
Mitochondrial Glutathione in Diabetic Nephropathy

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
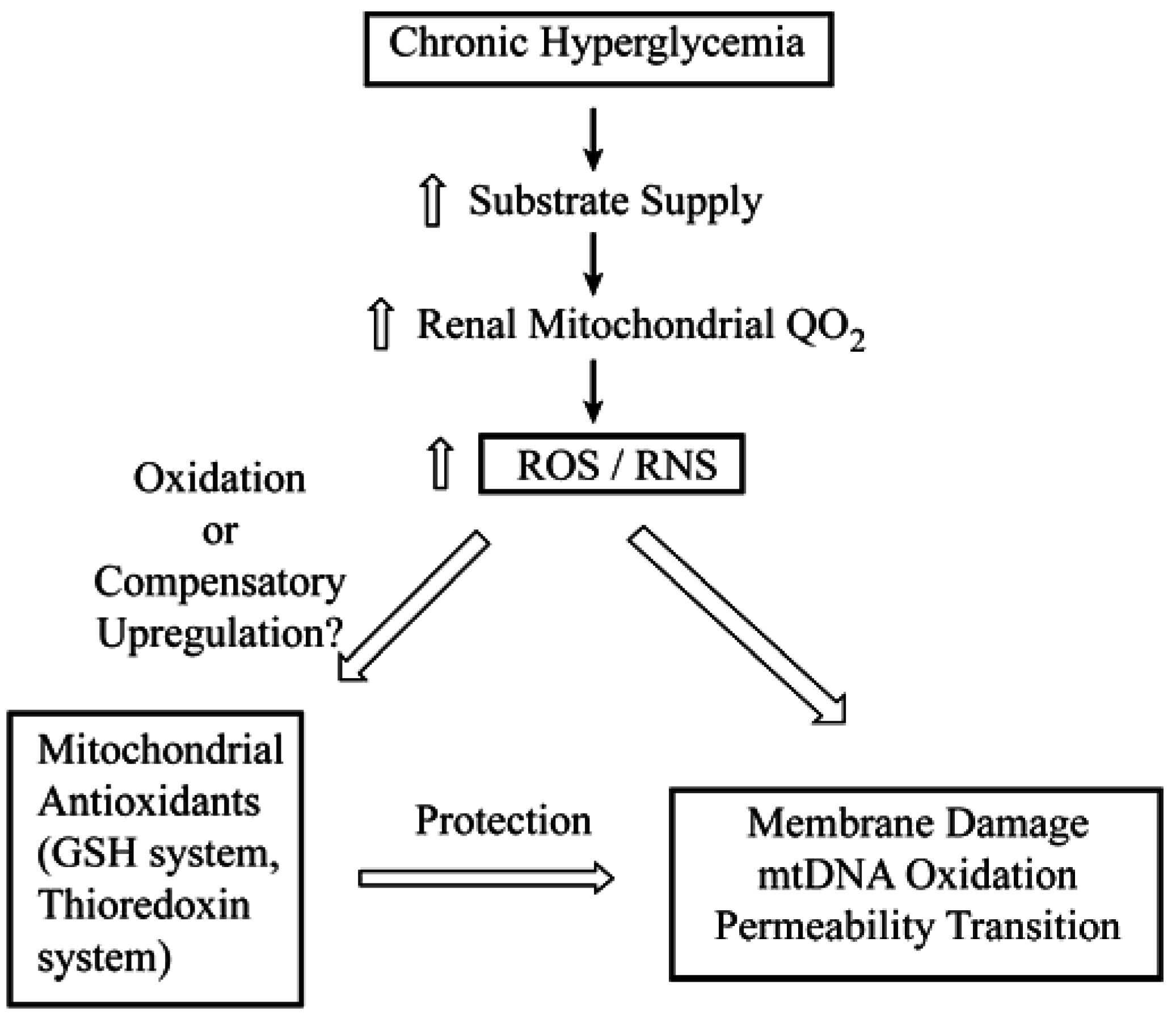
1. Introduction
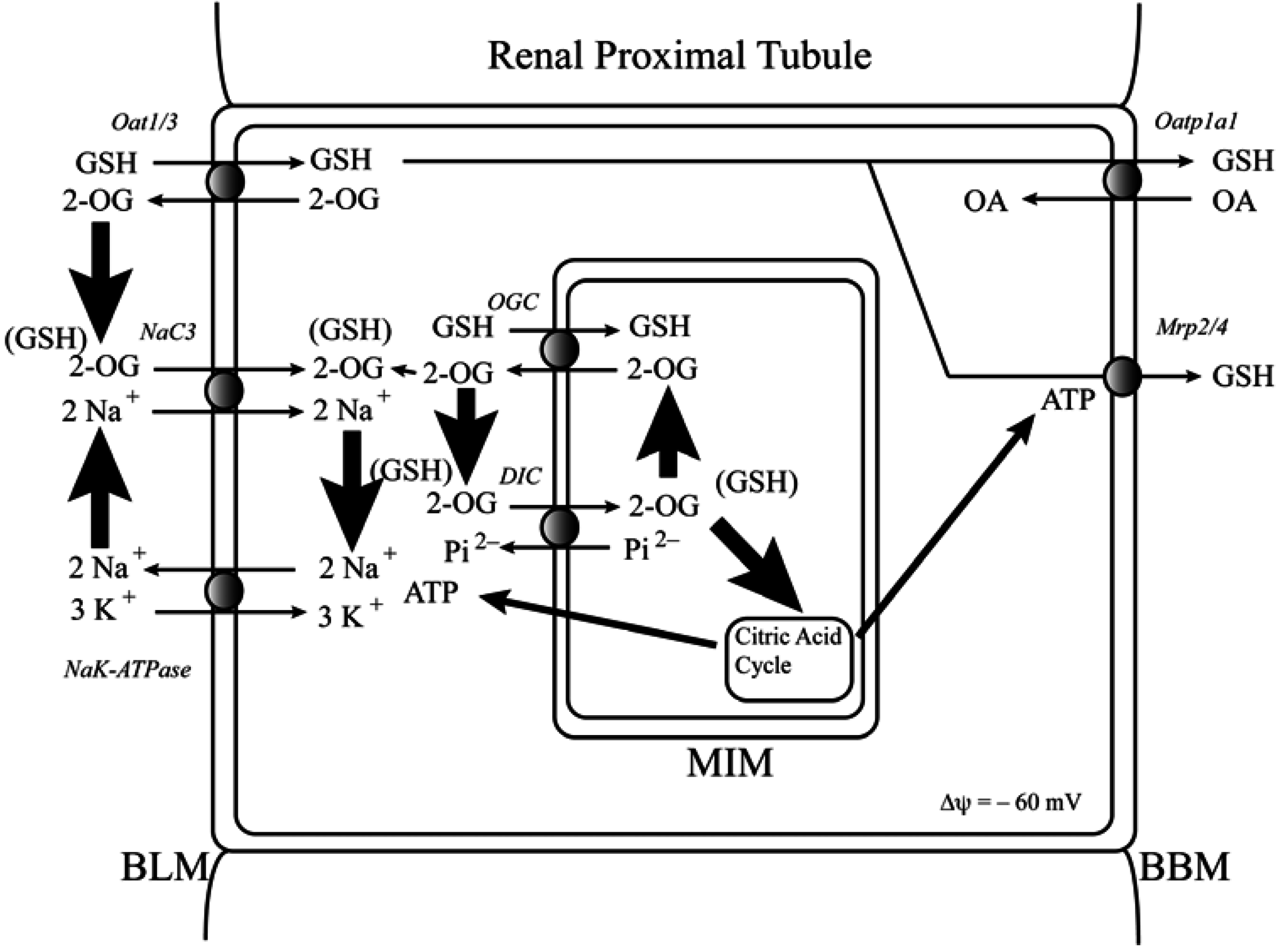
2. Determination of mtGSH Status: Transport versus Synthesis and Identification of Carriers
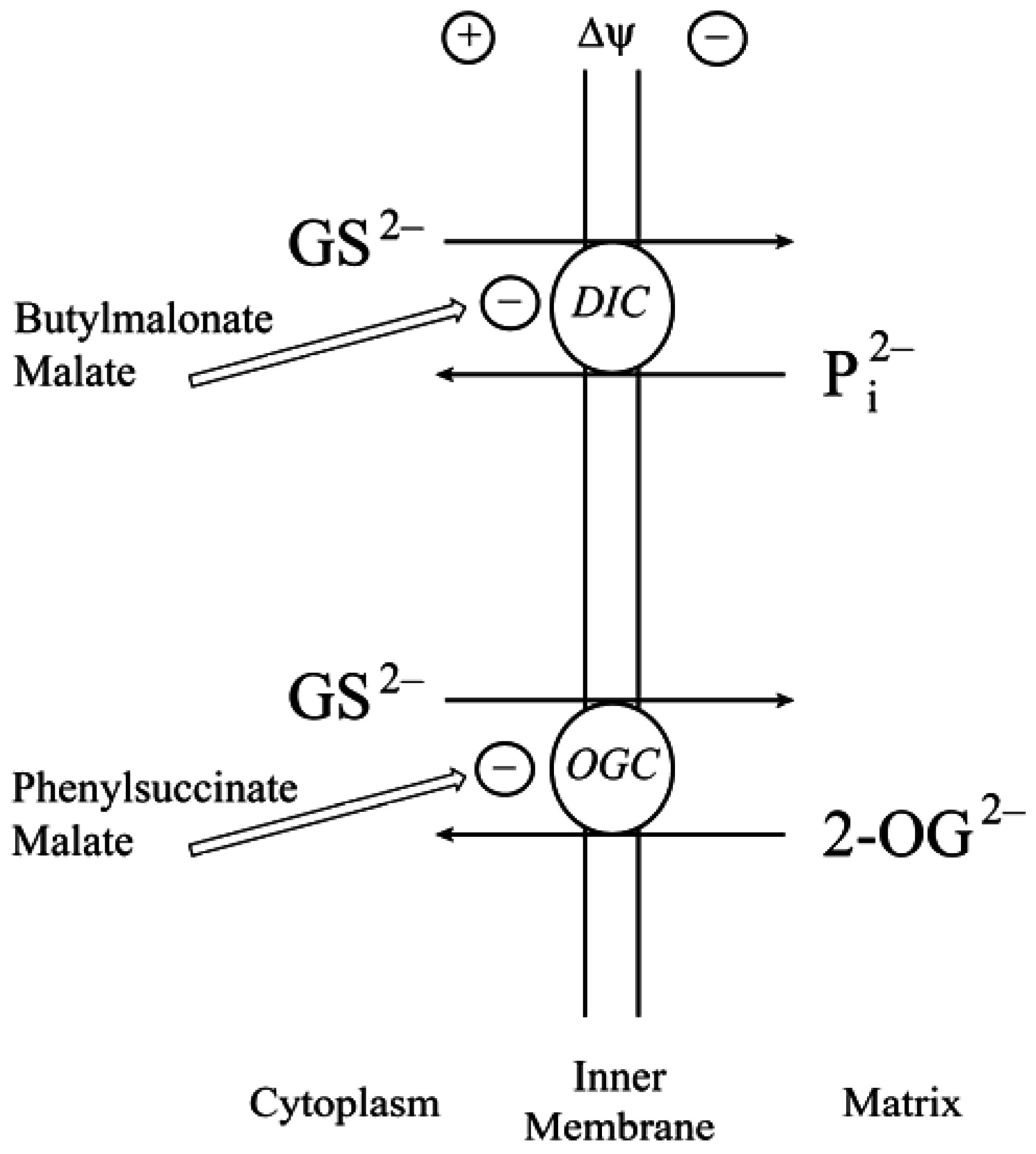
2.1. Background and Identification of mtGSH Carriers in Renal Mitochondria
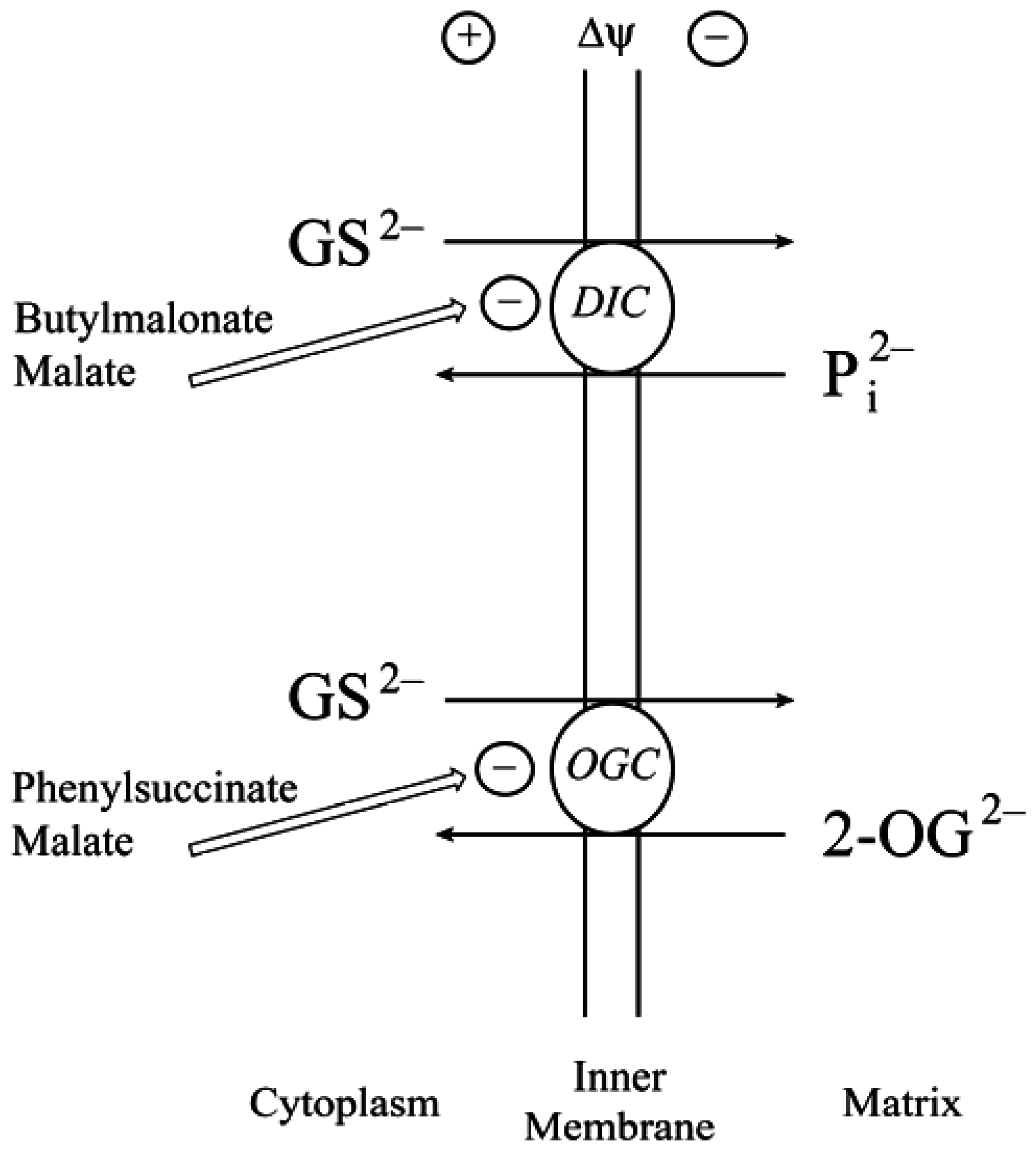
2.2. Function of the DIC and OGC Demonstrated in Mitochondria from Other Tissues
2.3. Do the DIC and OGC Really Transport GSH into Mitochondria?
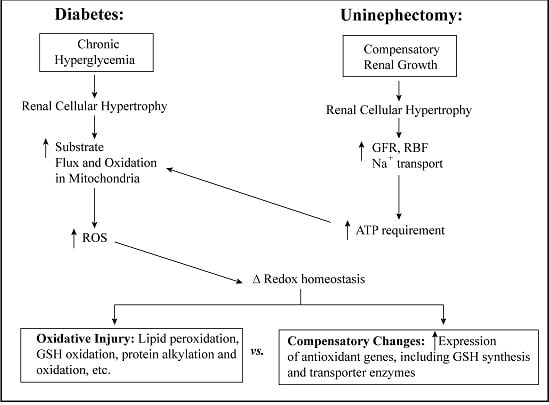
3. Cellular and mtGSH in Diabetic Nephropathy and Other Renal Diseases Involving Tubular Epithelial Cell Hypertrophy
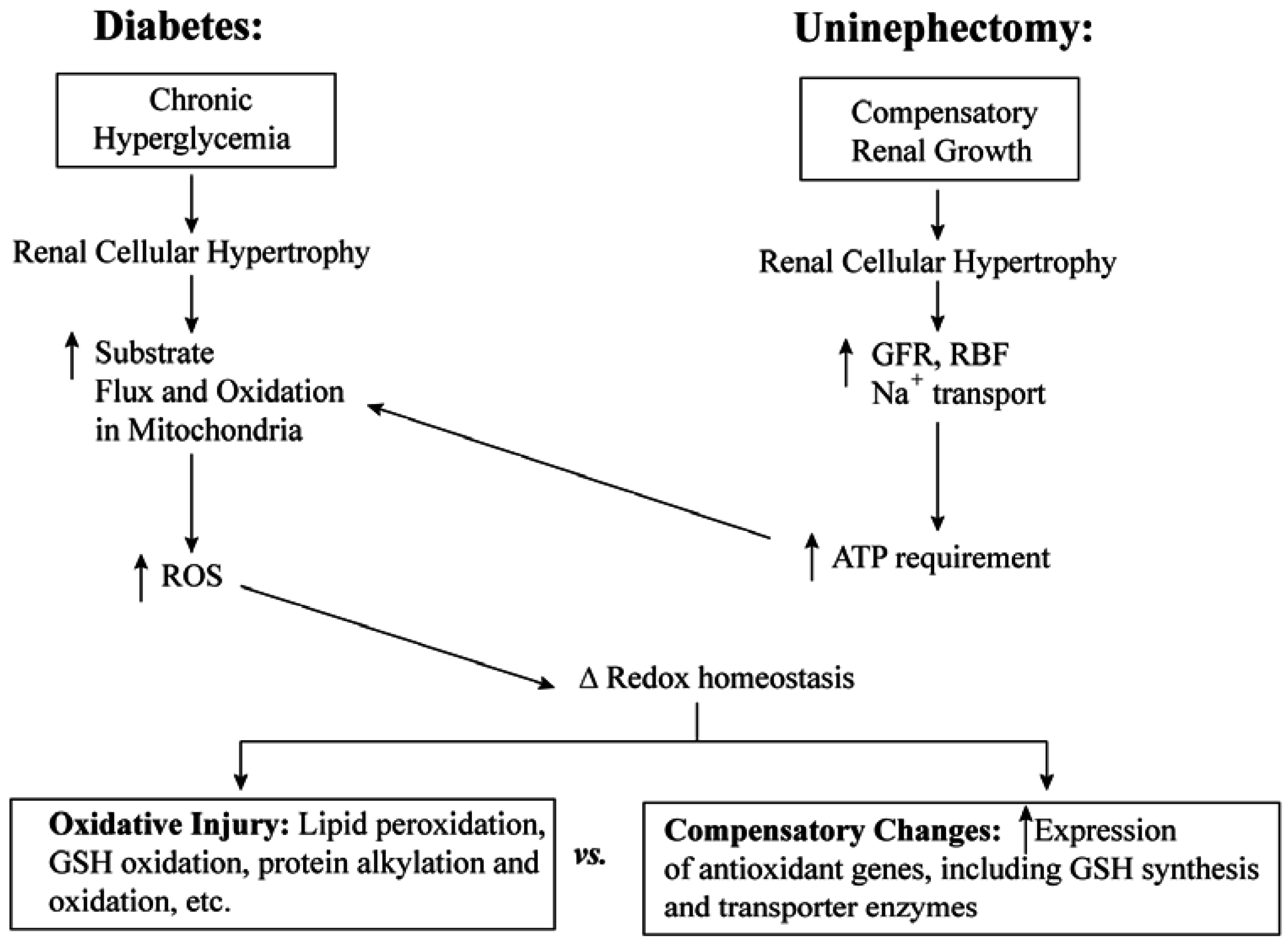
3.1. Diabetic Nephropathy
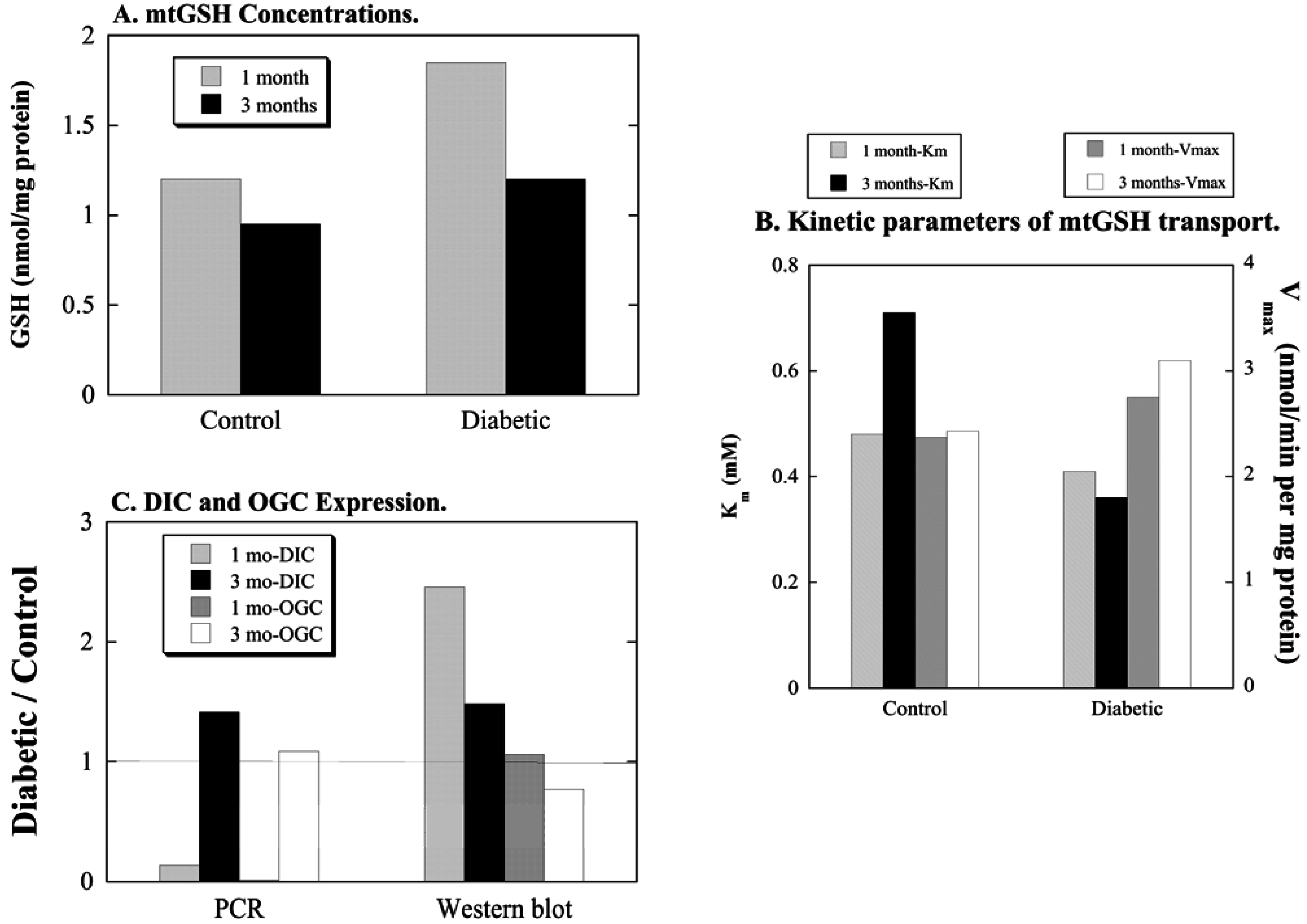
3.2. Uninephrectomy and Compensatory Renal Hypertrophy
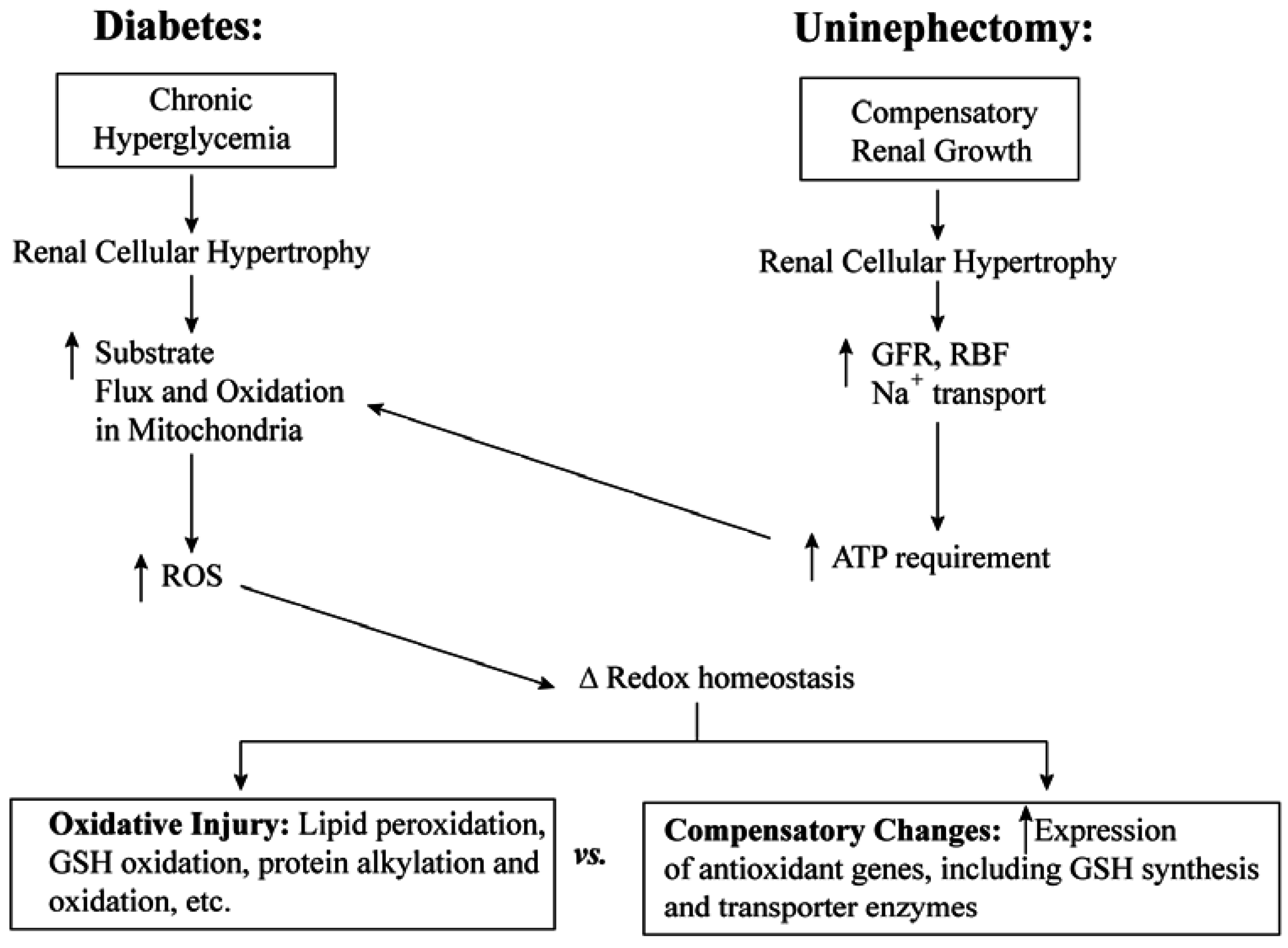
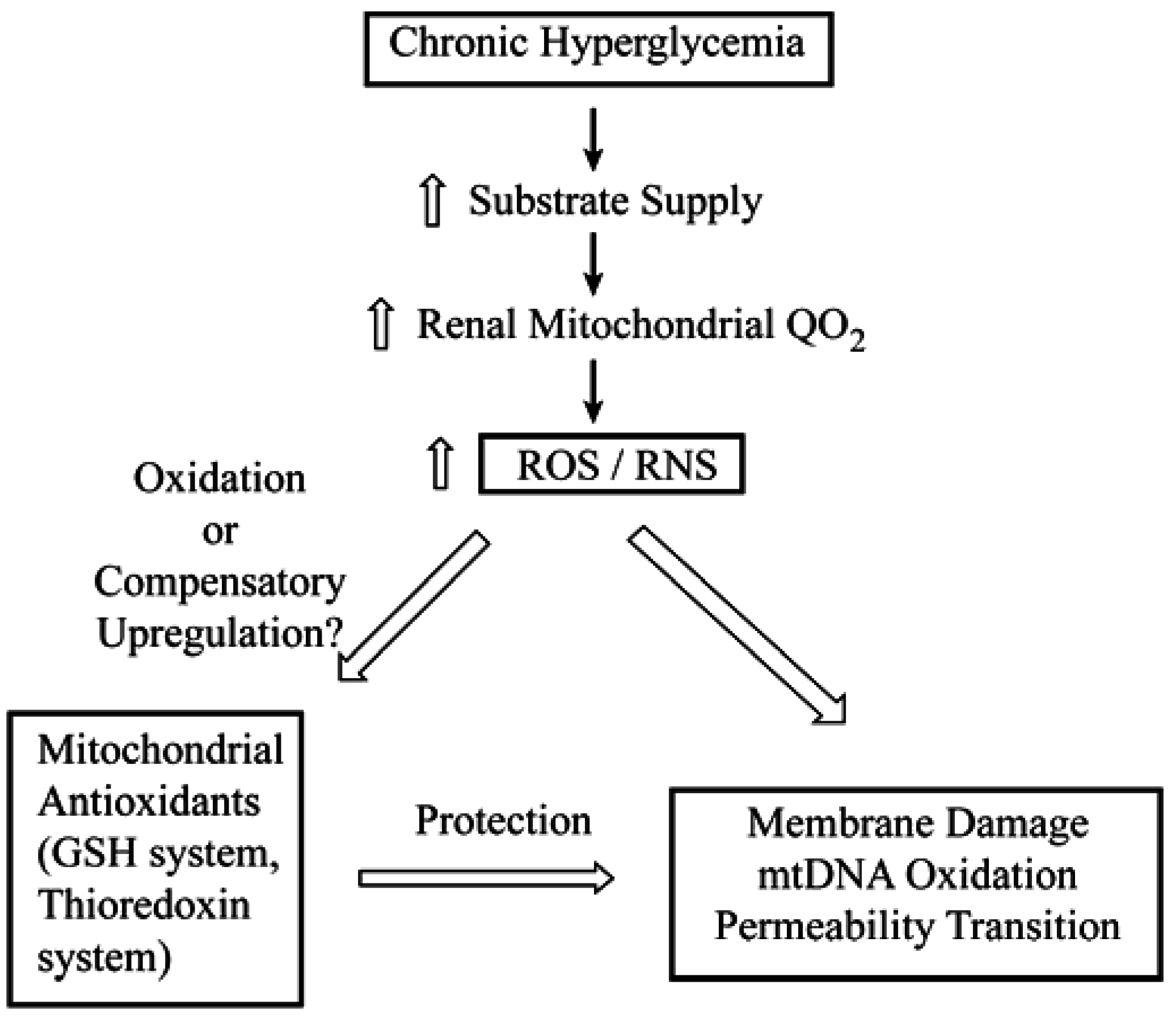
4. Toxicological Impact of Varying mtGSH Transport
5. Summary and Conclusions

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ibrahim, H.N.; Hostetter, T.H. Diabetic nephropathy. J. Am. Soc. Nephrol. 1997, 8, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Remuzzi, G.; Macia, M.; Ruggenenti, P. Prevention and treatment of diabetic renal disease in Type 2 diabetes: The BENEDICT study. J. Am. Soc. Nephrol. 2006, 17, S90–S97. [Google Scholar] [CrossRef] [PubMed]
- Geiss, L.S.; Herman, W.H.; Goldschmid, M.G.; DeStefano, F.; Eberhardt, M.S.; Ford, E.S.; German, R.R.; Newman, J.M.; Olson, D.R.; Sepe, S.J. Surveillance for diabetes mellitus: United States, 1980–1989. MMWR CDC Surveill. Summ. 1993, 42, 1–20. [Google Scholar] [PubMed]
