy through a tet-tnfalpha-p38-mapk signaling axis. Cancer Res. 2015, 75, 3912–3924. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.H.; Weng, W.H.; Li, Z. The effect of tet1 on the proliferation of renal cancer 786-o cells and its related mechanism. Tumor 2012, 32, 955–961. [Google Scholar]
- Fujiki, R.; Hashiba, W.; Sekine, H.; Yokoyama, A.; Chikanishi, T.; Ito, S.; Imai, Y.; Kim, J.; He, H.H.; Igarashi, K.; et al. Glcnacylation of histone h2b facilitates its monoubiquitination. Nature 2011, 480, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase iii study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Gattermann, N.; Germing, U.; Sanz, G.; List, A.F.; Gore, S.; Seymour, J.F.; et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, B.; Aquino-Parsons, C.; Raleigh, J.A.; Stanbridge, E.J.; Durand, R.E.; Banath, J.P.; MacPhail, S.H.; Olive, P.L. Comparison between pimonidazole binding, oxygen electrode measurements, and expression of endogenous hypoxia markers in cancer of the uterine cervix. Cytom. B Clin. Cytom. 2006, 70, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Masunaga, S.; Ono, K.; Abe, M. The detection and modification of the hypoxic fraction in quiescent cell populations in murine solid tumours. Br. J. Radiol. 1993, 66, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Koomagi, R.; Mattern, J.; Volm, M. Glucose-related protein (grp78) and its relationship to the drug-resistance proteins p170, gst-pi, lrp56 and angiogenesis in non-small cell lung carcinomas. Anticancer Res. 1999, 19, 4333–4336. [Google Scholar] [PubMed]
- Carlson, D.J.; Stewart, R.D.; Semenenko, V.A. Effects of oxygen on intrinsic radiation sensitivity: A test of the relationship between aerobic and hypoxic linear-quadratic (lq) model parameters. Med. Phys. 2006, 33, 3105–3115. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.J.; Kim, W.Y. Targeting tumor hypoxia with hypoxia-activated prodrugs. J. Clin. Oncol. 2015, 33, 1505–1508. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kao, S.-H.; Wu, K.-J.; Lee, W.-H. Hypoxia, Epithelial-Mesenchymal Transition, and TET-Mediated Epigenetic Changes. J. Clin. Med. 2016, 5, 24. https://doi.org/10.3390/jcm5020024
Kao S-H, Wu K-J, Lee W-H. Hypoxia, Epithelial-Mesenchymal Transition, and TET-Mediated Epigenetic Changes. Journal of Clinical Medicine. 2016; 5(2):24. https://doi.org/10.3390/jcm5020024
Chicago/Turabian StyleKao, Shih-Han, Kou-Juey Wu, and Wen-Hwa Lee. 2016. "Hypoxia, Epithelial-Mesenchymal Transition, and TET-Mediated Epigenetic Changes" Journal of Clinical Medicine 5, no. 2: 24. https://doi.org/10.3390/jcm5020024
APA StyleKao, S.-H., Wu, K.-J., & Lee, W.-H. (2016). Hypoxia, Epithelial-Mesenchymal Transition, and TET-Mediated Epigenetic Changes. Journal of Clinical Medicine, 5(2), 24. https://doi.org/10.3390/jcm5020024