
Hyponatremia in Patients with Cirrhosis of the Liver


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Epidemiology and Prognostic Importance of Hyponatremia in Cirrhosis
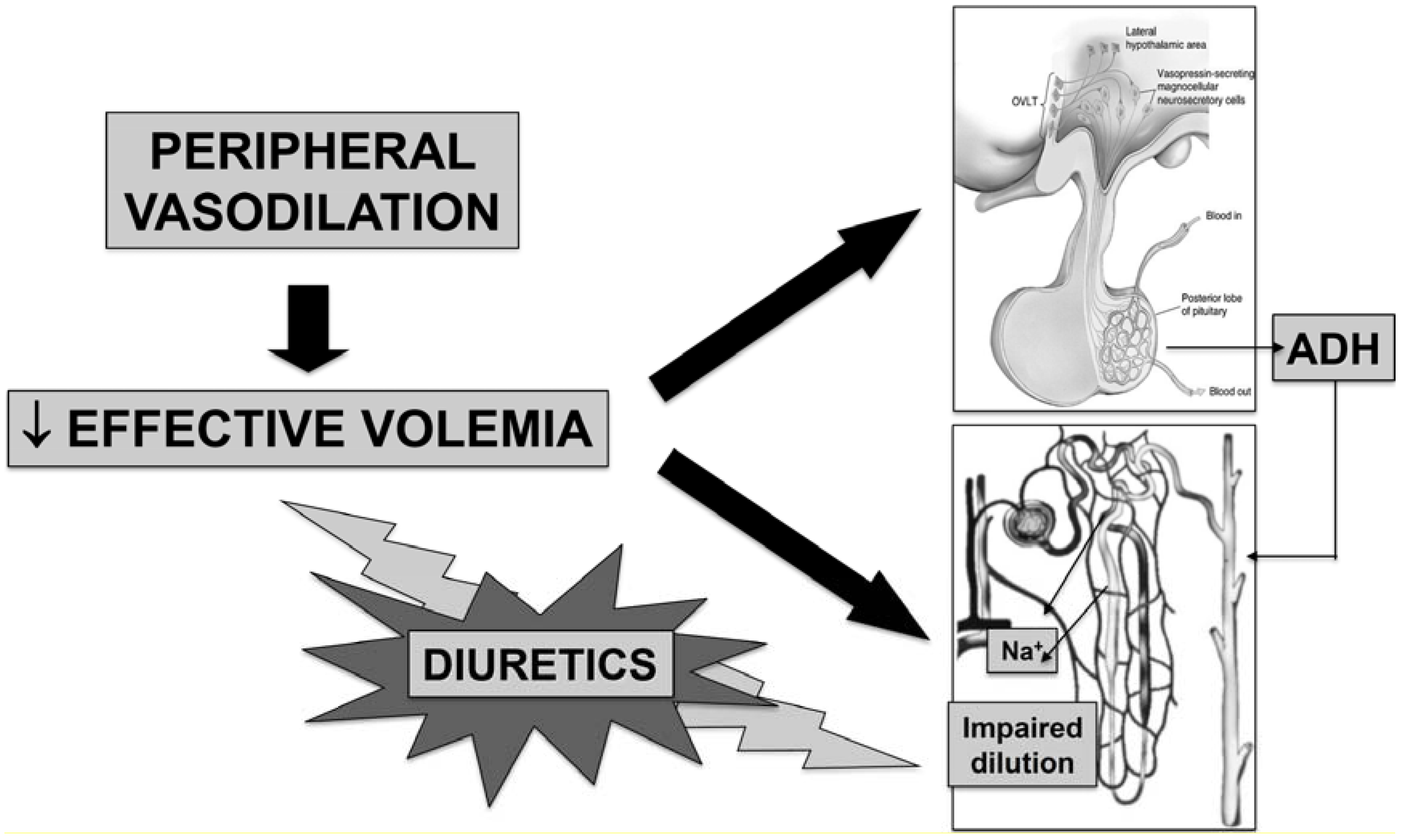
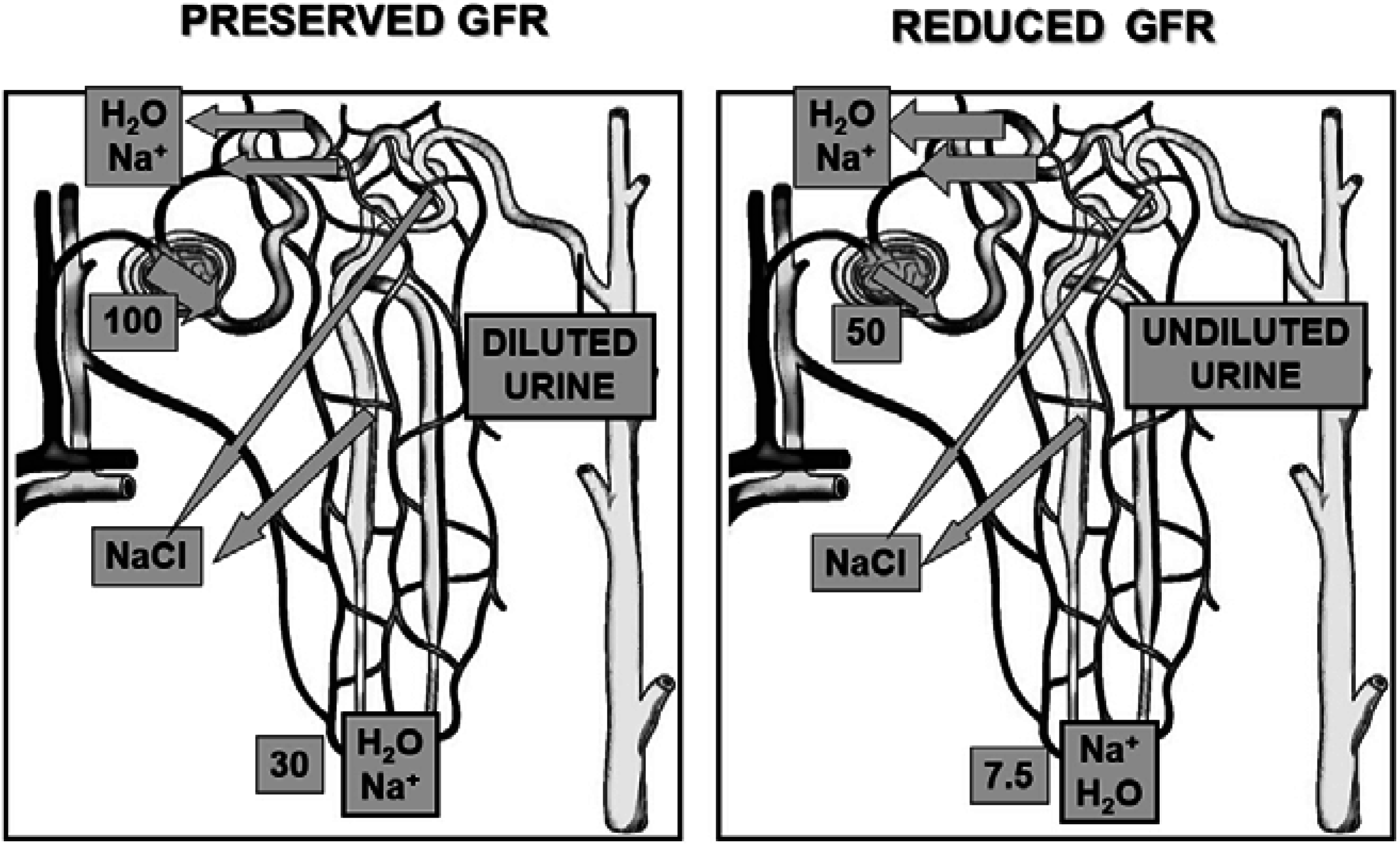
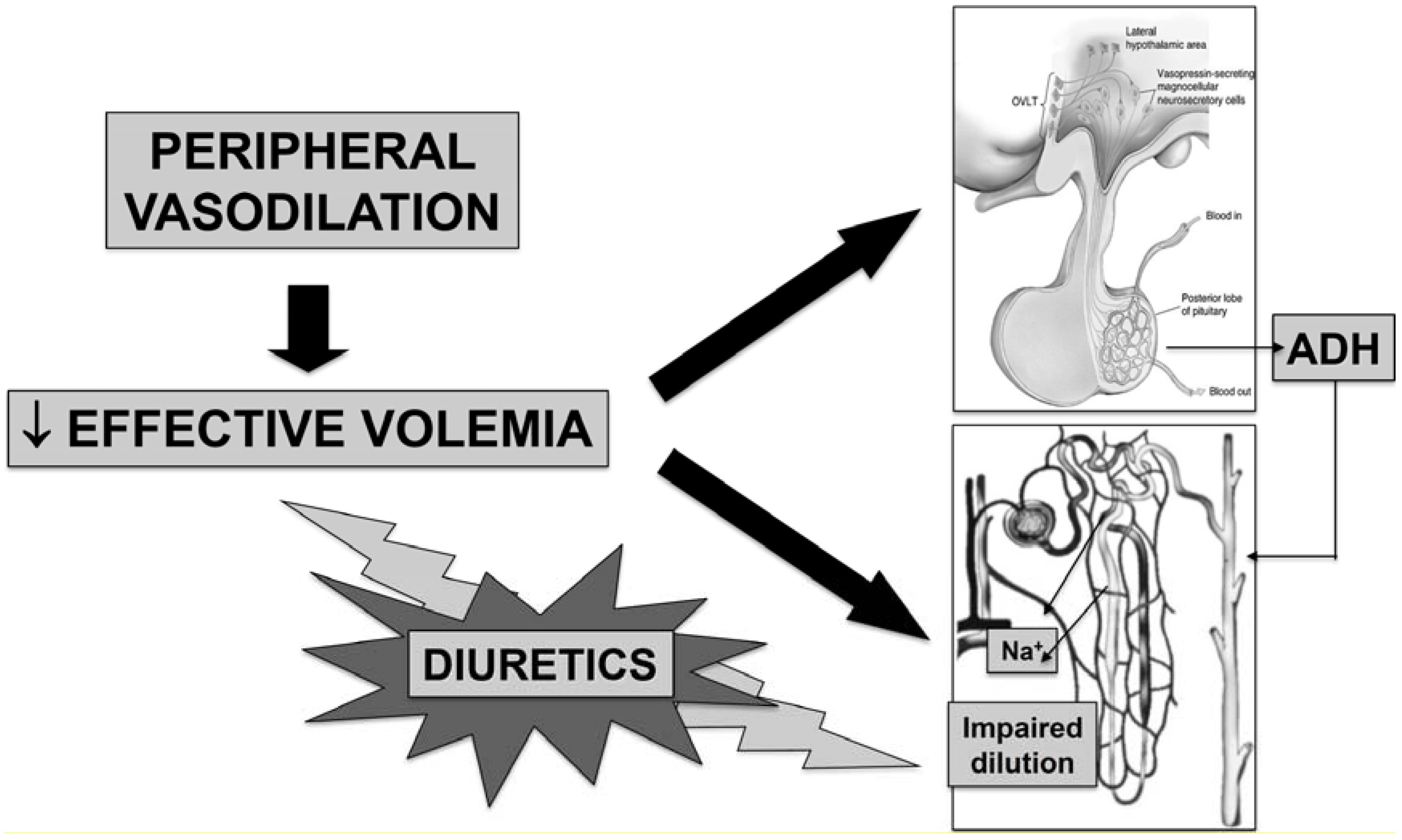
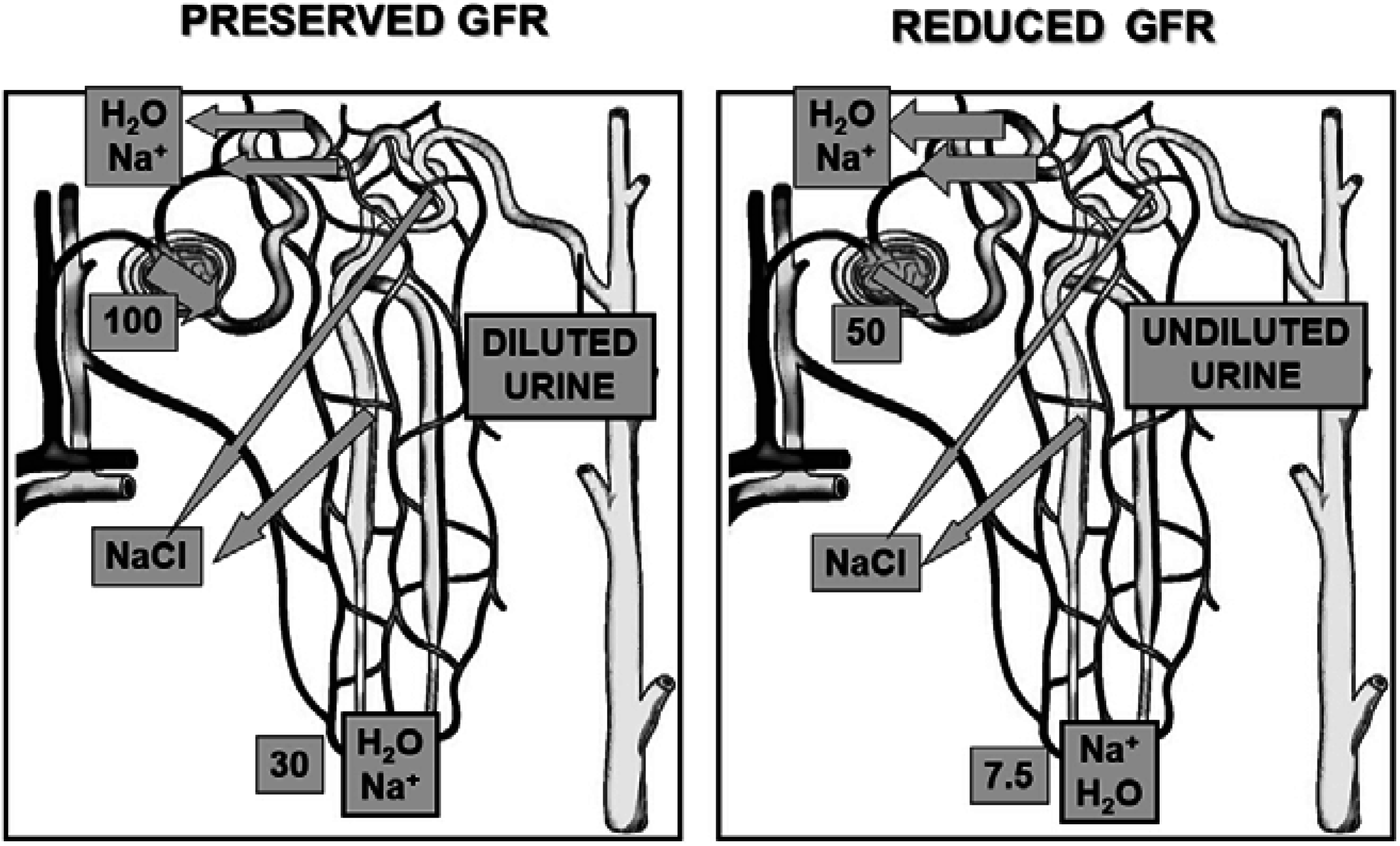