- Sedor, J.R. Frontiers in diabetic nephropathy: Can we predict who will get sick? J. Am. Soc. Nephrol. 2006, 17, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Susztak, K.; Böttinger, E.P. Diabetic nephropathy: A frontier for personalized medicine. J. Am. Soc. Nephrol. 2006, 17, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Atlas of Chronic Kidney Disease in the United States. 2013 USRDS Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Am. J. Kidney Dis. 2014, 63 (Suppl. 1), A1–A7, e1–e478. [PubMed]
- Fioretto, P.; Bruseghin, M.; Berto, I.; Gallina, P.; Manzato, E.; Mussap, M. Renal protection in diabetes: Role of glycemic control. J. Am. Soc. Nephrol. 2006, 17, S86–S89. [Google Scholar] [CrossRef] [PubMed]
- Alderson, N.L.; Chachich, M.E.; Frizzell, N.; Canning, P.; Metz, T.O.; Januszewski, A.S.; Youssef, N.N.; Stitt, A.W.; Baynes, J.W.; Thorpe, S.R. Effect of antioxidants and ACE inhibition on chemical modification of proteins and progression of nephropathy in the streptozotocin diabetic rat. Diabetologia 2004, 47, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, P.J.; Drummond, K.S.; Nelson, R.G.; Howell, S.K.; Szwergold, B.S.; Mauer, M. Susceptibility to diabetic nephropathy is related to dicarbonyl and oxidative stress. Diabetes 2005, 54, 3274–3281. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Chander, P.N.; Gealekman, O.; Brodsky, S.V.; Elitok, S.; Tojo, A.; Crabtree, M.; Gross, S.S.; Goligorsky, M.S. Nephropathy in Zucker diabetic fat rat is associated with oxidative and nitrosative stress: Prevention by chronic therapy with a peroxynitrite scavenger ebselen. J. Am. Soc. Nephrol. 2004, 15, 2391–2403. [Google Scholar] [CrossRef] [PubMed]
- Hakim, F.A.; Pflueger, A. Role of oxidative stress in diabetic kidney disease. Med. Sci. Monit. 2010, 16, RA37–RA48. [Google Scholar] [PubMed]
- Obrosova, I.G.; Fathallah, L.; Liu, E.; Nourooz-Zadeh, J. Early oxidative stress in the diabetic kidney: Effect of DL-α-lipoic acid. Free Rad. Biol. Med. 2003, 34, 186–195. [Google Scholar] [CrossRef]
- Prabhakar, S.; Starnes, J.; Shi, S.; Lonis, B.; Tran, R. Diabetic nephropathy is associated with oxidative stress and decreased renal nitric oxide production. J. Am. Soc. Nephrol. 2007, 18, 2945–2952. [Google Scholar] [CrossRef] [PubMed]
