
Scalable Electrophysiological Investigation of iPS Cell-Derived Cardiomyocytes Obtained by a Lentiviral Purification Strategy


Abstract
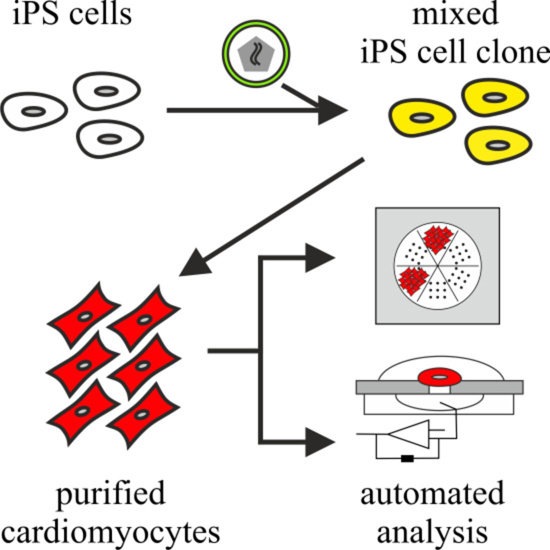
:
1. Introduction
2. Experimental Section
2.3. Differentiation of iPS Cells and Purification of Cardiomyocytes
2.4. Immunocytochemistry
2.5. Conventional Manual Patch Clamp Analysis
2.6. Automated Planar Patch Clamp Analysis
2.7. Microelectrode Array Analysis
2.8. Statistics
3. Results
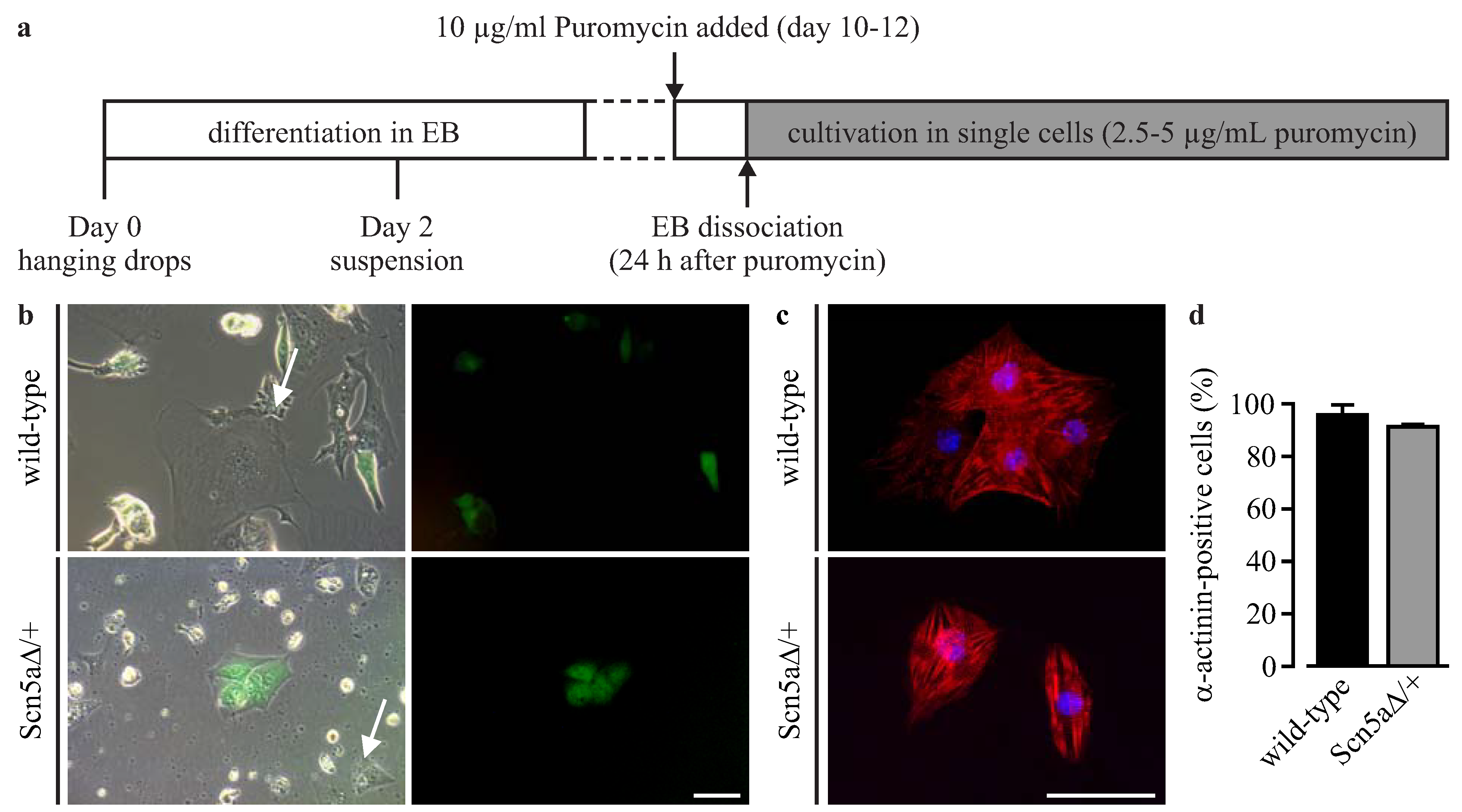
3.1. Lentiviral Strategy for Purification of iPS Cell-Derived Cardiomyocytes
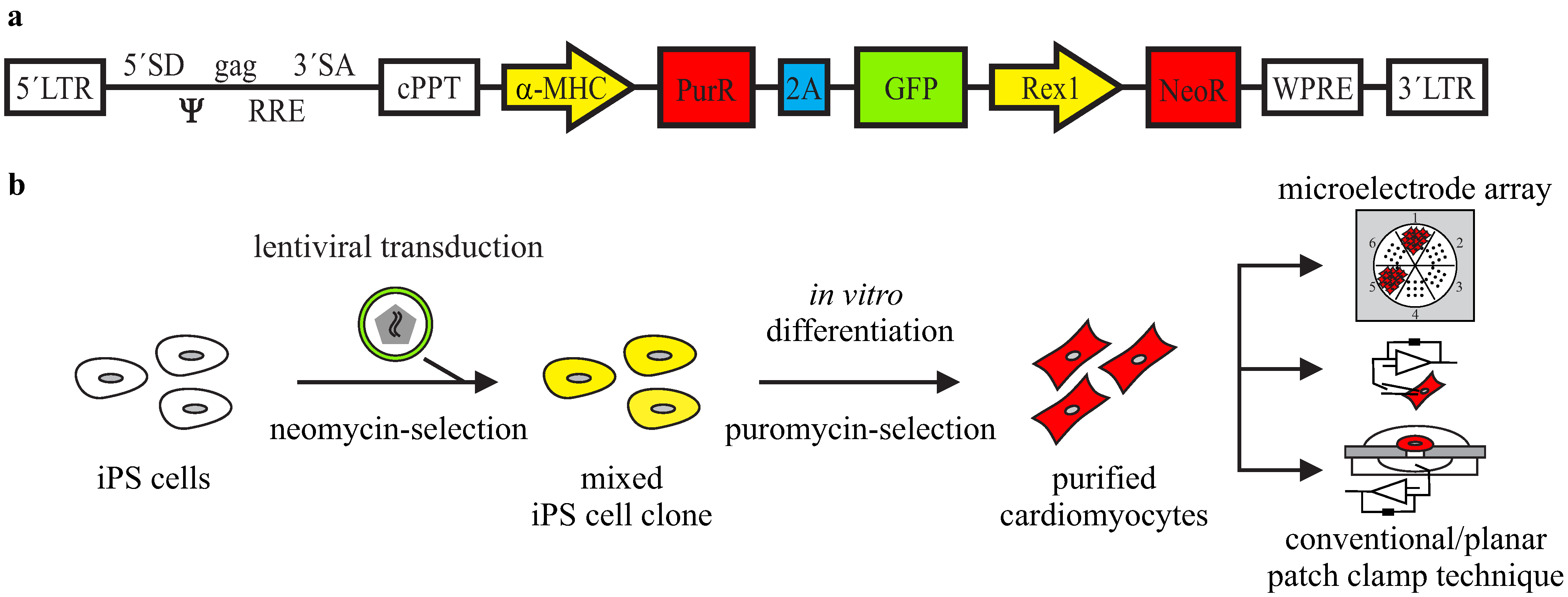
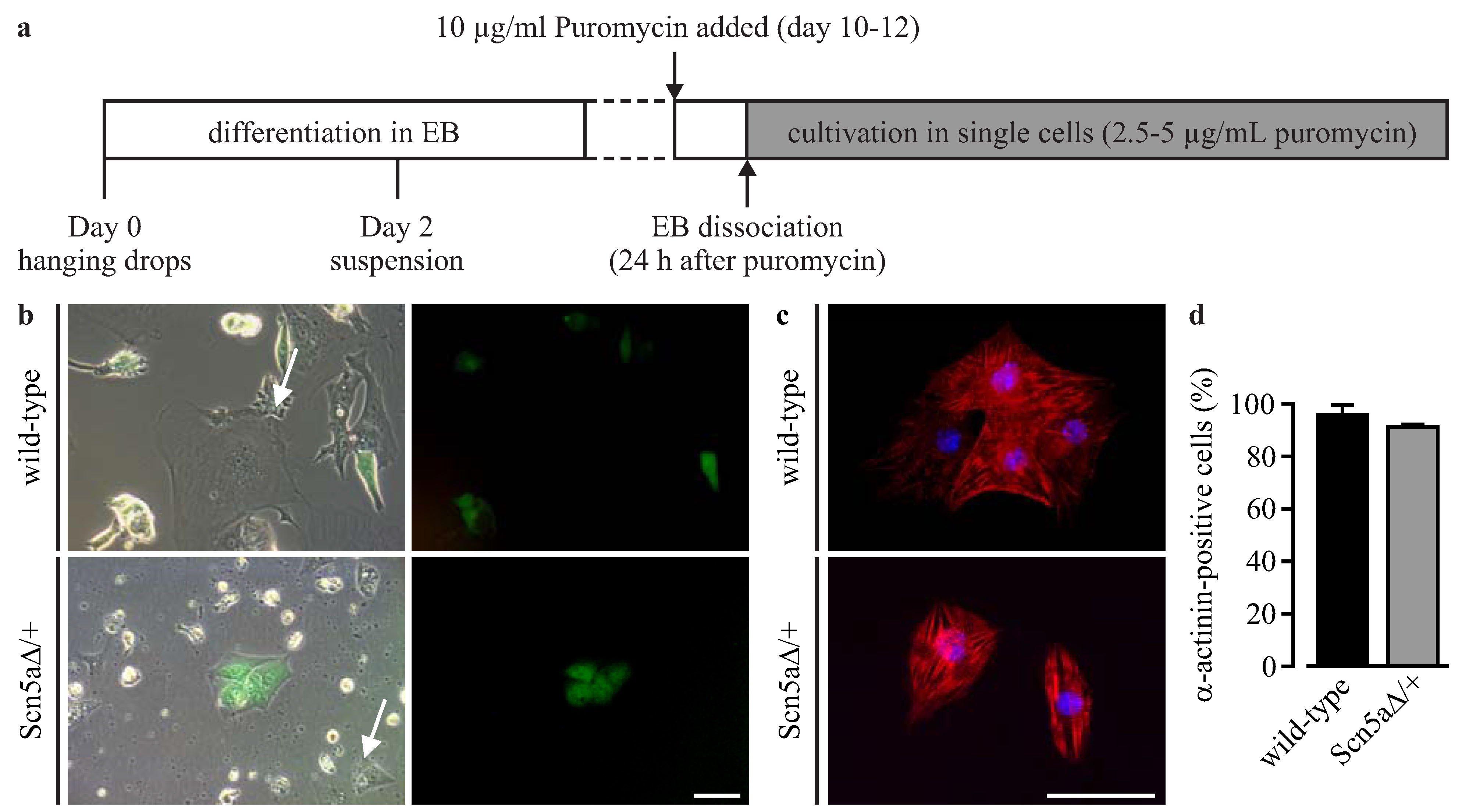
3.2. Purification of αPaG-RexNeo iPS Cell-Derived Cardiomyocytes
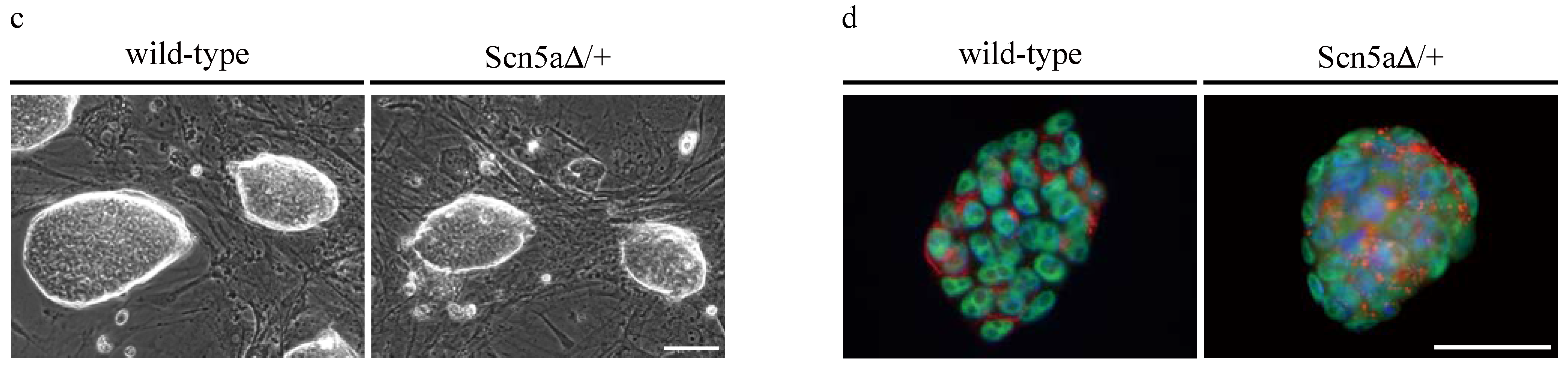
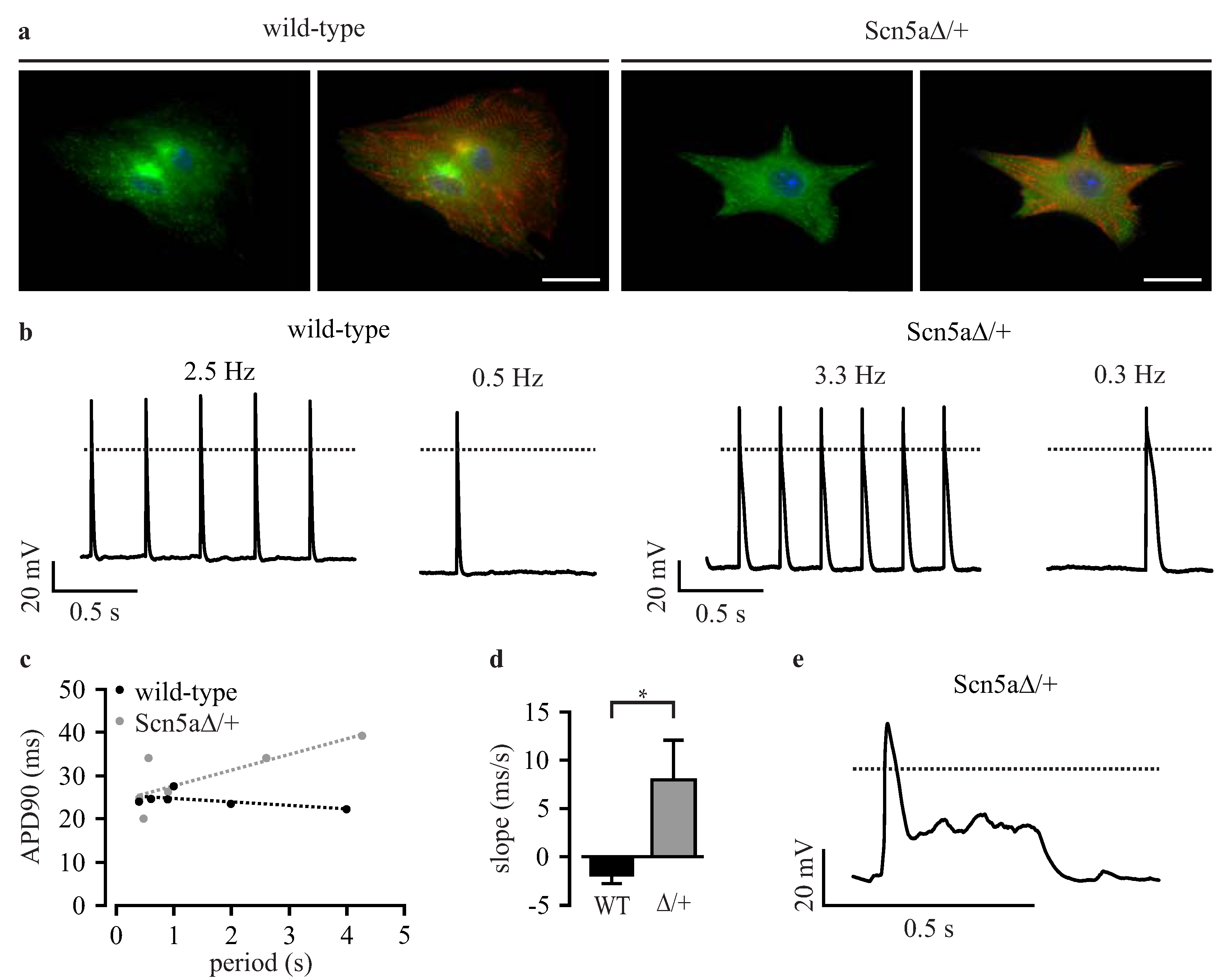
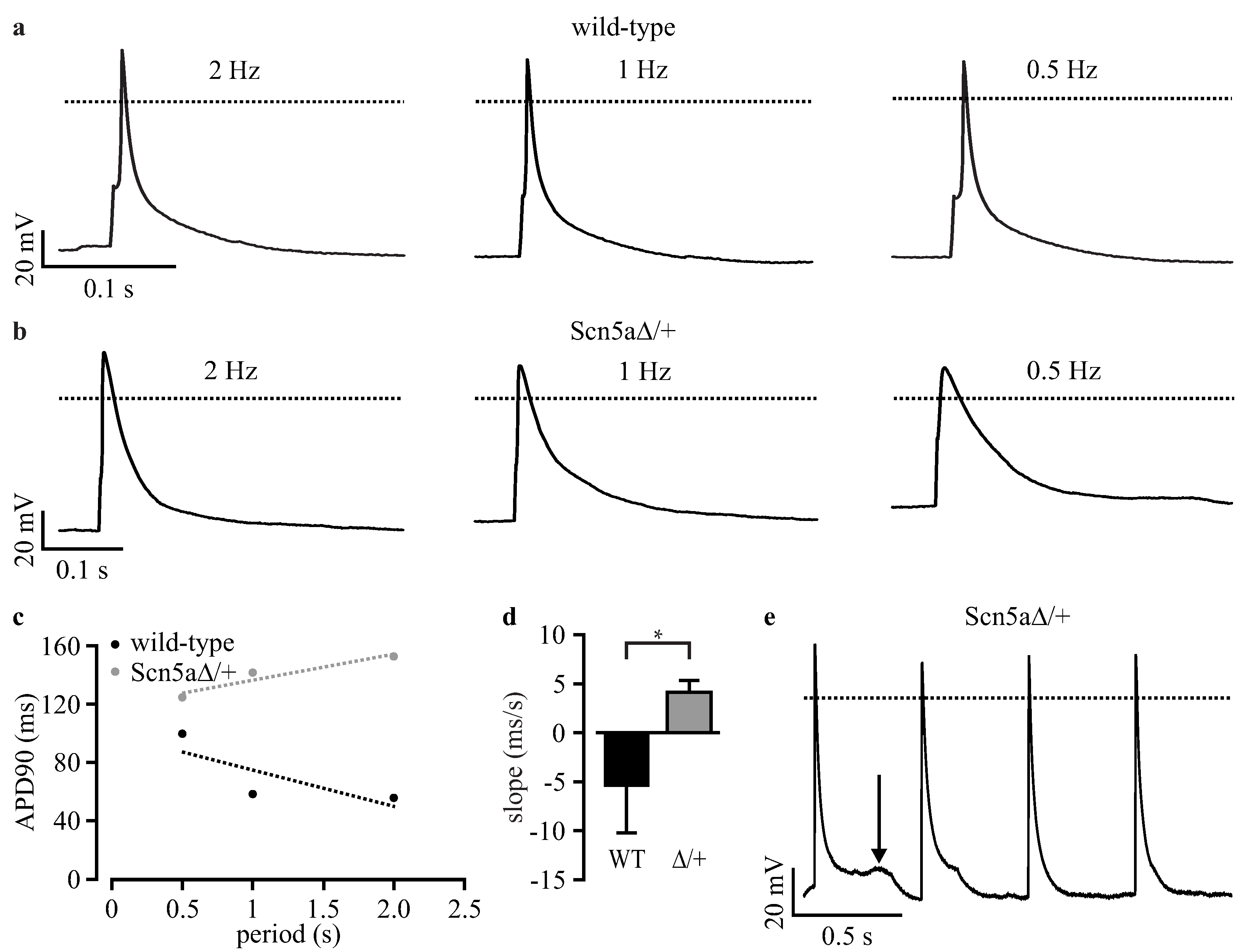
3.3. Phenotyping of Purified LQTS 3-Specific Cardiomyocytes from Scn5a∆/+ iPS Cells
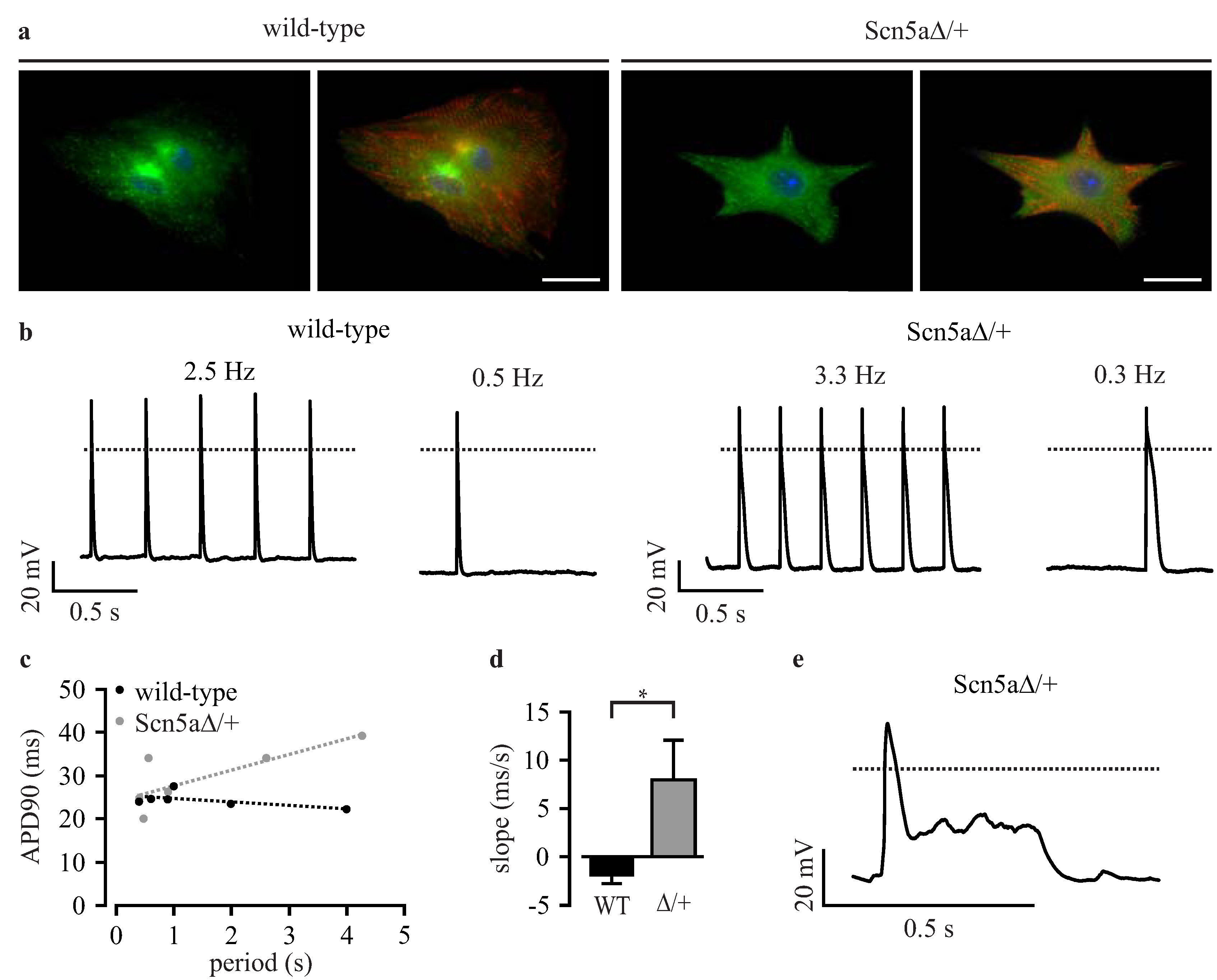

{kind=link}
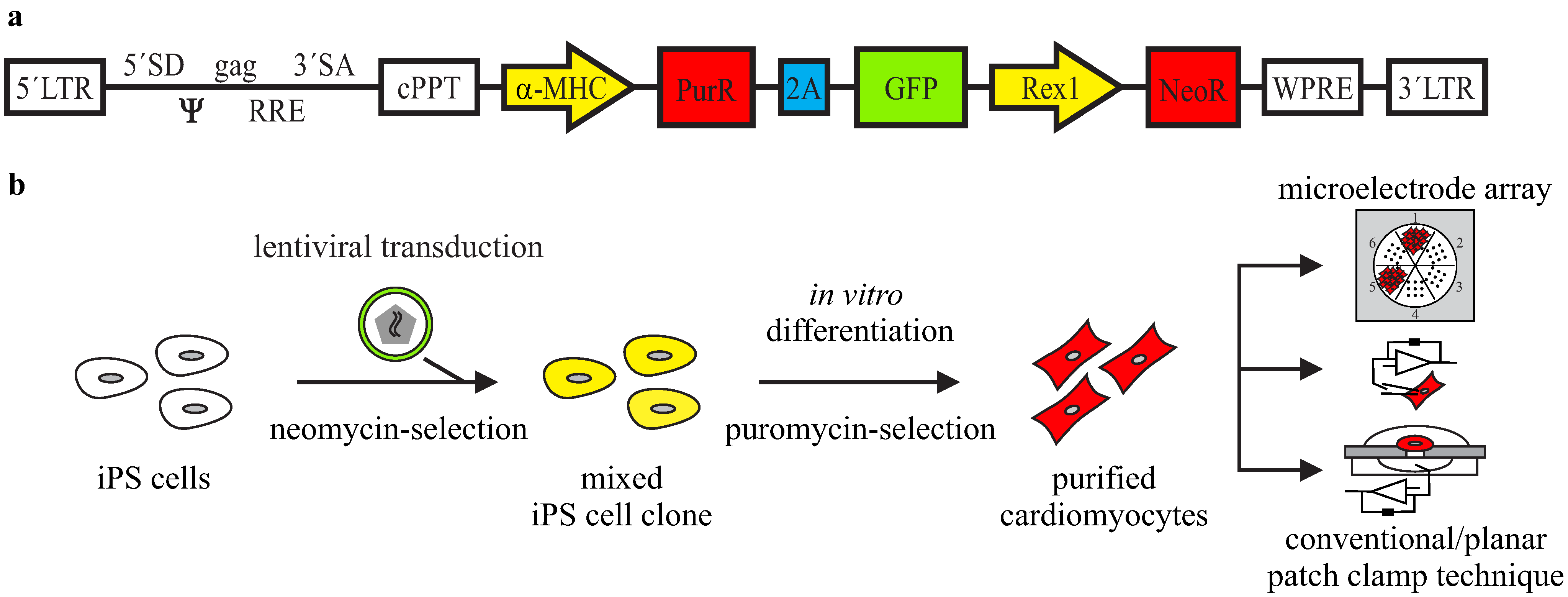{kind=link}
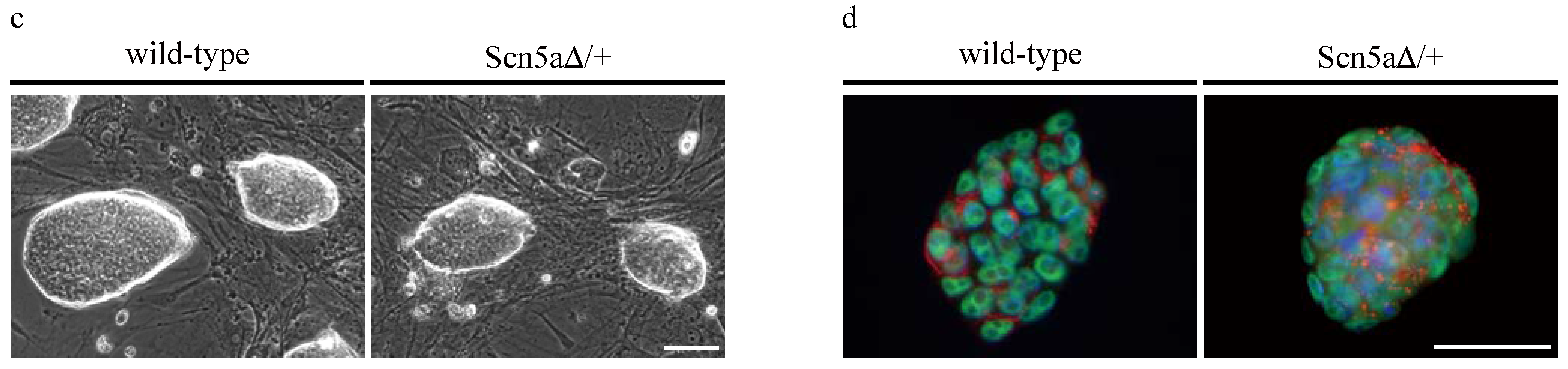{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Manual Patch Clamp | Automated Planar Patch Clamp | ||||
|---|---|---|---|---|---|---|
| Genotype | Wild-Type | Scn5aΔ/+ | p-Value | Wild-Type | Scn5aΔ/+ | p-Value |
| RMP at 1 Hz | −77.4 ± 4.9 | −74.7 ± 3.5 | 0.6589 | −69.4 ± 6.4 | −78.2 ± 3.0 | 0.2040 |
| (mV) | n = 10 | n = 17 | n = 7 | n = 9 | ||
| APA 1 Hz | 106.2 ± 5.7 | 103.3 ± 5.7 | 0.7207 | 81.1 ± 13.2 | 85.8 ± 10.1 | 0.7746 |
| (mV) | n = 10 | n = 17 | n = 7 | n = 9 | ||