2. Pathophysiology
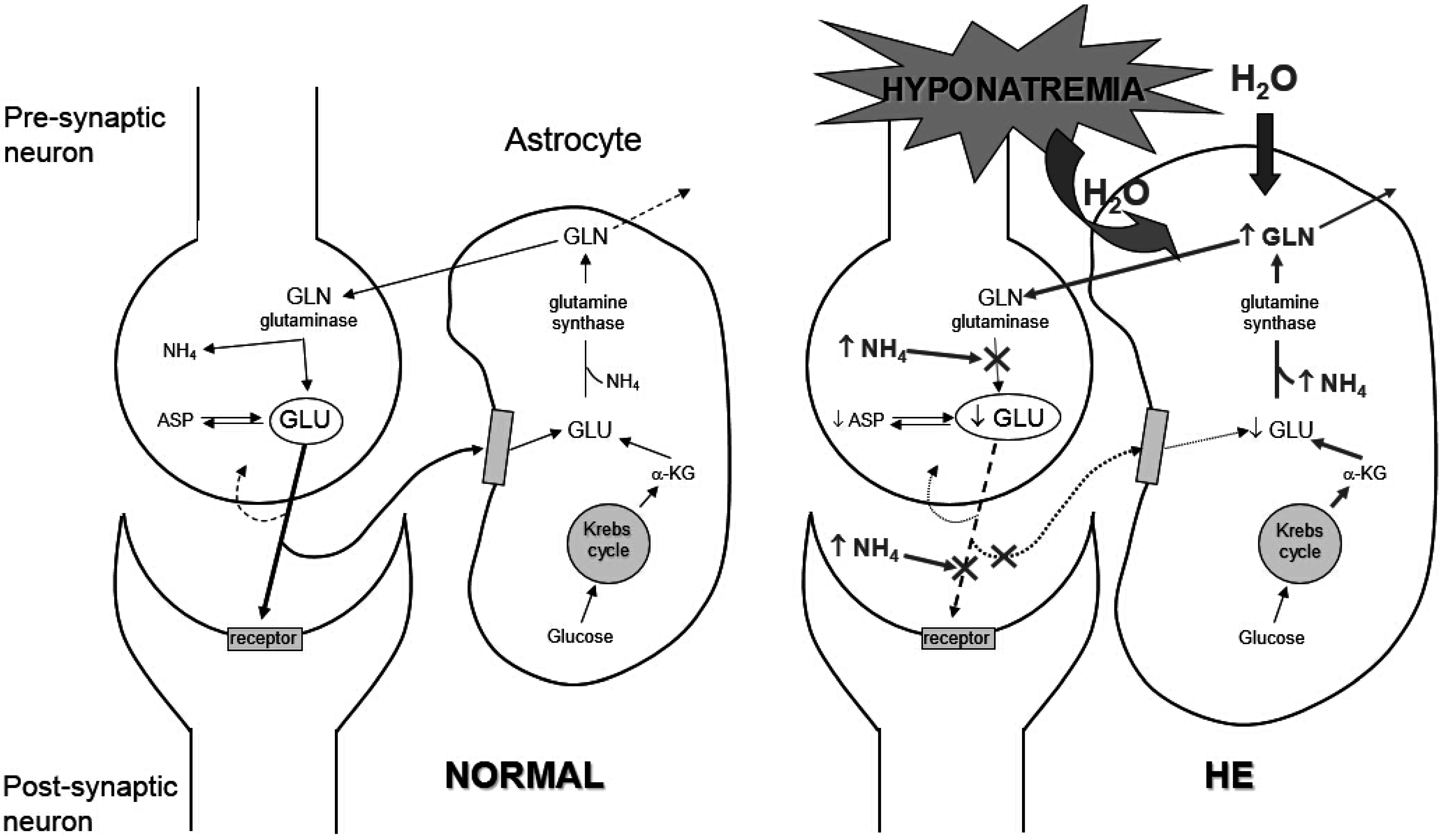
3. Clinical Features of Hyponatremia in Cirrhosis
3.1. Clinical Types
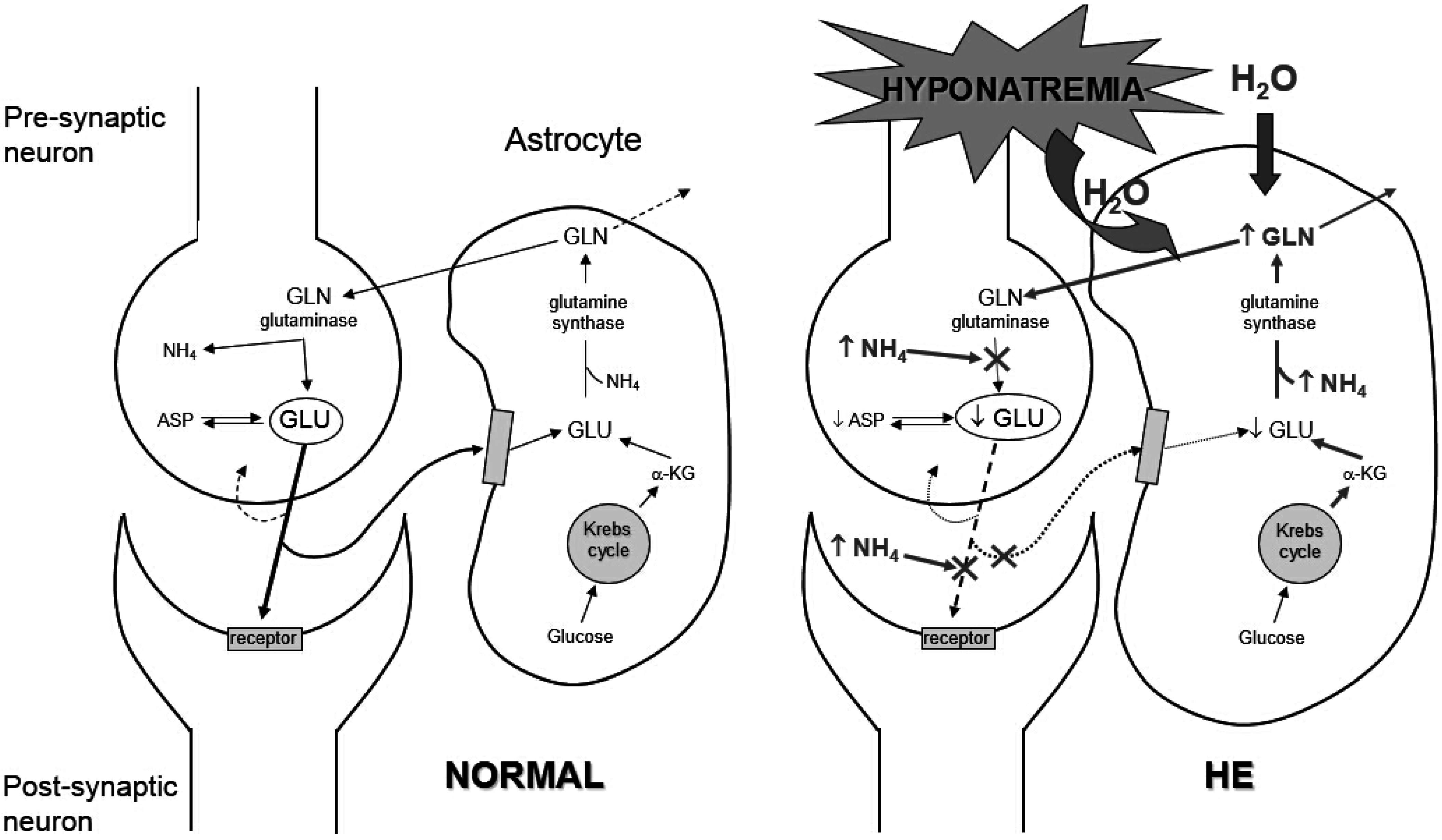
3.2. Clinical Manifestations
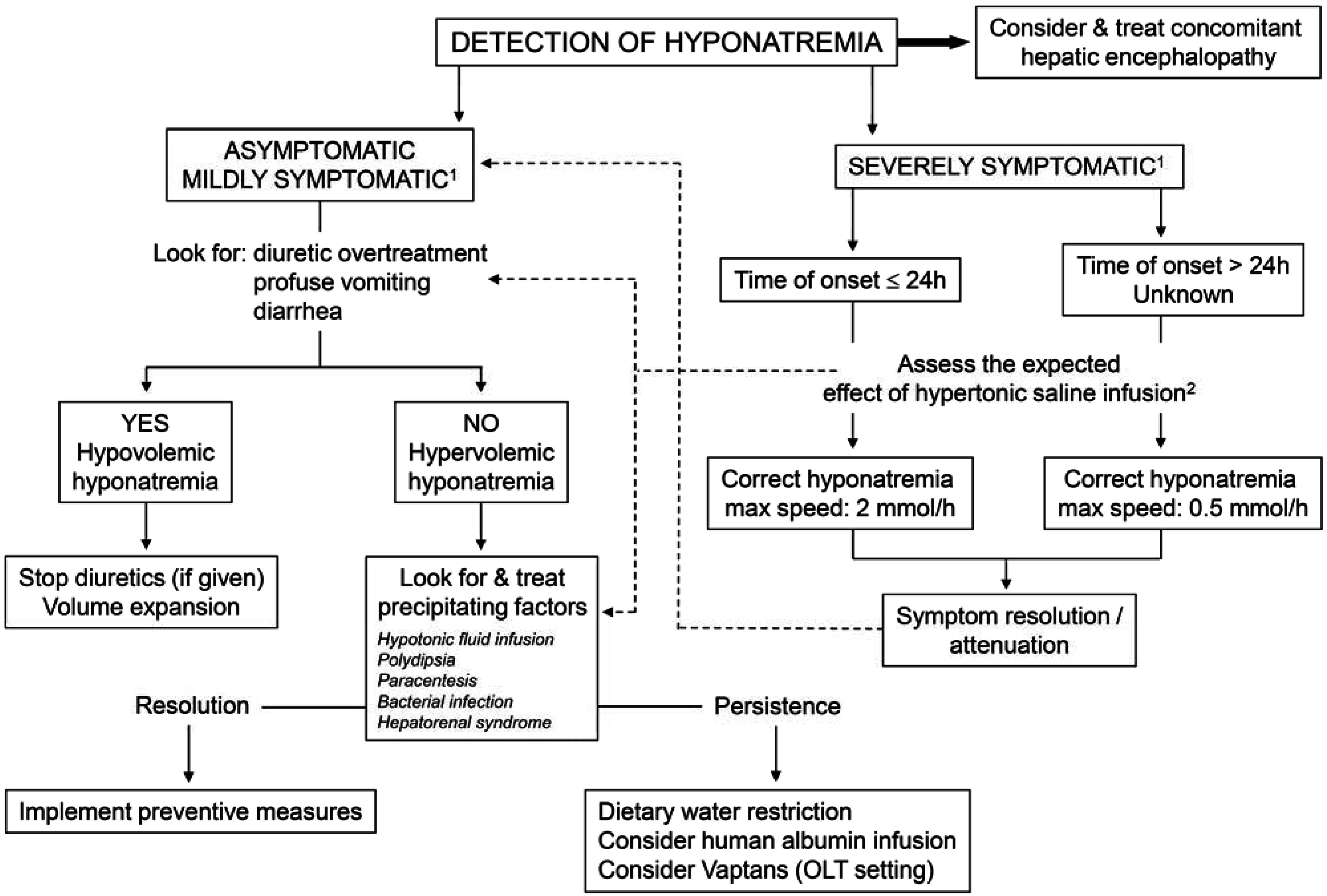
4. Management of Hyponatremia in Cirrhosis
4.1. Prevention
- (1)
- Prevention of hypovolemic hyponatremia mainly consists in avoiding marked fluid losses in excess of sodium. The most frequent cause of hypovolemic hyponatremia in patients with cirrhosis and ascites is represented by diuretic overtreatment. Therefore, great care has to be paid in avoiding a markedly negative fluid balance. In practice, daily body weight reduction under diuretic treatment should not exceed 500–800 g [38]. Patients with peripheral edema appear to be protected from these effects because of the preferential mobilization of edema and may safely undergo diuresis at a more rapid rate (up to 2 kg/day) until edema disappears [38].