- Winiarska, K.; Drozak, J.; Wegryznowicz, M.; Fraczyk, T.; Bryla, J. Diabetes-induced changes in glucose synthesis, intracellular glutathione status and hydroxyl free radical generation in rabbit kidney cortex tubules. Mol. Cell. Biochem. 2004, 261, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y.; Kizaki, M.; Nakafiri, R.; Kamiya, T.; Sumi, H.; Osawa, T. Dietary glutathione protects rats from diabetic nephropathy and neuropathy. J. Nutr. 2002, 132, 897–900. [Google Scholar] [PubMed]
- Kakkar, R.; Mantha, S.V.; Radhi, J.; Prasad, K.; Kaira, J. Antioxidant defense system in diabetic kidney: A time course study. Life Sci. 1997, 60, 667–679. [Google Scholar] [CrossRef]
- Yue, K.K.M.; Leung, S.-N.; Man, P.-M.; Yeung, W.-F.; Chung, W.-S.; Lee, K.-W.; Leung, A.W.N.; Cheng, C.H.K. Alterations in antioxidant enzyme activities in the eyes, aorta and kidneys of diabetic rats relevant to the onset of oxidative stress. Life Sci. 2005, 77, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.P.; Palmeira, C.M. Diabetes and mitochondrial function: Role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharmacol. 2006, 212, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H. Role of glutathione transport processes in kidney function. Toxicol. Appl. Pharmacol. 2005, 204, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H. Mitochondrial glutathione transport: Physiological, pathological and toxicological implications. Chem.-Biol. Interact. 2006, 163, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H. Renal glutathione transport: Identification of carriers, physiological functions, and controversies. BioFactors 2009, 35, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H. Renal membrane transport of glutathione in toxicology and disease. Vet. Pathol. 2011, 48, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H. Mitochondrial glutathione in toxicology and disease of the kidneys. Toxicol. Res. 2012, 1, 39–46. [Google Scholar] [CrossRef]