| V max at 1 Hz | 93.7 ± 11.1 | 71.6 ± 9.6 | 0.1484 | 56.6 ± 15.5 | 52.6 ± 10.2 | 0.8248 |
| (V/s) | n = 10 | n = 17 | n = 7 | n = 9 | ||
| APD90 at 2 Hz | 36.2 ± 3.5 | 39.5 ± 5.1 | 0.6004 | 78.0 ± 28.7 | 64.9 ± 17.1 | 0.7019 |
| (ms) | n = 10 | n = 14 | n = 7 | n = 7 | ||
| APD90 at 1 Hz | 35.5 ± 3.3 | 45.8 ± 6.8 | 0.1913 | 70.1 ± 26.6 | 76.7 ± 20.4 | 0.8454 |
| (ms) | n = 10 | n = 15 | n = 7 | n = 9 | ||
| APD90 at 0.2 Hz (manual) or 0.5 Hz (automated) (ms) | 39.7 ± 2.2 | 46.8 ± 6.7 | 0.1761 | 69.6 ± 24.3 | 91.5 ± 25.0 | 0.5440 |
| n = 5 | n = 5 | n = 7 | n = 8 | |||
| Slope | −1.85 ± 0.73 | 7.94 ± 4.05 | 0.0287 | −5.39 ± 4.82 | 4.13 ± 1.20 | 0.0494 |
| (ms/s) | n = 10 | n = 18 | n = 7 | n = 9 | ||
| Genotype | Wild-Type | Scn5aΔ/+ | ||||
|---|---|---|---|---|---|---|
| Passage | Early Passage | Late Passage | p-Value | Early Passage | Late Passage | p-Value |
| RMP at 1 Hz | −71.8 ± 2.3 | −68.3 ± 9.7 | 0.7061 | −71.5 ± 7.3 | −67.3 ± 5.6 | 0.6593 |
| (mV) | n = 4 | n = 3 | n = 4 | n = 7 | ||
| APA 1 Hz | 105.5 ± 6.9 | 99.7 ± 16.5 | 0.7312 | 97.3 ± 9.0 | 91.3 ± 8.1 | 0.6513 |
| (mV) | n = 4 | n = 3 | n = 4 | n = 7 | ||
| V max at 1 Hz | 98.8 ± 6.8 | 88.3 ± 31.7 | 0.7215 | 81.8 ± 15.6 | 59.6 ± 17.6 | 0.4230 |
| (V/s) | n = 4 | n = 3 | n = 4 | n = 7 | ||
| APD90 at 1 Hz | 33.1 ± 3.6 | 27.7 ± 0.7 | 0.2657 | 49.1 ± 15.7 | 52.8 ± 11.3 | 0.8499 |
| (ms) | n = 4 | n = 3 | n = 4 | n = 7 | ||
| Slope | −1.10 ± 0.32 | −1.10 ± 0.57 | 1.00 | 14.75 ± 13.12 | 8.94 ± 6.68 | 0.7106 |
| (ms/s) | n = 4 | n = 3 | n = 4 | n = 8 | ||
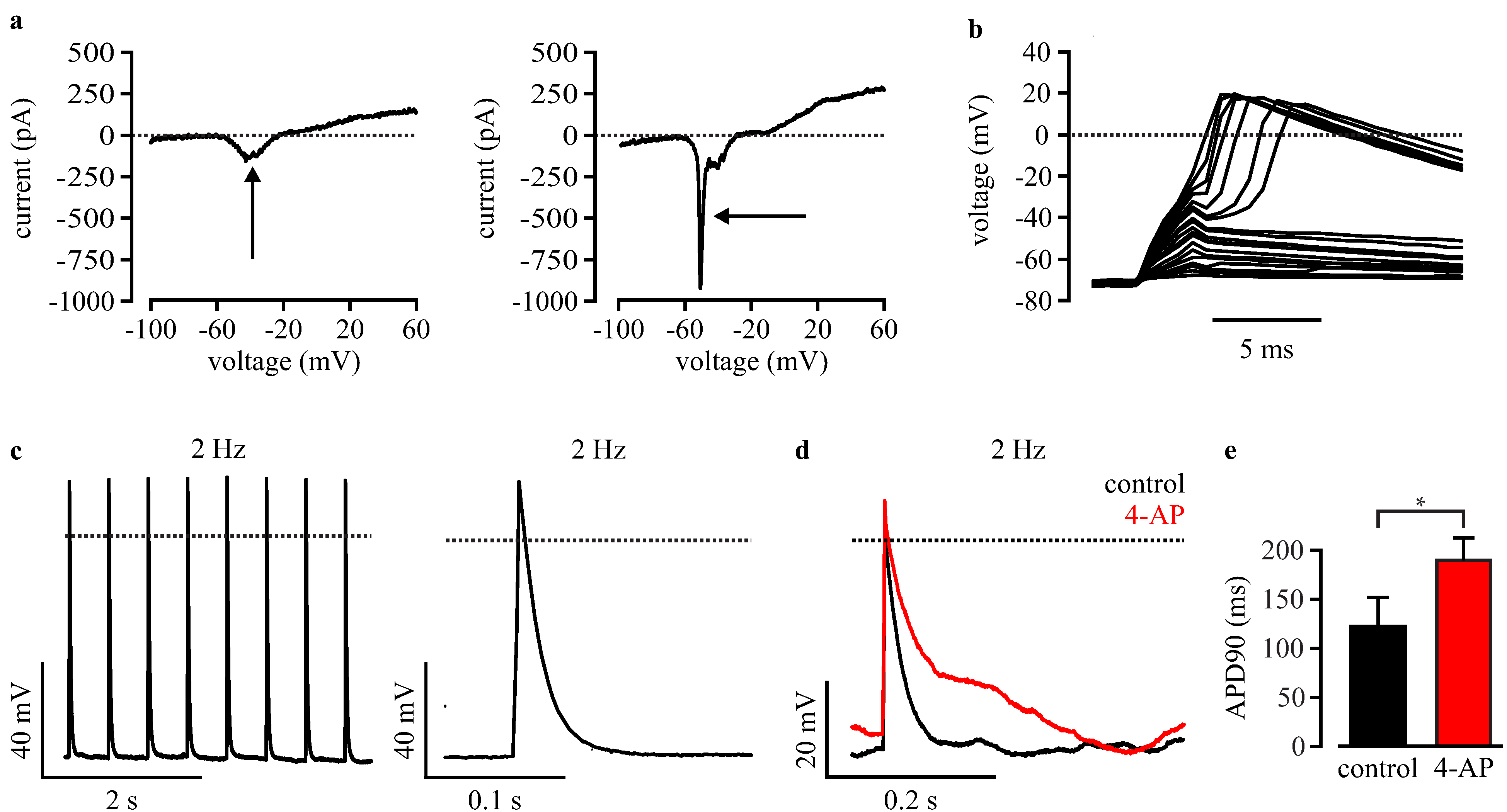