- (2)
- In the majority of patients with advanced cirrhosis, hypervolemic hyponatremia develops in the setting of expanded extracellular fluid volume secondary to renal fluid retention in excess of water with respect to sodium. There are some measures that can help in preventing hypervolemic hyponatremia. First, it is inadvisable to administer hypotonic fluids to patient with ascites and impaired renal perfusion due to altered renal water metabolism. This includes the utilization of branched chain amino acids and glucose containing solution. Second, in case of complications that can acutely reduce effective volemia, there are established treatments aimed at preventing renal impairment and, therefore, dilutional hyponatremia. This is the case of post-paracentesis circulatory dysfunction (PPCD), which results from a further arterial vasodilation [39]. This complication may be effectively prevented with the administration of 8 g of human albumin/L of tapped ascites after the completion of large-volume paracentesis (>5 L) [29]. Indeed, this procedure not only prevents PPCD, but also its consequences, such hyponatremia and death [40]. Another condition that often induces an acute impairment of renal function, due to an infection-induced storm of pro-inflammatory cytokines, is represented by spontaneous bacterial peritonitis [41]. An albumin load (1.5 g/kg of body weight at diagnosis plus 1 g/kg b.w. on day three) in addition to antibiotic treatment significantly reduces the incidence of renal impairment and in-hospital and 3 month mortality [42]. Finally, treatment of hepatorenal syndrome with terlipressin and albumin also lead to an improvement of serum sodium concentration [43,44]. Indeed, terlipressin can induce hyponatremia because of V2 receptor-mediated water reabsorption in the collecting duct. However, this has been reported in patients with cirrhosis without renal failure receiving this drug because of variceal bleeding [45]. In hepatorenal syndrome the amelioration of effective volemia induced by V1 receptor stimulation by terlipressin overcomes its potential effects on V2 receptors [45].
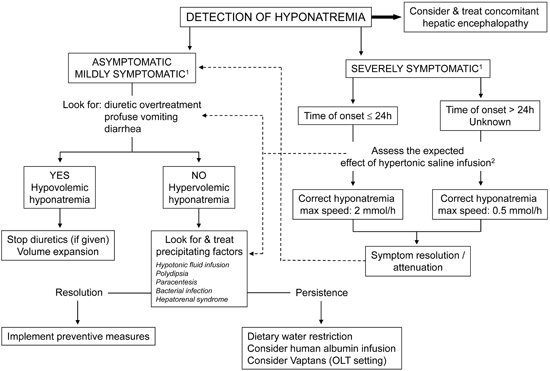
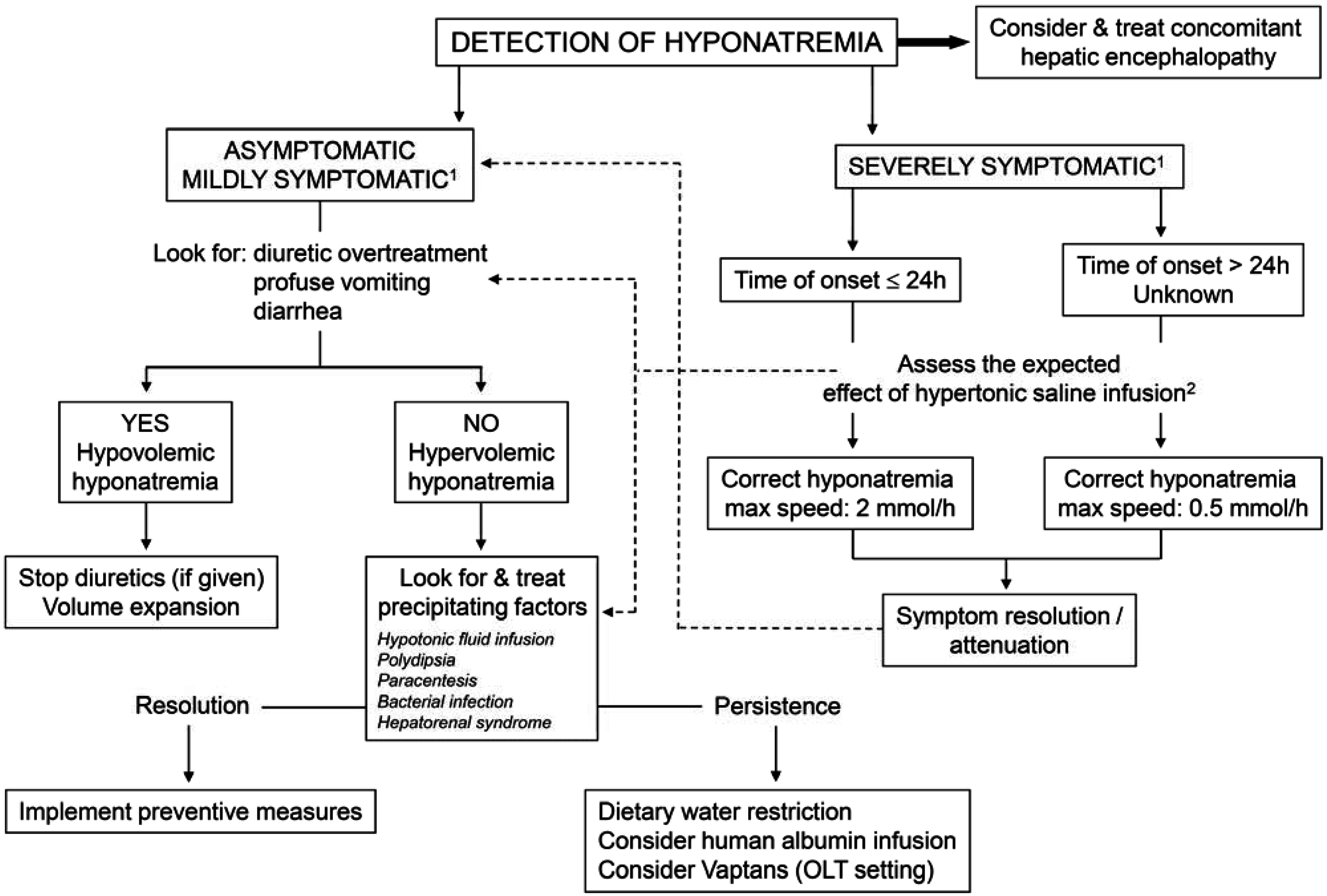
4.2. Treatment (Figure 4)
- (1)
- Hypovolemic hyponatremia cannot always be easily recognized in cirrhosis. Thus, the assessment of the clinical context where hyponatremia has ensued is crucial. The management consists of administration of normal saline and of identification and removal of the precipitating factor, which is often represented by diuretic overtreatment [3].