- Schnellmann, R.G. Renal mitochondrial glutathione transport. Life Sci. 1991, 49, 393–398. [Google Scholar] [CrossRef]
- Griffith, O.W.; Meister, A. Origin and turnover of mitochondrial glutathione. Proc. Natl. Acad. Sci. USA 1985, 82, 4668–4672. [Google Scholar] [CrossRef] [PubMed]
- McKernan, T.B.; Woods, E.B.; Lash, L.H. Uptake of glutathione by renal cortical mitochondria. Arch. Biochem. Biophys. 1991, 288, 653–663. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflugers Arch. 2004, 447, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lash, L.H. Evidence for mitochondrial uptake of glutathione by dicarboxylate and 2-oxoglutarate carriers. J. Pharmacol. Exp. Ther. 1998, 285, 608–618. [Google Scholar] [PubMed]
- Chen, Z.; Putt, D.A.; Lash, L.H. Enrichment and functional reconstitution of glutathione transport activity from rabbit kidney mitochondria: Further evidence for the role of the dicarboxylate and 2-oxoglutarate carriers in mitochondrial glutathione transport. Arch. Biochem. Biophys. 2000, 373, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H.; Putt, D.A.; Hueni, S.E.; Cao, W.; Xu, F.; Kulidjian, S.J.; Horwitz, J.P. Cellular energetics and glutathione status in NRK-52E cells: Toxicological implications. Biochem. Pharmacol. 2002, 64, 1533–1546. [Google Scholar] [CrossRef]
- Lash, L.H.; Putt, D.A.; Matherly, L.H. Protection of NRK-52E cells, a rat renal proximal tubular cell line, from chemical induced apoptosis by overexpression of a mitochondrial glutathione transporter. J. Pharmacol. Exp. Ther. 2002, 303, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Putt, D.A.; Matherly, L.H.; Lash, L.H. Modulation of expression of rat mitochondrial 2-oxoglutarate carrier in NRK-52E cells alters mitochondrial transport and accumulation of glutathione and susceptibility to chemically induced