3.4. Automated Electrophysiological Investigation and AP Measurements of Purified Cardiomyocytes with a Planar Patch Clamp System
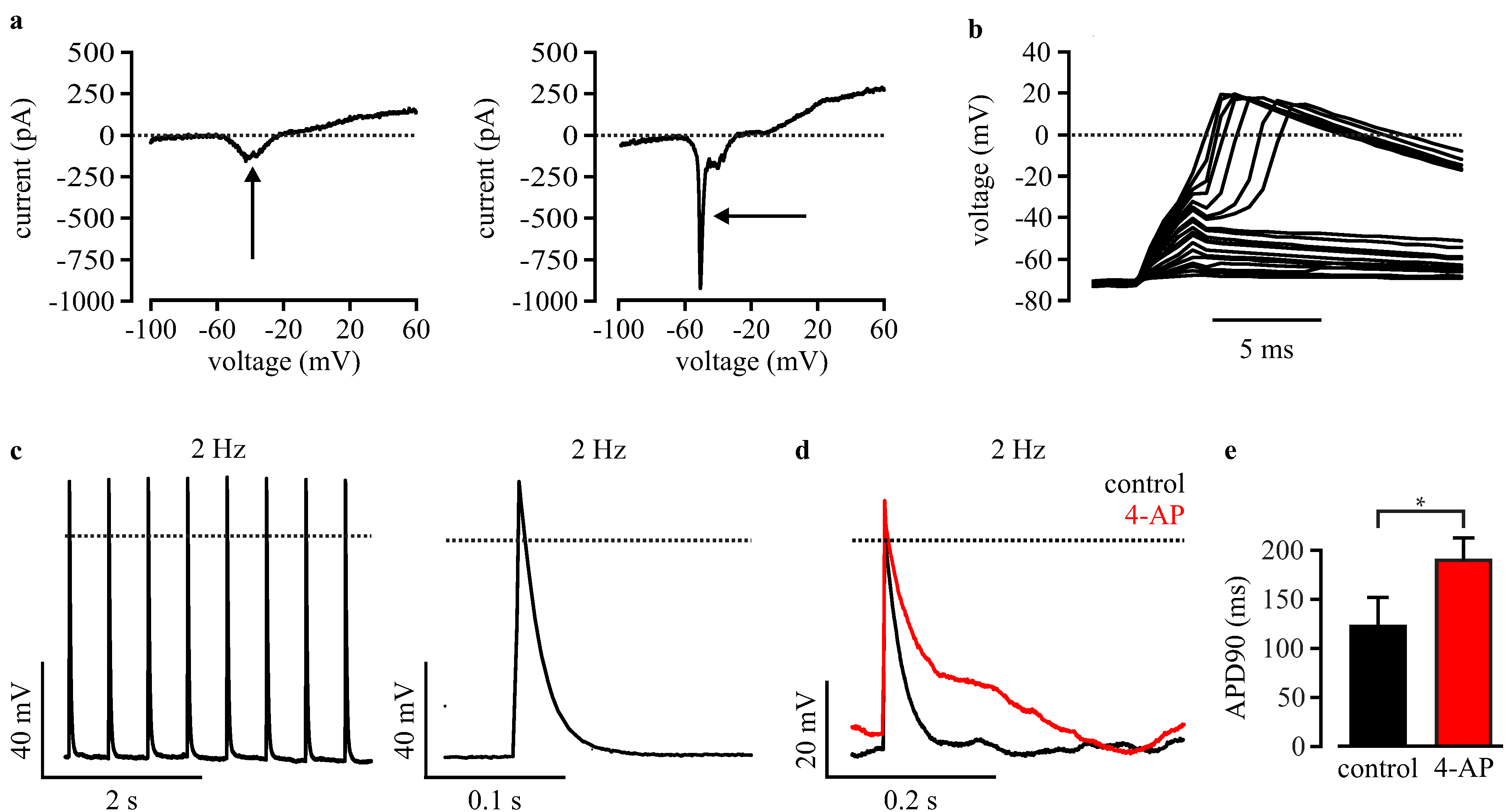
3.5. Automated Phenotypic Characterization of LQTS 3-Specific Purified Cardiomyocytes from Scn5a∆/+ iPS Cells with a Planar Patch Clamp Robot

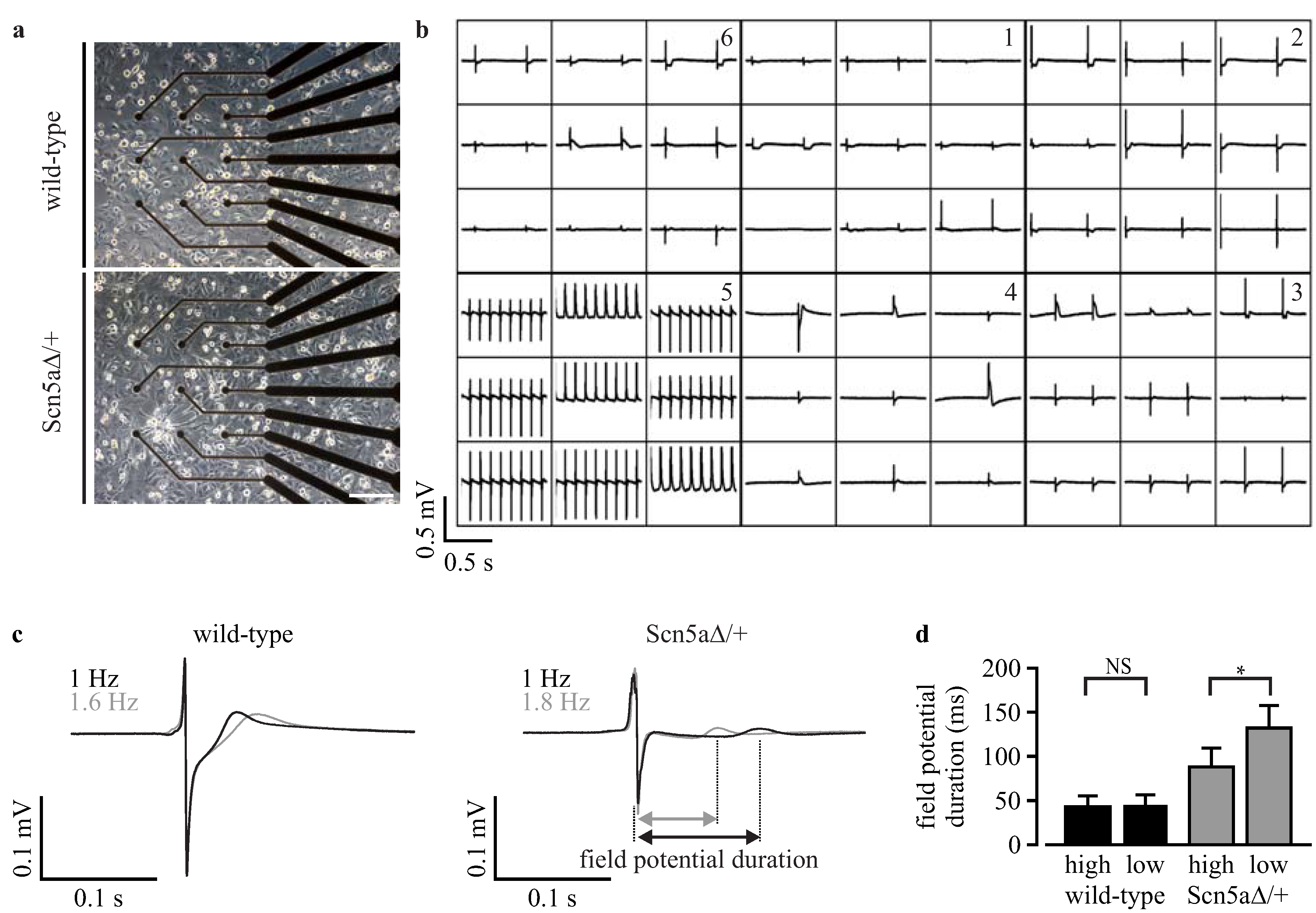
3.6. Analysis of Field Potentials from Purified iPS Cell-Derived Cardiomyocytes with Microelectrode Arrays
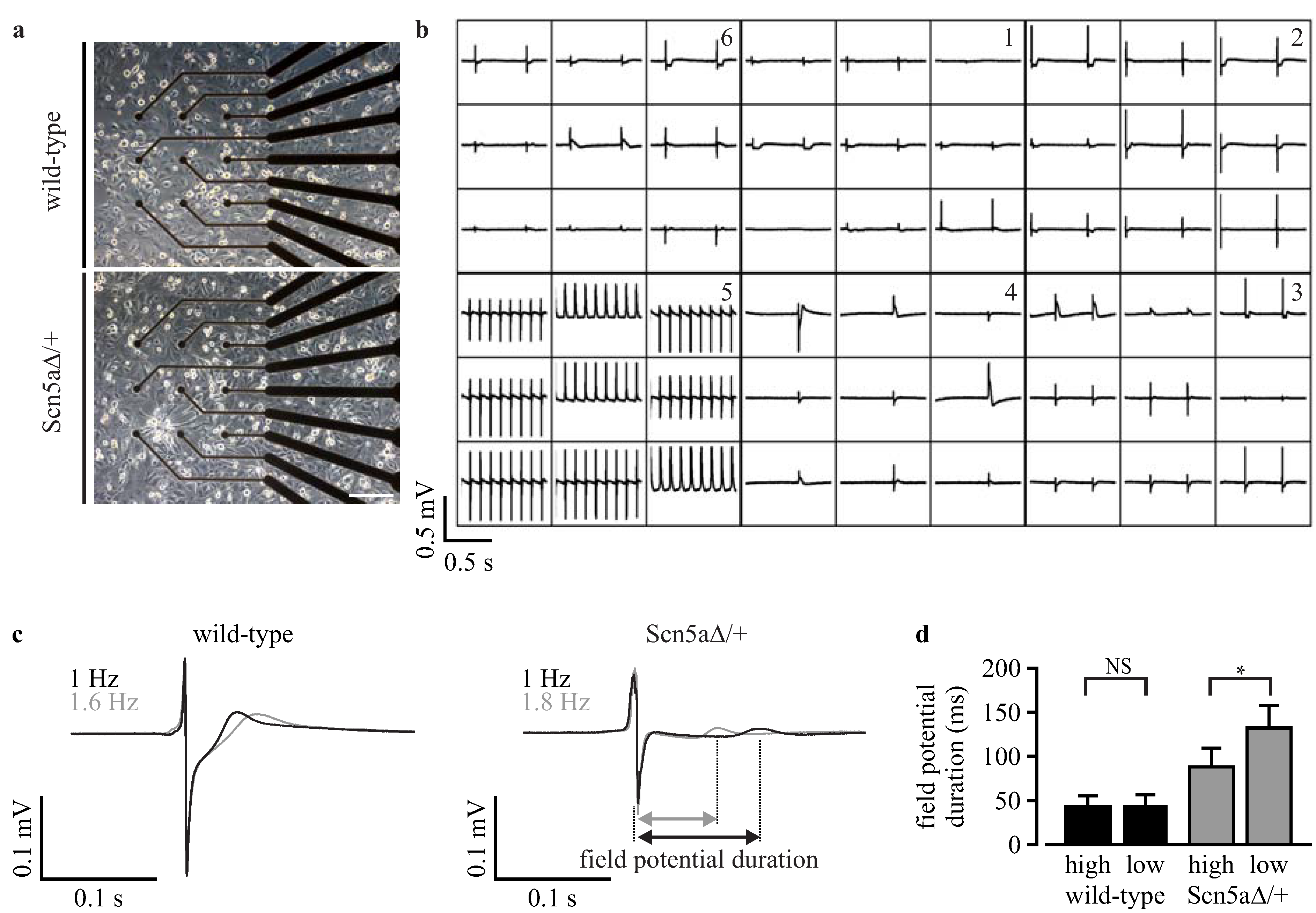
4. Discussion
4.2. Choice of a Cardiac-Specific Promoter
4.3. Automatable and Scalable Electrophysiological Screening
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schwartz, P.J.; Priori, S.G.; Spazzolini, C.; Moss, A.J.; Vincent, G.M.; Napolitano, C.; Denjoy, I.; Guicheney, P.; Breithardt, G.; Keating, M.T.; et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation 2001, 103, 89–95. [Google Scholar] [PubMed]
- Bennett, P.B.; Yazawa, K.; Makita, N.; George, A.L., Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature 1995, 376, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.; Starmer, C.F.; Grant, A.O. Multiple effects of KPQ deletion mutation on gating of human cardiac Na+ channels expressed in mammalian cells. Am. J. Physiol. 1998, 274, H1643–H1654. [Google Scholar] [PubMed]
- Charpentier, F.; Bourge, A.; Merot, J. Mouse models of SCN5A-related cardiac arrhythmias. Prog. Biophys. Mol. Biol. 2008, 98, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Malan, D.; Friedrichs, S.; Fleischmann, B.K.; Sasse, P. Cardiomyocytes obtained from induced pluripotent stem cells with long-QT syndrome 3 recapitulate typical disease-specific features in vitro. Circ. Res. 2011, 109, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Matsa, E.; Rajamohan, D.; Dick, E.; Young, L.; Mellor, I.; Staniforth, A.; Denning, C. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long qt syndrome type 2 mutation. Eur. Heart J. 2011, 32, 952–962. [Google Scholar] [CrossRef]
- Lahti, A.L.; Kujala, V.J.; Chapman, H.; Koivisto, A.P.; Pekkanen-Mattila, M.; Kerkela, E.; Hyttinen, J.; Kontula, K.; Swan, H.; Conklin, B.R.; et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis. Model. Mech. 2012, 5, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.P.; Casini, S.; van den Berg, C.W.; Hoekstra, M.; Remme, C.A.; Dambrot, C.; Salvatori, D.; Oostwaard, D.W.; Wilde, A.A.; Bezzina, C.R.; et al. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation 2012, 125, 3079–3091. [Google Scholar] [CrossRef] [PubMed]
- Egashira, T.; Yuasa, S.; Suzuki, T.; Aizawa, Y.; Yamakawa, H.; Matsuhashi, T.; Ohno, Y.; Tohyama, S.; Okata, S.; Seki, T.; et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovasc. Res. 2012, 95, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flugel, L.; Dorn, T.; Goedel, A.; Hohnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Stoelzle, S.; Haythornthwaite, A.; Kettenhofen, R.; Kolossov, E.; Bohlen, H.; George, M.; Bruggemann, A.; Fertig, N. Automated patch clamp on mesc-derived cardiomyocytes for cardiotoxicity prediction. J. Biomol. Screen. 