
Switching Roles of TGF-β in Cancer Development: Implications for Therapeutic Target and Biomarker Studies


{kind=link}
Abstract
:1. Introduction
2. The Context-Dependent Functions of TGF-β in Normal Tissues
3. The Context-Dependent Functions of TGF-β in Cancer Development
4. Challenges in Current Cancer Therapy Targeting TGF-β and Implications for Biomarker Studies
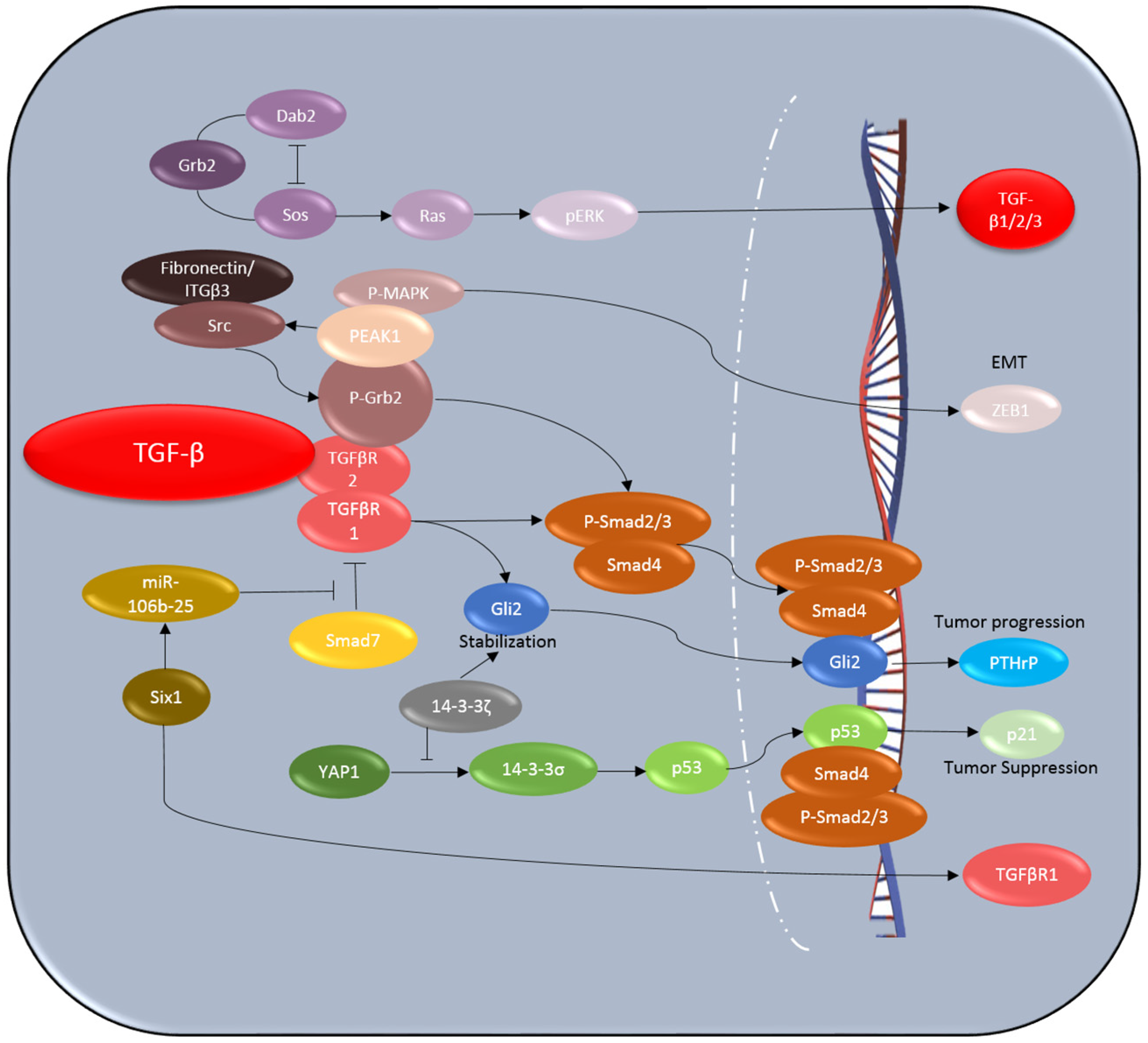
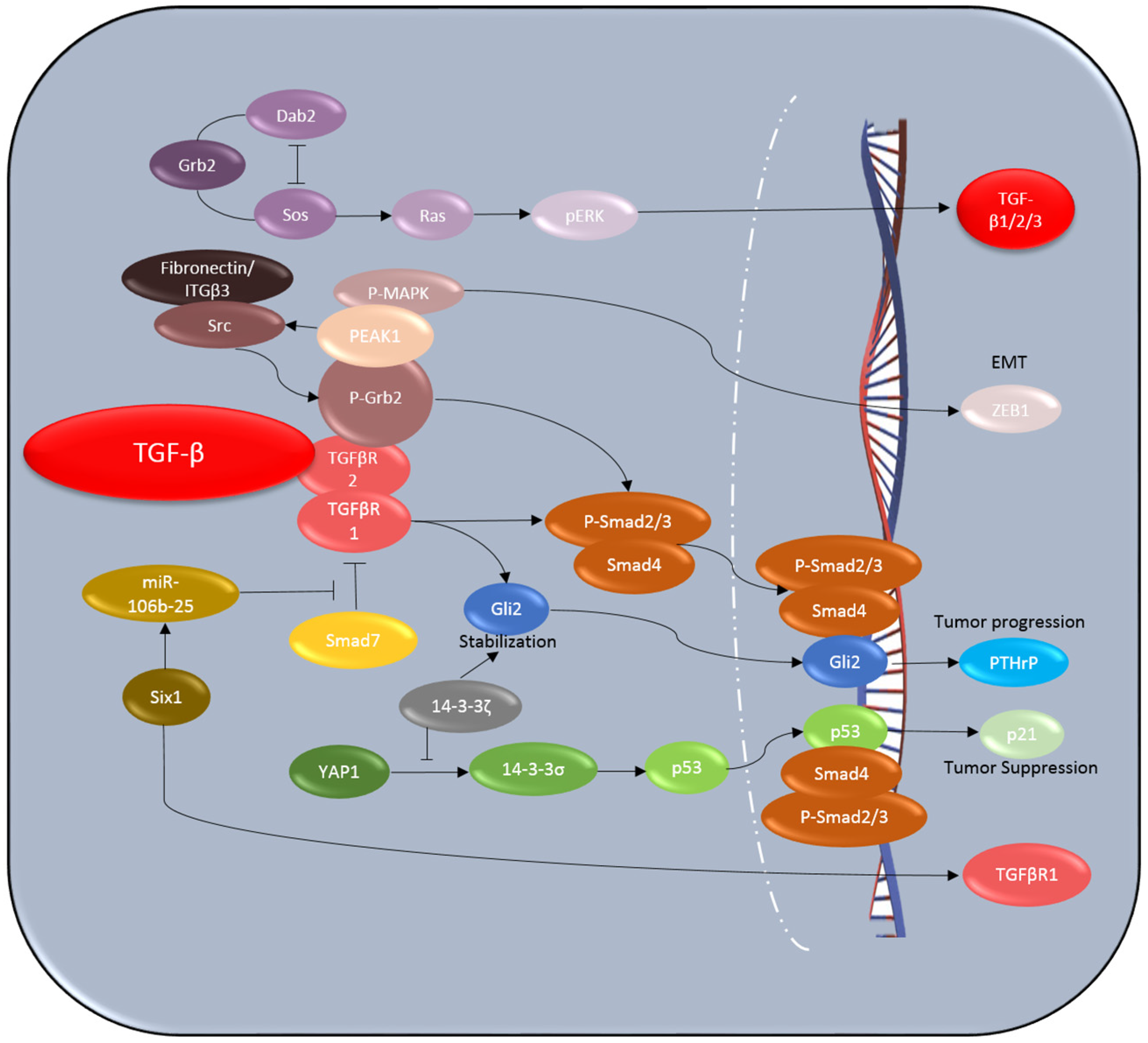
4.1. Six1
4.2. Dab2
4.3. 14-3-3ζ
4.4. PEAK1
4.5. p53
4.6. Gli2
4.7. Circulating TGF-β and TGF-β-Associated Markers
5. Conclusions and Perspectives
Conflicts of Interest
References
- Massague, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massagué, J.; Mundy, G.R.; Guise, T.A. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Massague, J. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Pouponnot, C.; Staller, P.; Schader, M.; Eilers, M.; Massague, J. TGFβ influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat. Cell Biol. 2001, 3, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Le, H.V.; Shen, L.; Anderson, S.A.; Massague, J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef]
- Pardali, K.; Kurisaki, A.; Moren, A.; ten Dijke, P.; Kardassis, D.; Moustakas, A. Role of Smad proteins and transcription factor Sp1 in p21(Waf1/Cip1) regulation by transforming growth factor-β. J. Biol. Chem. 2000, 275, 29244–29256. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Kardassis, D. Regulation of the human p21/WAF1/Cip1 promoter in hepatic cells by functional interactions between Sp1 and Smad family members. Proc. Natl. Acad. Sci. USA 1998, 95, 6733–6738. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.R.; Kang, Y.; Siegel, P.M.; Massague, J. E2F4/5 and p107 as Smad cofactors linking the TGFβ receptor to c-myc repression. Cell 2002, 110, 19–32. [Google Scholar] [CrossRef]
- Claassen, G.F.; Hann, S.R. A role for transcriptional repression of p21CIP1 by c-Myc in overcoming transforming growth factor β-induced cell-cycle arrest. Proc. Natl. Acad. Sci. USA 2000, 97, 9498–9503. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.R.; Kang, Y.; Massague, J. Defective repression of c-myc in breast cancer cells: A loss at the core of the transforming growth factor β growth arrest program. Proc. Natl. Acad. Sci. USA 2001, 98, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Iwanaga, R.; Drasin, D.J.; Micalizzi, D.S.; Vartuli, R.L.; Tan, A.C.; Ford, H.L. The miR-106b-25 cluster targets Smad7, activates TGF-β signaling, and induces EMT and tumor initiating cell characteristics downstream of Six1 in human breast cancer. Oncogene 2012, 31, 5162–5171. [Google Scholar] [CrossRef] [PubMed]