apoptosis. J. Pharmacol. Exp. Ther. 2006, 316, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.; Bisaccia, F.; Capobianco, L.; Dolce, V.; Fiermonte, G.; Iacobazzi, V.; Indiveri, C.; Palmieri, L. Mitochondrial metabolite transporters. Biochim. Biophys. Acta 1996, 1275, 127–132. [Google Scholar] [CrossRef]
- Zhong, Q.; Putt, D.A.; Xu, F.; Lash, L.H. Hepatic mitochondrial transport of glutathione: Studies in isolated rat liver mitochondria and H4IIE rat hepatoma cells. Arch. Biochem. Biophys. 2008, 474, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Coll, O.; Garcia-Ruiz, C.; Kaplowitz, N.; Fernandez-Checa, J.C. Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion. Hepatology 2003, 38, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Checa, J.; Kaplowitz, N. Hepatic mitochondrial glutathione: Transport and role in disease and toxicity. Toxicol. Appl. Pharmacol. 2005, 204, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Kamga, C.K.; Zhang, S.X.; Wang, Y. Dicarboxylate carrier-mediated glutathione transport is essential for reactive oxygen species homeostasis and normal respiration in rat brain mitochondria. Am. J. Physiol. 2010, 299, C497–C505. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Marquardt, K.; Lash, L.H.; Linseman, D.A. Bcl-2 is a novel interacting partner for the 2-oxoglutarate carrier and a key regulator of mitochondrial glutathione. Free Rad. Biol. Med. 2012, 52, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Kirchhof, D.; Manning, E.; Joseph, J.W.; Linseman, D.A. Mitochondrial glutathione transport is a key determinant of neuronal susceptibility to oxidative and nitrosative stress. J. Biol. Chem. 2013, 288, 5091–5101. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Rodriguez, C.; Maloney, R.; Moyer, M.P.; Aw, T.Y. Contribution of mitochondrial GSH transport to matrix GSH status and colonic epithelial cell apoptosis. Free Rad. Biol. Med. 2008, 44, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Moyer, M.P.; Harrison, L.; Aw, T.Y. Contribution of glutathione status to oxidant-induced mitochondrial DNA damage in colonic epithelial cells. Free Rad. Biol. Med. 2009, 47, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Booty, L.M.; King, M.S.; Thangaratnarajah, C.; Majd, H.; James, A.M.; Kunji, E.R.S.; Murphy, M.P. The mitochondrial dicarboxylate and 2-oxoglutarate carriers do not transport glutathione. FEBS Lett. 2015, 589, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.S.; Pedersen, P.L. Isolation and reconstitution of the n-butylmalonate-sensitive dicarboxylate transporter from rat liver mitochondria. J. Biol. Chem. 1985, 260, 10293–10298. [Google Scholar] [PubMed]
- Runswick, M.J.; Walker, J.E.; Bisaccia, F.; Iacobazzi, V.; Palmieri, F. Sequence of the bovine 2-oxoglutarate/malate carrier protein: Structural relationship to other mitochondrial transport proteins. Biochemistry 1990, 29, 11033–11040. [Google Scholar] [CrossRef] [PubMed]
- Bisaccia, F.; Capobianco, L.; Mazzeo, M.; Palmieri, F. The mitochondrial oxoglutarate carrier protein contains a disulfide bridge between intramembranous cysteines 221 and 224. FEBS Lett. 1996, 392, 54–58. [Google Scholar] [CrossRef]
- Stipani, I.; Manguillo, G.; Stipani, V.; Daddabbo, L.; Natuzzi, D.; Palmieri, F. Inhibition of the reconstituted mitochondrial oxoglutarate carrier by arginine-specific reagents. Arch. Biochem. Biophys. 1996, 331, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Kakhniashvili, D.; Major, J.A.; Gremse, D.A.; Xu, Y.; Kaplan, R.S. Identification of a novel gene encoding the yeast mitochondrial dicarboxylate transport protein via overexpression, purification, and characterization of its protein product. J. Biol. Chem. 1997, 272, 4516–4521. [Google Scholar] [CrossRef] [PubMed]
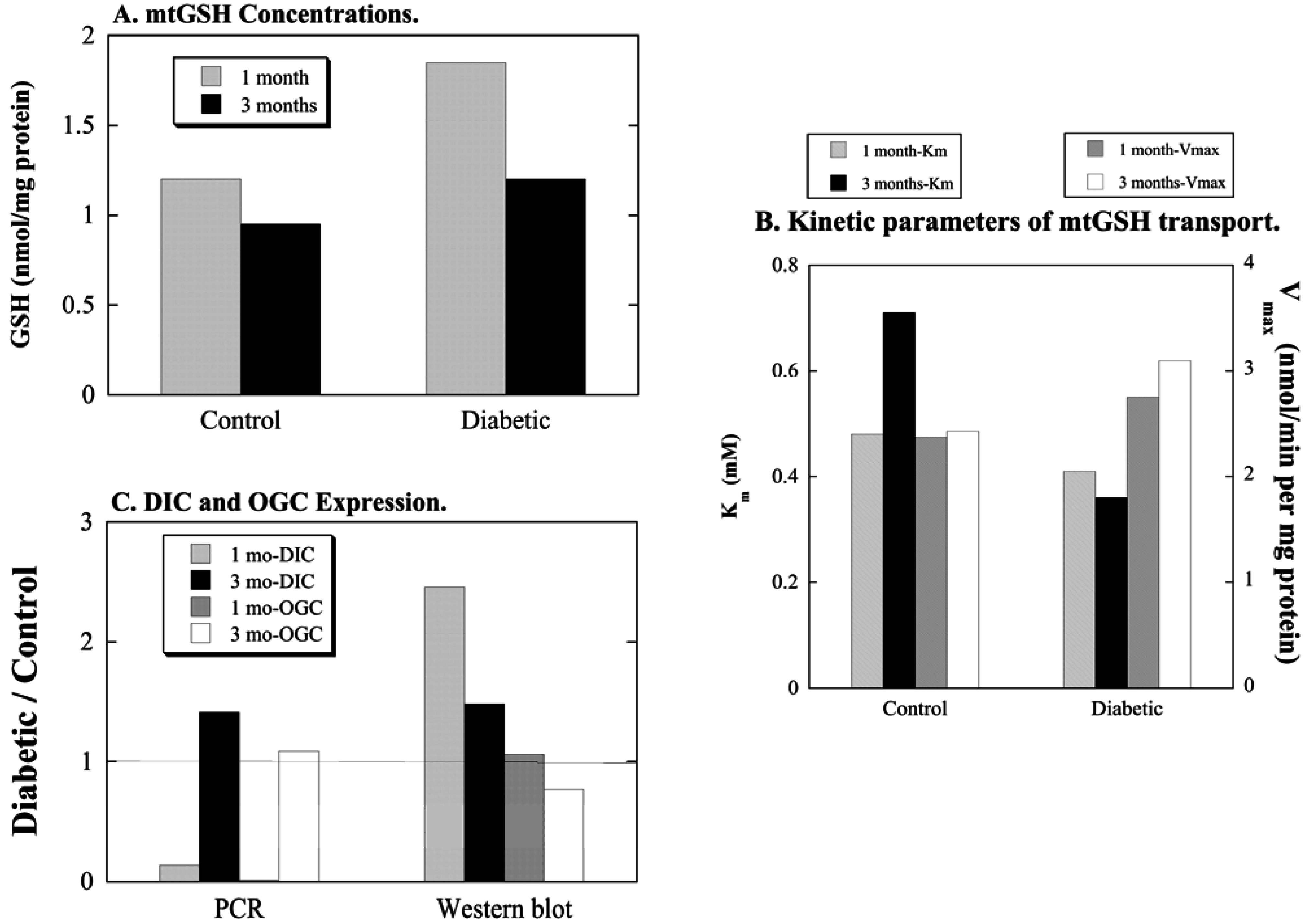