2011, 16, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Mauritz, C.; Schwanke, K.; Reppel, M.; Neef, S.; Katsirntaki, K.; Maier, L.S.; Nguemo, F.; Menke, S.; Haustein, M.; Hescheler, J.; et al. Generation of functional murine cardiac myocytes from induced pluripotent stem cells. Circulation 2008, 118, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Huber, I.; Itzhaki, I.; Caspi, O.; Arbel, G.; Tzukerman, M.; Gepstein, A.; Habib, M.; Yankelson, L.; Kehat, I.; Gepstein, L. Identification and selection of cardiomyocytes during human embryonic stem cell differentiation. FASEB J. 2007, 21, 2551–2563. [Google Scholar] [CrossRef] [PubMed]
- Hattori, F.; Chen, H.; Yamashita, H.; Tohyama, S.; Satoh, Y.S.; Yuasa, S.; Li, W.; Yamakawa, H.; Tanaka, T.; Onitsuka, T.; et al. Nongenetic method for purifying stem cell-derived cardiomyocytes. Nat. Methods 2010, 7, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Kolossov, E.; Bostani, T.; Roell, W.; Breitbach, M.; Pillekamp, F.; Nygren, J.M.; Sasse, P.; Rubenchik, O.; Fries, J.W.; Wenzel, D.; et al. Engraftment of engineered ES cell-derived cardiomyocytes but not bm cells restores contractile function to the infarcted myocardium. J. Exp. Med. 2006, 203, 2315–2327. [Google Scholar] [CrossRef] [PubMed]
- Kita-Matsuo, H.; Barcova, M.; Prigozhina, N.; Salomonis, N.; Wei, K.; Jacot, J.G.; Nelson, B.; Spiering, S.; Haverslag, R.; Kim, C.; et al. Lentiviral vectors and protocols for creation of stable hesc lines for fluorescent tracking and drug resistance selection of cardiomyocytes. PLoS One 2009, 4, e5046. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Qian, J.J.; Yi, S.; Harding, T.C.; Tu, G.H.; VanRoey, M.; Jooss, K. Stable antibody expression at therapeutic levels using the 2A peptide. Nat. Biotechnol. 2005, 23, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, A.; Hofmann, A. Lentiviral transgenesis. Methods Mol. Biol. 2009, 530, 391–405. [Google Scholar] [PubMed]