- Chu, I.M.; Lai, W.C.; Aprelikova, O.; El Touny, L.H.; Kouros-Mehr, H.; Green, J.E. Expression of GATA3 in MDA-MB-231 triple-negative breast cancer cells induces a growth inhibitory response to TGFss. PLoS ONE 2013, 8, e61125. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.H.; Derynck, R. Specificity and versatility in tgf-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S. p53 regulation orchestrates the TGF-β response. Cell 2008, 133, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Cordenonsi, M.; Dupont, S.; Maretto, S.; Insinga, A.; Imbriano, C.; Piccolo, S. Links between tumor suppressors: p53 is required for TGF-β gene responses by cooperating with Smads. Cell 2003, 113, 301–314. [Google Scholar] [CrossRef]
- Cordenonsi, M.; Montagner, M.; Adorno, M.; Zacchigna, L.; Martello, G.; Mamidi, A.; Soligo, S.; Dupont, S.; Piccolo, S. Integration of TGF-β and Ras/MAPK signaling through p53 phosphorylation. Science 2007, 315, 840–843. [Google Scholar] [CrossRef] [PubMed]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.F. Transforming growth factor β induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.C.; Orlando, D.A.; Newman, J.J.; Loven, J.; Kumar, R.M.; Bilodeau, S.; Reddy, J.; Guenther, M.G.; DeKoter, R.P.; Young, R.A. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 2011, 147, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Trompouki, E.; Bowman, T.V.; Lawton, L.N.; Fan, Z.P.; Wu, D.C.; DiBiase, A.; Martin, C.S.; Cech, J.N.; Sessa, A.K.; Leblanc, J.L.; et al. Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell 2011, 147, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.T.; Charney Le, R.; Blitz, I.L.; Fish, M.B.; Li, Y.; Biesinger, J.; Xie, X.; Cho, K.W. Genome-wide view of TGFβ/Foxh1 regulation of the early mesendoderm program. Development 2014, 141, 4537–4547. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Pouponnot, C.; Massague, J. Dual role of the Smad4/DPC4 tumor suppressor in TGFβ-inducible transcriptional complexes. Genes Dev. 1997, 11, 3157–3167. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V.B.; Wong, E.; Orlov, Y.L.; Zhang, W.; Jiang, J.; et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Ying, Q.L.; Nichols, J.; Chambers, I.; Smith, A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 2003, 115, 281–292. [Google Scholar] [CrossRef]
- Gomis, R.R.; Alarcon, C.; Nadal, C.; Van Poznak, C.; Massague, J. C/EBPβ at the core of the TGFβ cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell 2006, 10, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Schier, A.F. Nodal morphogens. Cold Spring Harb. Perspect. Biol. 2009, 1, a003459. [Google Scholar] [CrossRef] [PubMed]