- Putt, D.A.; Zhong, Q.; Lash, L.H. Adaptive changes in renal mitochondrial redox status in diabetic nephropathy. Toxicol. Appl. Pharmacol. 2012, 258, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Terlecky, S.R.; Lash, L.H. Diabetes increases susceptibility of rat proximal tubular cells to chemical injury. Toxicol. Appl. Pharmacol. 2009, 241, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.K.; Christiansen, J.S.; Steven, K.; Parving, H.H. Renal function in streptozotocin-diabetic rats. Diabetologia 1981, 21, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.H.; Allen, T.J. Rodent models of streptozotocin-induced diabetic nephropathy. Nephrology 2007, 12, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Walrand, S.; Valeix, S.; Rodriguez, C.; Ligot, P.; Chassagne, J.; Vasson, M.-P. Flow cytometry studies of polymorphonuclear neutrophil oxidative burst: A comparison of three fluorescent probes. Clin. Chim. Acta 2003, 331, 103–110. [Google Scholar] [CrossRef]
- Zalups, R.K.; Lash, L.H. Effects of uninephrectomy and mercuric chloride on renal glutathione homeostasis. J. Pharmacol. Exp. Ther. 1990, 254, 962–970. [Google Scholar] [PubMed]
- Lash, L.H.; Zalups, R.K. Activities of enzymes involved in renal cellular glutathione metabolism after uninephrectomy in the rat. Arch. Biochem. Biophys. 1994, 309, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H.; Putt, D.A.; Horky, S.J., III; Zalups, R.K. Functional and toxicological characteristics of isolated renal mitochondria: Impact of compensatory renal growth. Biochem. Pharmacol. 2001, 62, 383–395. [Google Scholar] [CrossRef]
- Lash, L.H.; Putt, D.A.; Zalups, R.K. Biochemical and functional characteristics of cultured renal epithelial cells from uninephrectomized rats: Factors influencing nephrotoxicity. J. Pharmacol. Exp. Ther. 2001, 296, 243–251. [Google Scholar] [PubMed]