- Hosler, B.A.; LaRosa, G.J.; Grippo, J.F.; Gudas, L.J. Expression of REX-1, a gene containing zinc finger motifs, is rapidly reduced by retinoic acid in F9 teratocarcinoma cells. Mol. Cell. Biol. 1989, 9, 5623–5629. [Google Scholar] [PubMed]
- Wobus, A.M.; Wallukat, G.; Hescheler, J. Pluripotent mouse embryonic stem cells are able to differentiate into cardiomyocytes expressing chronotropic responses to adrenergic and cholinergic agents and Ca2+ channel blockers. Differentiation 1991, 48, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Friedrichs, S.; Malan, D.; Sasse, P. Modeling long QT syndromes using induced pluripotent stem cells: Current progress and future challenges. Trends Cardiovasc. Med. 2013, 23, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Nuyens, D.; Stengl, M.; Dugarmaa, S.; Rossenbacker, T.; Compernolle, V.; Rudy, Y.; Smits, J.F.; Flameng, W.; Clancy, C.E.; Moons, L.; et al. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat. Med. 2001, 7, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Halbach, M.; Egert, U.; Hescheler, J.; Banach, K. Estimation of action potential changes from field potential recordings in multicellular mouse cardiac myocyte cultures. Cell. Physiol. Biochem. 2003, 13, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ramezani, A.; Lewis, R.; Hawley, R.G.; Thomson, J.A. High-level sustained transgene expression in human embryonic stem cells using lentiviral vectors. Stem Cells 2003, 21, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Fontes, A. Cloning technologies. Methods Mol. Biol. 2013, 997, 253–261. [Google Scholar] [PubMed]