- (2)
- The management of hypervolemic hyponatremia, persisting after the correction of possible precipitating events or apparently spontaneous, may be difficult.
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ginés, P.; Berl, T.; Bernardi, M.; Bichet, D.G.; Hamon, G.; Jiménez, W.; Liard, J.F.; Martin, P.Y.; Schrier, R.W. Hyponatremia in cirrhosis: From pathogenesis to treatment. Hepatology 1998, 28, 851–864. [Google Scholar] [CrossRef]
- Angeli, P.; Wong, F.; Watson, H.; Gines, P.; CAPPS Investigators. Hyponatremia in cirrhosis: Results of a patient population survey. Hepatology 2006, 44, 1535–1542. [Google Scholar] [CrossRef]
- Ginès, P.; Guevara, M. Hyponatremia in cirrhosis: Pathogenesis, clinical significance, and management. Hepatology 2008, 48, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Pugh, R.N.; Murray-Lyon, I.M.; Dawson, J.L.; Pietroni, M.C.; Williams, R. Transection of the oesophagus for bleeding oesophageal varices. Br. J. Surg. 1973, 60, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Ginès, A.; Escorsell, A.; Ginès, P.; Saló, J.; Jiménez, W.; Inglada, L.; Navasa, M.; Clària, J.; Rimola, A.; Arroyo, V.; et al. Incidence, predictive factors, and prognosis of the hepatorenal syndrome in cirrhosis with ascites. Gastroenterology 1993, 105, 229–236. [Google Scholar]
- Guevara, M.; Baccaro, M.E.; Torre, A.; Gómez-Ansón, B.; Ríos, J.; Torres, F.; Rami, L.; Monté-Rubio, G.C.; Martín-Llahí, M.; Arroyo, V.; et al. Hyponatremia is a risk factor of hepatic encephalopathy in patients with cirrhosis: A prospective study with time-dependent analysis. Am. J. Gastroenterol. 2009, 104, 1382–1389. [Google Scholar] [CrossRef]
- Sersté, T.; Gustot, T.; Rautou, P.E.; Francoz, C.; Njimi, H.; Durand, F.; Valla, D.; Lebrec, D.; Moreau, R. Severe hyponatremia is a better predictor of mortality than MELDNa in patients with cirrhosis and refractory ascites. J. Hepatol. 2012, 57, 274–280. [Google Scholar] [CrossRef]
- Kamath, P.S.; Wiesner, R.H.; Malinchoc, M.; Kremers, W.; Therneau, T.M.; Kosberg, C.L.; D’Amico, G.; Dickson, E.R.; Kim, W.R. A model to predict survival in patients with end-stage liver disease. Hepatology 2001, 33, 464–470. [Google Scholar] [CrossRef]
- Biggins, S.W.; Kim, W.R.; Terrault, N.A.; Saab, S.; Balan, V.; Schiano, T.; Benson, J.; Therneau, T.; Kremers, W.; Wiesner, R.; et al. Evidence-based incorporation of serum sodium concentration into MELD. Gastroenterology 2006, 130, 1652–1660. [Google Scholar] [CrossRef]
- Kim, W.R.; Biggins, S.W.; Kremers, W.K.; Wiesner, R.H.; Kamath, P.S.; Benson, J.T.; Edwards, E.; Therneau, T.M. Hyponatremia and mortality among patients on the liver-transplant waiting list. N. Engl. J. Med. 2008, 359, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Biselli, M.; Gitto, S.; Gramenzi, A.; Di Donato, R.; Brodosi, L.; Ravaioli, M.; Grazi, G.L.; Pinna, A.D.; Andreone, P.; Bernardi, M. Six score systems to evaluate candidates with advanced cirrhosis for orthotopic liver transplant: Which is the winner? Liver Transplant. 2010, 16, 964–973. [Google Scholar] [CrossRef]
- Londoño, M.C.; Cárdenas, A.; Guevara, M.; Quintó, L.; de Las Heras, D.; Navasa, M.; Rimola, A.; Garcia-Valdecasas, J.C.; Arroyo, V.; Ginès, P. MELD score and serum sodium in the prediction of survival of patients with cirrhosis awaiting liver transplantation. Gut 2007, 56, 1283–9120. [Google Scholar] [CrossRef]
- Abbasoglu, O.; Goldstein, R.M.; Vodapally, M.S.; Jennings, L.W.; Levy, M.F.; Husberg, B.S.; Klintmalm, G.B. Liver transplantation in hyponatremic patients with emphasis on central pontine myelinolysis. Clin. Transplant. 1998, 12, 263–269. [Google Scholar]
- Londoño, M.C.; Guevara, M.; Rimola, A.; Navasa, M.; Taurà, P.; Mas, A.; García-Valdecasas, J.C.; Arroyo, V.; Ginès, P. Hyponatremia impairs early post-transplantation outcome in patients with cirrhosis undergoing liver transplantation. Gastroenterology 2006, 130, 1135–1143. [Google Scholar] [CrossRef]
- Nielsen, S.; Frokiaer, J.; Marples, D.; Kwon, T.H.; Agre, P.; Knepper, M.A. Aquaporins in the kidney: From molecules to medicine. Physiol. Rev. 2002, 82, 205–244. [Google Scholar] [PubMed]
- Bichet, D.G. Posterior pituitary. In The Pituitary, 1st ed.; Melmed, S., Ed.; Blackwell Scientific Publications, Inc.: Cambridge, MA, USA, 1995; pp. 277–306. [Google Scholar]
- Berl, T.; Schrier, R.W. Disorders of water metabolism. In Renal and Electrolyte Disorders, 5th ed.; Schrier, R.W., Ed.; Lippincott-Raven: Boston, MA, USA, 1997; pp. 1–71. [Google Scholar]