- Gomis, R.R.; Alarcon, C.; He, W.; Wang, Q.; Seoane, J.; Lash, A.; Massague, J. A FoxO-Smad synexpression group in human keratinocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 12747–12752. [Google Scholar] [CrossRef] [PubMed]
- Koinuma, D.; Tsutsumi, S.; Kamimura, N.; Taniguchi, H.; Miyazawa, K.; Sunamura, M.; Imamura, T.; Miyazono, K.; Aburatani, H. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor β signaling. Mol. Cell. Biol. 2009, 29, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Singha, P.K.; Yeh, I.T.; Venkatachalam, M.A.; Saikumar, P. Transforming growth factor-β (TGF-β)-inducible gene TMEPAI converts TGF-β from a tumor suppressor to a tumor promoter in breast cancer. Cancer Res. 2010, 70, 6377–6383. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Qu, S.; Liu, B.; Pan, S.; Jiao, Y.; Nie, Y.; Su, F.; Liu, Q.; Song, E. MiR-106b expression determines the proliferation paradox of TGF-β in breast cancer cells. Oncogene 2015, 34, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Johansson, J.; Berg, T.; Kurzejamska, E.; Pang, M.F.; Tabor, V.; Jansson, M.; Roswall, P.; Pietras, K.; Sund, M.; Religa, P.; et al. MiR-155-mediated loss of C/EBPβ shifts the TGF-β response from growth inhibition to epithelial-mesenchymal transition, invasion and metastasis in breast cancer. Oncogene 2013, 32, 5614–5624. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Elston, R.; Inman, G.J. Crosstalk between p53 and TGF-β Signalling. J. Signal Transduct. 2012, 2012, 294097. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Zacchigna, L.; Adorno, M.; Soligo, S.; Volpin, D.; Piccolo, S.; Cordenonsi, M. Convergence of p53 and TGF-β signaling networks. Cancer Lett. 2004, 213, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Termen, S.; Tan, E.J.; Heldin, C.H.; Moustakas, A. p53 regulates epithelial-mesenchymal transition induced by transforming growth factor β. J. Cell. Physiol. 2013, 228, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Chao, C.H.; Xia, W.; Yang, J.Y.; Xiong, Y.; Li, C.W.; Yu, W.H.; Rehman, S.K.; Hsu, J.L.; Lee, H.H.; et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. 2011, 13, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.Y.; et al. An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Ohainmhire, E.; Quartuccio, S.M.; Cheng, W.; Ahmed, R.A.; King, S.M.; Burdette, J.E. Mutation or loss of p53 differentially modifies TGFβ action in ovarian cancer. PLoS ONE 2014, 9, e89553. [Google Scholar]
- Kalo, E.; Buganim, Y.; Shapira, K.E.; Besserglick, H.; Goldfinger, N.; Weisz, L.; Stambolsky, P.; Henis, Y.I.; Rotter, V. Mutant p53 attenuates the SMAD-dependent transforming growth factor β1 (TGF-β1) signaling pathway by repressing the expression of TGF-β receptor type II. Mol. Cell. Biol. 2007, 27, 8228–8242. [Google Scholar] [CrossRef] [PubMed]