- Benipal, B.; Lash, L.H. Influence of renal compensatory hypertrophy on mitochondrial energetics and redox status. Biochem. Pharmacol. 2011, 81, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Benipal, B.; Lash, L.H. Modulation of mitochondrial glutathione status and cellular energetics in primary cultures of proximal tubular cells from remnant kidney of uninephrectomized rats. Biochem. Pharmacol. 2013, 85, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H.; Zalups, R.K. Mercuric chloride-induced cytotoxicity and compensatory hypertrophy in rat kidney proximal tubular cells. J. Pharmacol. Exp. Ther. 1992, 261, 819–829. [Google Scholar] [PubMed]
- Lash, L.H.; Putt, D.A.; Zalups, R.K. Influence of compensatory renal growth on susceptibility of primary cultures of renal cells to chemically induced injury. Toxicol. Sci. 2006, 94, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F. Diseases caused by defects of mitochondrial carriers: A review. Biochim. Biophys. Acta 2008, 1777, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Masuram, S.; Fredriksson, R.; Schiöth, H.B. Solute carriers as drug targets: Current use, clinical trials and perspective. Mol. Aspects Med. 2013, 34, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Sugiyama, Y. Transporter biology in drug approval: Regulatory aspects. Mol. Aspects Med. 2013, 34, 711–718. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Bentz, S.; Hediger, M.A.; Anderle, P. Solute carriers (SLCs) in cancer. Mol. Aspects Med. 2013, 34, 719–734. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lash, L.H. Mitochondrial Glutathione in Diabetic Nephropathy. J. Clin. Med. 2015, 4, 1428-1447. https://doi.org/10.3390/jcm4071428
Lash LH. Mitochondrial Glutathione in Diabetic Nephropathy. Journal of Clinical Medicine. 2015; 4(7):1428-1447. https://doi.org/10.3390/jcm4071428
Chicago/Turabian StyleLash, Lawrence H. 2015. "Mitochondrial Glutathione in Diabetic Nephropathy" Journal of Clinical Medicine 4, no. 7: 1428-1447. https://doi.org/10.3390/jcm4071428
APA StyleLash, L. H. (2015). Mitochondrial Glutathione in Diabetic Nephropathy. Journal of Clinical Medicine, 4(7), 1428-1447. https://doi.org/10.3390/jcm4071428