- Willmann, C.A.; Hemeda, H.; Pieper, L.A.; Lenz, M.; Qin, J.; Joussen, S.; Sontag, S.; Wanek, P.; Denecke, B.; Schuler, H.M.; et al. To clone or not to clone? Induced pluripotent stem cells can be generated in bulk culture. PLoS One 2013, 8, e65324. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004, 2, e234. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral vectors: A look back and ahead on gene transfer technology. New Microbiol. 2013, 36, 1–22. [Google Scholar] [PubMed]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Malan, D.; Sasse, P. University of Bonn: Bonn, Germany, Unpublished work. 2014.
- Zandstra, P.W.; Bauwens, C.; Yin, T.; Liu, Q.; Schiller, H.; Zweigerdt, R.; Pasumarthi, K.B.; Field, L.J. Scalable production of embryonic stem cell-derived cardiomyocytes. Tissue Eng. 2003, 9, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.Q.; Zweigerdt, R.; Soo, S.Y.; Ngoh, Z.X.; Tham, S.C.; Wang, S.T.; Graichen, R.; Davidson, B.; Colman, A.; Sun, W.; et al. Highly enriched cardiomyocytes from human embryonic stem cells. Cytotherapy 2008, 10, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Ni, T.T.; Hou, J.; Rellinger, E.; Zhong, T.P. Promoter analysis of ventricular myosin heavy chain (VMHC) in zebrafish embryos. Dev. Dyn. 2009, 238, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Reiser, P.J.; Portman, M.A.; Ning, X.H.; Schomisch Moravec, C. Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1814–H1820. [Google Scholar] [PubMed]