- Her, N.G.; Jeong, S.I.; Cho, K.; Ha, T.K.; Han, J.; Ko, K.P.; Park, S.K.; Lee, J.H.; Lee, M.G.; Ryu, B.K.; et al. PPARdelta promotes oncogenic redirection of TGF-β1 signaling through the activation of the ABCA1-Cav1 pathway. Cell Cycle 2013, 12, 1521–1535. [Google Scholar] [CrossRef] [PubMed]
- Leight, J.L.; Wozniak, M.A.; Chen, S.; Lynch, M.L.; Chen, C.S. Matrix rigidity regulates a switch between TGF-β1-induced apoptosis and epithelial-mesenchymal transition. Mol. Biol. Cell 2012, 23, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGFβ: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Wrzesinski, S.H.; Wan, Y.Y.; Flavell, R.A. Transforming growth factor-β and the immune response: Implications for anticancer therapy. Clin. Cancer Res. 2007, 13, 5262–5270. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulos, P.; Zizi-Sermpetzoglou, A.; Rizos, S.; Kostakis, A.; Nikiteas, N.; Papavassiliou, A.G. TGF-β signalling in colon carcinogenesis. Cancer Lett. 2012, 314, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of TGF-β targeted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Padua, D.; Massague, J. Roles of TGFβ in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L. Inhibition of TGFβ signaling in cancer therapy. Curr. Opin. Genet. Dev. 2006, 16, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.S.; Ilaria, R., Jr.; de Alwis, D.P.; Mendelson, D.S.; McKane, S.; Wagner, M.M.; Look, K.Y.; LoRusso, P.M. A phase I study of tasisulam sodium (LY573636 sodium), a novel anticancer compound, administered as a 24-h continuous infusion in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Sheen, Y.Y.; Kim, M.J.; Park, S.A.; Park, S.Y.; Nam, J.S. Targeting the Transforming Growth Factor-β Signaling in Cancer Therapy. Biomol. Ther. (Seoul) 2013, 21, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Forrester, E.; Chytil, A.; Bierie, B.; Aakre, M.; Gorska, A.E.; Sharif-Afshar, A.R.; Muller, W.J.; Moses, H.L. Effect of conditional knockout of the type II TGF-β receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res. 2005, 65, 2296–2302. [Google Scholar] [CrossRef] [PubMed]
- Novitskiy, S.V.; Forrester, E.; Pickup, M.W.; Gorska, A.E.; Chytil, A.; Aakre, M.; Polosukhina, D.; Owens, P.; Yusupova, D.R.; Zhao, Z.; et al. Attenuated transforming growth factor β signaling promotes metastasis in a model of HER2 mammary carcinogenesis. Breast Cancer Res. 2014, 16, 425. [Google Scholar] [CrossRef] [PubMed]