- Oliet, S.H.; Bourque, C.W. Mechano-sensitive channels transduce osmosensitivity in supraoptic neurons. Nature 1993, 364, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.S.; Bhat, R.V.; Preston, G.M.; Guggino, W.B.; Baraban, J.M.; Agre, P. Molecular characterisation of an aquaporin cDNA from brain: Candidate osmoreceptor and regulator of water balance. Proc. Natl. Acad. Sci. USA 1994, 91, 13052–13056. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W.; Arroyo, V.; Bernardi, M.; Epstein, M.; Henriksen, J.H.; Rodés, J. Peripheral vasodilation hypothesis: A proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology 1988, 8, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y.; Groszmann, R.J. The hyperdynamic circulation of chronic liver diseases: From the patient to the molecule. Hepatology 2006, 43 (Suppl. S1), 121–131. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.Y.; Ohara, M.; Ginès, P.; Xu, D.L.; St John, J.; Niederberger, M.; Schrier, R.W. Nitric oxide synthase (NOS) inhibition for one week improves renal sodium and water excretion in cirrhotic rats with ascites. J. Clin. Investig. 1998, 101, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Esteva-Font, C.; Baccaro, M.E.; Fernández-Llama, P.; Sans, L.; Guevara, M.; Ars, E.; Jiménez, W.; Arroyo, V.; Ballarín, J.A.; Ginès, P. Aquaporin-1 and aquaporin-2 urinary excretion in cirrhosis: Relationship with ascites and hepatorenal syndrome. Hepatology 2006, 44, 1555–1563. [Google Scholar] [CrossRef]
- Asahina, Y.; Izumi, N.; Enomoto, N.; Sasaki, S.; Fushimi, K.; Marumo, F.; Sato, C. Increased gene expression of water channel in cirrhotic rat kidneys. Hepatology 1995, 21, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Llama, P.; Jimenez, W.; Bosch-Marce, M.; Arroyo, V.; Nielsen, S.; Knepper, M.A. Dysregulation of renal aquaporins and Na-Cl cotransporter in CCl4-induced cirrhosis. Kidney Int. 2000, 58, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Verbalis, J.G. Whole-body volume regulation and escape from antidiuresis. Am. J. Med. 2006, 119 (Suppl. S1), 21–29. [Google Scholar]
- Schedl, H.P.; Bartter, F.C. An explanation for and experimental correction of the abnormal water diuresis in cirrhosis. J. Clin. Investig. 1960, 39, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-del-Arbol, L.; Monescillo, A.; Jimenéz, W.; Garcia-Plaza, A.; Arroyo, V.; Rodés, J. Paracentesis-induced circulatory dysfunction: Mechanism and effect on hepatic hemodynamics in cirrhosis. Gastroenterology 1997, 113, 579–586. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL clinical practice guidelines on the management of ascites, spontaneous bacterial peritonitis, and hepatorenal syndrome in cirrhosis. J. Hepatol. 2010, 53, 397–417. [Google Scholar]
- Adrogué, H.J.; Madias, N.E. Hyponatremia. N. Engl. J. Med. 2000, 342, 1581–1589. [Google Scholar] [CrossRef]
- Córdoba, J; Mìnguez, B. Hepatic encephalopathy. Semin. Liver Dis. 2008, 28, 70–80. [Google Scholar] [CrossRef]
- Häussinger, D. Low grade cerebral edema and the pathogenesis of hepatic encephalopathy in cirrhosis. Hepatology 2006, 43, 1187–1190. [Google Scholar] [CrossRef]
- Restuccia, T.; Gómez-Ansón, B.; Guevara, M.; Alessandria, C.; Torre, A.; Alayrach, M.E.; Terra, C.; Martín, M.; Castellví, M.; Rami, L.; et al. Effects of dilutional hyponatremia on brain organic osmolytes and water content in patients with chirrosis. Hepatology 2004, 39, 1613–1622. [Google Scholar] [CrossRef]
- Heins, J.; Zwingmann, C. Organic osmolytes in hyponatremia and ammonia toxicity. Metab. Brain Dis. 2010, 25, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Córdoba, J.; García-Martinez, R.; Simón-Talero, M. Hyponatremic and hepatic encephalopathies: Similarities, differences and coexistence. Metab. Brain Dis. 2010, 25, 73–80. [Google Scholar] [CrossRef]
- Amodio, P.; Del Piccolo, F.; Pettenò, E.; Mapelli, D.; Angeli, P.; Iemmolo, R.; Muraca, M.; Musto, C.; Gerunda, G.; Rizzo, C.; et al. Prevalence and prognostic value of quantified electroencephalogram (EEG) alterations in cirrhotic patients. J. Hepatol. 2001, 35, 37–45. [Google Scholar] [CrossRef]
- Yun, B.C.; Kim, W.R.; Benson, J.T.; Biggins, S.W.; Therneau, T.M.; Kremers, W.K.; Rosen, C.B.; Klintmalm, G.B. Impact of pretransplant hyponatremia on outcome following liver transplantation. Hepatology 2009, 49, 1610–1605. [Google Scholar] [CrossRef] [PubMed]
- Pockros, P.J.; Reynolds, T.B. Rapid diuresis in patients with ascites from chronic liver disease: The importance of peripheral edema. Gastroenterology 1986, 90, 1827–1833. [Google Scholar]
- Saló, J.; Ginès, A.; Ginès, P.; Piera, C.; Jiménez, W.; Guevara, M.; Fernández-Esparrach, G.; Sort, P.; Bataller, R.; Arroyo, V.; et al. Effect of therapeutic paracentesis on plasma volume and transvascular escape rate of albumin in patients with cirrhosis. J. Hepatol. 1997, 27, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, M.; Caraceni, P.; Navickis, R.J.; Wilkes, M.M. Albumin infusion in patient undergoing large-volume paracentesis: A meta-analysis of randomized trials. Hepatology 2012, 55, 172–181. [Google Scholar] [CrossRef]
- Follo, A.; Llovet, J.M.; Navasa, M.; Planas, R.; Forns, X.; Francitorra, A.; Rimola, A.; Gassull, M.A.; Arroyo, V.; Rodès, J. Renal impairment after spontaneous bacterial peritonitis in cirrhosis: Incidence, clinical course, predictive factors and prognosis. Hepatology 1994, 20, 1495–1501. [Google Scholar] [CrossRef]