- Meyer, T.; Leisgen, C.; Gonser, B.; Gunther, E. QT-screen: High-throughput cardiac safety pharmacology by extracellular electrophysiology on primary cardiac myocytes. Assay Drug Dev. Technol. 2004, 2, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Bruegmann, T.; Malan, D.; Hesse, M.; Beiert, T.; Fuegemann, C.J.; Fleischmann, B.K.; Sasse, P. Optogenetic control of heart muscle in vitro and in vivo. Nat. Methods 2010, 7, 897–900. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friedrichs, S.; Malan, D.; Voss, Y.; Sasse, P. Scalable Electrophysiological Investigation of iPS Cell-Derived Cardiomyocytes Obtained by a Lentiviral Purification Strategy. J. Clin. Med. 2015, 4, 102-123. https://doi.org/10.3390/jcm4010102
Friedrichs S, Malan D, Voss Y, Sasse P. Scalable Electrophysiological Investigation of iPS Cell-Derived Cardiomyocytes Obtained by a Lentiviral Purification Strategy. Journal of Clinical Medicine. 2015; 4(1):102-123. https://doi.org/10.3390/jcm4010102
Chicago/Turabian StyleFriedrichs, Stephanie, Daniela Malan, Yvonne Voss, and Philipp Sasse. 2015. "Scalable Electrophysiological Investigation of iPS Cell-Derived Cardiomyocytes Obtained by a Lentiviral Purification Strategy" Journal of Clinical Medicine 4, no. 1: 102-123. https://doi.org/10.3390/jcm4010102
APA StyleFriedrichs, S., Malan, D., Voss, Y., & Sasse, P. (2015). Scalable Electrophysiological Investigation of iPS Cell-Derived Cardiomyocytes Obtained by a Lentiviral Purification Strategy. Journal of Clinical Medicine, 4(1), 102-123. https://doi.org/10.3390/jcm4010102