- Muraoka-Cook, R.S.; Kurokawa, H.; Koh, Y.; Forbes, J.T.; Roebuck, L.R.; Barcellos-Hoff, M.H.; Moody, S.E.; Chodosh, L.A.; Arteaga, C.L. Conditional overexpression of active transforming growth factor β1 in vivo accelerates metastases of transgenic mammary tumors. Cancer Res. 2004, 64, 9002–9011. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, S.; White, R.; Malkoski, S.; Oka, M.; Han, G.; Cleaver, T.; Reh, D.; Andersen, P.; Gross, N.; Olson, S.; et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J. Clin. Investig. 2009, 119, 3408–3419. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.L.; Herrington, H.; Reh, D.; Weber, S.; Bornstein, S.; Wang, D.; Li, A.G.; Tang, C.F.; Siddiqui, Y.; Nord, J.; et al. Loss of transforming growth factor-β type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006, 20, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J. TGF β signaling in health and disease. Nat. Genet. 2004, 36, 790–792. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.H.; Saunier, E.F.; de Koning, J.P.; McKinnon, M.M.; Higgins, M.N.; Nicklas, K.; Yang, H.T.; Balmain, A.; Akhurst, R.J. Genetic variants of Tgfb1 act as context-dependent modifiers of mouse skin tumor susceptibility. Proc. Natl. Acad. Sci. USA 2006, 103, 8125–8130. [Google Scholar] [CrossRef] [PubMed]
- Freimuth, J.; Clermont, F.F.; Huang, X.; DeSapio, A.; Tokuyasu, T.A.; Sheppard, D.; Akhurst, R.J. Epistatic interactions between Tgfb1 and genetic loci, Tgfbm2 and Tgfbm3, determine susceptibility to an asthmatic stimulus. Proc. Natl. Acad. Sci. USA 2012, 109, 18042–18047. [Google Scholar] [CrossRef] [PubMed]
- Malkoski, S.P.; Haeger, S.M.; Cleaver, T.G.; Rodriguez, K.J.; Li, H.; Lu, S.L.; Feser, W.J.; Baron, A.E.; Merrick, D.; Lighthall, J.G.; et al. Loss of transforming growth factor β type II receptor increases aggressive tumor behavior and reduces survival in lung adenocarcinoma and squamous cell carcinoma. Clin. Cancer Res. 2012, 18, 2173–2183. [Google Scholar] [CrossRef] [PubMed]
- McCoy, E.L.; Iwanaga, R.; Jedlicka, P.; Abbey, N.S.; Chodosh, L.A.; Heichman, K.A.; Welm, A.L.; Ford, H.L. Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 2663–2677. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Christensen, K.L.; Jedlicka, P.; Coletta, R.D.; Baron, A.E.; Harrell, J.C.; Horwitz, K.B.; Billheimer, D.; Heichman, K.A.; Welm, A.L.; et al. The Six1 homeoprotein induces human mammary carcinoma cells to undergo epithelial-mesenchymal transition and metastasis in mice through increasing TGF-β signaling. J. Clin. Investig. 2009, 119, 2678–2690. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Wang, C.A.; Farabaugh, S.M.; Schiemann, W.P.; Ford, H.L. Homeoprotein Six1 increases TGF-β type I receptor and converts TGF-β signaling from suppressive to supportive for tumor growth. Cancer Res. 2010, 70, 10371–10380. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.H.; Liu, D.; Deng, Y.T.; Zhang, X.X.; Wan, D.Y.; Xi, B.X.; Huang, W.; Chen, Q.; Li, M.C.; Wang, M.W.; et al. SIX1 coordinates with TGFβ signals to induce epithelial-mesenchymal transition in cervical cancer. Oncol. Lett. 2016, 12, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Hocevar, B.A.; Smine, A.; Xu, X.X.; Howe, P.H. The adaptor molecule Disabled-2 links the transforming growth factor β receptors to the Smad pathway. EMBO J. 2001, 20, 2789–2801. [Google Scholar] [CrossRef] [PubMed]