- Sort, P.; Navasa, M.; Arroyo, V.; Aldeguer, X.; Planas, R.; Ruiz-del-Arbol, L.; Castells, L.; Vargas, V.; Soriano, G.; Guevara, M.; et al. Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N. Engl. J. Med. 1999, 341, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Boyer, T.; Garcia-Tsao, G.; Regenstein, F.; Rossaro, L.; Appenrodt, B.; Blei, A.; Gilber, V.; Sigal, S.; Teuber, P.; et al. A randomized, prospective, double-blind, placebo-controlled trial of terlipressin for type 1 hepatorenal syndrome. Gastroenterology 2008, 134, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Martín-Llahí, M.; Pépin, M.N.; Guevara, M.; Díaz, F.; Torre, A.; Monescillo, A.; Soriano, G.; Terra, C.; Fábrega, E.; Arroyo, V.; et al. Terlipressin and albumin vs. albumin in patients with cirrhosis and hepatorenal syndrome: A randomized study. Gastroenterology 2008, 134, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Prakoso, E.; Jones, C.; Koorey, D.J.; Strasser, S.I.; Bowen, D.; McCaughan, G.W.; Shackel, N.A. Terlipressin therapy for moderate-to-severe hyponatraemia in patients with liver failure. Intern. Med. J. 2013, 43, 240–246. [Google Scholar] [CrossRef] [PubMed]
- McCormick, P.A.; Mistry, P.; Kaye, G.; Burroughs, A.K.; McIntyre, N. Intravenous albumin infusion is an effective therapy for hyponatraemia in cirrhotic patients with ascites. Gut 1990, 31, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Spasovski, G.; Vanholder, R.; Allolio, B.; Annane, D.; Ball, S.; Bichet, D.; Decaux, G.; Fenske, W.; Hoorn, E.J.; Ichai, C.; et al. Clinical practice guideline on diagnosis and treatment of hyponatraemia. Intensive Care Med. 2014, 40, 320–331. [Google Scholar] [CrossRef]
- Holtkamp, M.; Othman, J.; Buchheim, K.; Meierkord, H. Predictors and prognosis of refractory status epilepticus treated in a neurological intensive care unit. J. Neurol. Neurosurg. Psychiatry 2005, 76, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Cagnin, A.; Taylor-Robinson, S.D.; Forton, D.M.; Banati, R.B. In vivo imaging of cerebral “peripheral benzodiazepine binding sites” in patients with hepatic encephalopathy. Gut 2006, 55, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. The astrocytic (“peripheral-type”) benzodiazepine receptor: Role in the pathogenesis of portal-systemic encephalopathy. Neurochem. Int. 2000, 36, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Branch, R.A.; Morgan, M.H.; James, J.; Read, A.E. Intravenous administration of diazepam in patients with chronic liver disease. Gut 1976, 17, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Carrilho, F.; Bosch, J.; Arroyo, V.; Mas, A.; Viver, J.; Rodes, J. Renal failure associated with demeclocycline in cirrhosis. Annu. Intern. Med. 1977, 87, 195–197. [Google Scholar] [CrossRef]
- Gadano, A.; Moreau, R.; Pessione, F.; Trombino, C.; Giuily, N.; Sinnassamy, P.; Valla, D.; Lebrec, D. Aquaretic effects of niravoline, a kappa-opioid agonist, in patients with cirrhosis. J. Hepatol. 2000, 32, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Quittnat, F.; Gross, P. Vaptans and the treatment of water-retaining disorders. Semin. Nephrol. 2006, 26, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, A.; Ginès, P.; Marotta, P.; Czerwiec, F.; Oyuang, J.; Guevara, M.; Afdhal, N.H. Tolvaptan, an oral vasopressin antagonist, in the treatment of hyponatremia in cirrhosis. J. Hepatol. 2012, 56, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Ginès, P.; Wong, F.; Watson, H.; Milutinovic, S.; del Arbol, L.R.; Olteanu, D.; HypoCAT Study Investigators. Effects of satavaptan, a selective vasopressin V(2) receptor antagonist, on ascites and serum sodium in cirrhosis with hyponatremia: A randomized trial. Hepatology 2008, 48, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Gerbes, A.L.; Gülberg, V.; Ginès, P.; Decaux, G.; Gross, P.; Gandjini, H.; Djian, J.; VPA Study Group. Therapy of hyponatremia in cirrhosis with a vasopressin receptor antagonist: A randomized double-blind multicenter trial. Gastroenterology 2003, 124, 933–939. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, J.G.; Favis, G. Conivaptan increases serum sodium in hyponatremic patients with end stage liver disease. Liver Transplant. 2009, 15, 1325–1329. [Google Scholar] [CrossRef]
- Wong, F.; Gines, P.; Watson, H.; Horsmans, Y.; Angeli, P.; Gow, P.; Minini, P.; Bernardi, M. Effects of a selective vasopressin V2 receptor antagonist, satavaptan, on ascites recurrence after paracentesis in patients with cirrhosis. J. Hepatol. 2010, 53, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.; Watson, H.; Gerbes, A.; Vilstrup, H.; Badalamenti, S.; Bernardi, M.; Ginès, P.; Satavaptan Investigators Group. Satavaptan for the management of ascites in cirrhosis: Efficacy and safety across the spectrum of ascites severity. Gut 2012, 61, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Perrone, R.D.; Krasa, H.B.; Ouyang, J.; Czerwiec, F.S.; et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2012, 20, 2407–2418. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernardi, M.; Ricci, C.S.; Santi, L. Hyponatremia in Patients with Cirrhosis of the Liver. J. Clin. Med. 2015, 4, 85-101. https://doi.org/10.3390/jcm4010085
Bernardi M, Ricci CS, Santi L. Hyponatremia in Patients with Cirrhosis of the Liver. Journal of Clinical Medicine. 2015; 4(1):85-101. https://doi.org/10.3390/jcm4010085
Chicago/Turabian StyleBernardi, Mauro, Carmen Serena Ricci, and Luca Santi. 2015. "Hyponatremia in Patients with Cirrhosis of the Liver" Journal of Clinical Medicine 4, no. 1: 85-101. https://doi.org/10.3390/jcm4010085
APA StyleBernardi, M., Ricci, C. S., & Santi, L. (2015). Hyponatremia in Patients with Cirrhosis of the Liver. Journal of Clinical Medicine, 4(1), 85-101. https://doi.org/10.3390/jcm4010085