- Prunier, C.; Howe, P.H. Disabled-2 (Dab2) is required for transforming growth factor β-induced epithelial to mesenchymal transition (EMT). J. Biol. Chem. 2005, 280, 17540–17548. [Google Scholar] [CrossRef] [PubMed]
- Hocevar, B.A.; Prunier, C.; Howe, P.H. Disabled-2 (Dab2) mediates transforming growth factor β (TGFβ)-stimulated fibronectin synthesis through TGFβ-activated kinase 1 and activation of the JNK pathway. J. Biol. Chem. 2005, 280, 25920–25927. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, A.; Hussey, G.S.; Ray, P.S.; Jin, G.; Fox, P.L.; Howe, P.H. TGF-β-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat. Cell Biol. 2010, 12, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Hannigan, A.; Smith, P.; Kalna, G.; Lo Nigro, C.; Orange, C.; O’Brien, D.I.; Shah, R.; Syed, N.; Spender, L.C.; Herrera, B.; et al. Epigenetic downregulation of human disabled homolog 2 switches TGF-β from a tumor suppressor to a tumor promoter. J. Clin. Investig. 2010, 120, 2842–2857. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.C.; Herbert, B.S.; Hocevar, B.A. Disabled-2 downregulation promotes epithelial-to-mesenchymal transition. Br. J. Cancer 2010, 103, 1716–1723. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhu, J.; Wu, Z. Loss of Dab2 expression in breast cancer cells impairs their ability to deplete TGF-β and induce Tregs development via TGF-β. PLoS ONE 2014, 9, e91709. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.L.; Chang, W.L.; Yang, H.B.; Wang, Y.C.; Chang, I.W.; Lee, C.T.; Chang, C.Y.; Lin, J.T.; Sheu, B.S. Low disabled-2 expression promotes tumor progression and determines poor survival and high recurrence of esophageal squamous cell carcinoma. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Danes, C.G.; Wyszomierski, S.L.; Lu, J.; Neal, C.L.; Yang, W.; Yu, D. 14-3-3ζ down-regulates p53 in mammary epithelial cells and confers luminal filling. Cancer Res. 2008, 68, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Neal, C.L.; Yao, J.; Yang, W.; Zhou, X.; Nguyen, N.T.; Lu, J.; Danes, C.G.; Guo, H.; Lan, K.H.; Ensor, J.; et al. 14-3-3ζ overexpression defines high risk for breast cancer recurrence and promotes cancer cell survival. Cancer Res. 2009, 69, 3425–3432. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Guo, H.; Treekitkarnmongkol, W.; Li, P.; Zhang, J.; Shi, B.; Ling, C.; Zhou, X.; Chen, T.; Chiao, P.J.; et al. 14-3-3ζ Cooperates with ErbB2 to promote ductal carcinoma in situ progression to invasive breast cancer by inducing epithelial-mesenchymal transition. Cancer Cell 2009, 16, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Acharya, S.; Sahin, O.; Zhang, Q.; Saito, Y.; Yao, J.; Wang, H.; Li, P.; Zhang, L.; Lowery, F.J.; et al. 14-3-3ζ turns TGF-β’s function from tumor suppressor to metastasis promoter in breast cancer by contextual changes of Smad partners from p53 to Gli2. Cancer Cell 2015, 27, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Kelber, J.A.; Klemke, R.L. PEAK1, a novel kinase target in the fight against cancer. Oncotarget 2010, 1, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kelber, J.A.; Tran Cao, H.S.; Cantin, G.T.; Lin, R.; Wang, W.; Kaushal, S.; Bristow, J.M.; Edgington, T.S.; Hoffman, R.M.; et al. Pseudopodium-enriched atypical kinase 1 regulates the cytoskeleton and cancer progression. Proc. Natl. Acad. Sci. USA 2010, 107, 10920–10925. [Google Scholar] [CrossRef] [PubMed]
- Kelber, J.A.; Reno, T.; Kaushal, S.; Metildi, C.; Wright, T.; Stoletov, K.; Weems, J.M.; Park, F.D.; Mose, E.; Wang, Y.; et al. KRas induces a Src/PEAK1/ErbB2 kinase amplification loop that drives metastatic growth and therapy resistance in pancreatic cancer. Cancer Res. 2012, 72, 2554–2564. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Qin, W.; Li, B.; Yang, H.; Guan, J.; Liu, Z.; Li, S. Analysis of a cytoskeleton-associated kinase PEAK1 and E-cadherin in gastric cancer. Pathol. Res. Pract. 2014, 210, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Agajanian, M.; Campeau, A.; Hoover, M.; Hou, A.; Brambilla, D.; Kim, S.L.; Klemke, R.L.; Kelber, J.A. PEAK1 acts as a molecular switch to regulate context-dependent TGFβ responses in breast cancer. PLoS ONE 2015, 10, e0135748. [Google Scholar] [CrossRef] [PubMed]
- Agajanian, M.; Runa, F.; Kelber, J.A. Identification of a PEAK1/ZEB1 signaling axis during TGFβ/fibronectin-induced EMT in breast cancer. Biochem. Biophys. Res. Commun. 2015, 465, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, L.; Huang, Q.; Jiang, Y.; Wang, J.; Zhang, L.; Tian, Y.; Yang, H. Radiosensitization of non-small cell lung cancer cells by inhibition of TGF-β1 signaling with SB431542 is dependent on p53 Status. Oncol. Res. 2016, 24, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, V.I.; Javelaud, D.; Van Kempen, L.C.; Mohammad, K.S.; Dennler, S.; Luciani, F.; Hoek, K.S.; Juarez, P.; Goydos, J.S.; Fournier, P.J.; et al. GLI2-mediated melanoma invasion and metastasis. J. Natl. Cancer Inst. 2010, 102, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, G.; Fan, Q.; Xie, J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene 2010, 29, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Andre, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-β: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Nguyen, M.P.; Padalecki, S.S.; Grubbs, B.G.; Merkel, A.R.; Oyajobi, B.O.; Matrisian, L.M.; Mundy, G.R.; Sterling, J.A. TGF-β promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer Res. 2011, 71, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Rothenberg, M.L.; Tabernero, J.; Seoane, J.; Daly, T.; Cleverly, A.; Berry, B.; Rhoades, S.K.; Ray, C.A.; Fill, J.; et al. TGF-β signalling-related markers in cancer patients with bone metastasis. Biomarkers 2008, 13, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Farrington, D.L.; Yingling, J.M.; Fill, J.A.; Yan, L.; Qian, Y.W.; Shou, J.; Wang, X.; Ehsani, M.E.; Cleverly, A.L.; Daly, T.M.; et al. Development and validation of a phosphorylated SMAD ex vivo stimulation assay. Biomarkers 2007, 12, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Dave, H.; Trivedi, S.; Shah, M.; Shukla, S. Transforming growth factor β 2: A predictive marker for breast cancer. Indian J. Exp. Biol. 2011, 49, 879–887. [Google Scholar] [PubMed]
- Dave, H.; Shah, M.; Trivedi, S.; Shukla, S. Prognostic utility of circulating transforming growth factor β 1 in breast cancer patients. Int. J. Biol. Markers 2012, 27, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, T.; Numajiri, M.; Nakazaki, H.; Okazaki, Y.; Kuwana, M. Induction of immune tolerance to platelet antigen by short-term thrombopoietin treatment in a mouse model of immune thrombocytopenia. Int. J. Hematol. 2014, 100, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Lyman, G.H.; Moses, H.L. Biomarker Tests for Molecularly Targeted Therapies—The Key to Unlocking Precision Medicine. N. Engl. J. Med. 2016, 375, 4–6. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, N.; Taguchi, A.; Hanash, S. Switching Roles of TGF-β in Cancer Development: Implications for Therapeutic Target and Biomarker Studies. J. Clin. Med. 2016, 5, 109. https://doi.org/10.3390/jcm5120109
Sun N, Taguchi A, Hanash S. Switching Roles of TGF-β in Cancer Development: Implications for Therapeutic Target and Biomarker Studies. Journal of Clinical Medicine. 2016; 5(12):109. https://doi.org/10.3390/jcm5120109
Chicago/Turabian StyleSun, Nan, Ayumu Taguchi, and Samir Hanash. 2016. "Switching Roles of TGF-β in Cancer Development: Implications for Therapeutic Target and Biomarker Studies" Journal of Clinical Medicine 5, no. 12: 109. https://doi.org/10.3390/jcm5120109
APA StyleSun, N., Taguchi, A., & Hanash, S. (2016). Switching Roles of TGF-β in Cancer Development: Implications for Therapeutic Target and Biomarker Studies. Journal of Clinical Medicine, 5(12), 109. https://doi.org/10.3390/jcm5120109