Induced Pluripotent Stem Cells Derived from Alzheimer’s Disease Patients: The Promise, the Hope and the Path Ahead


Abstract
:
1. Introduction
2. AD Pathology and Progression
3. Requirement for Further Basic Research into the Disease
4. Hope in Modeling Alzheimer’s Disease Using Patient-Specific Induced Pluripotent Stem Cells
5. Therapeutic Benefits
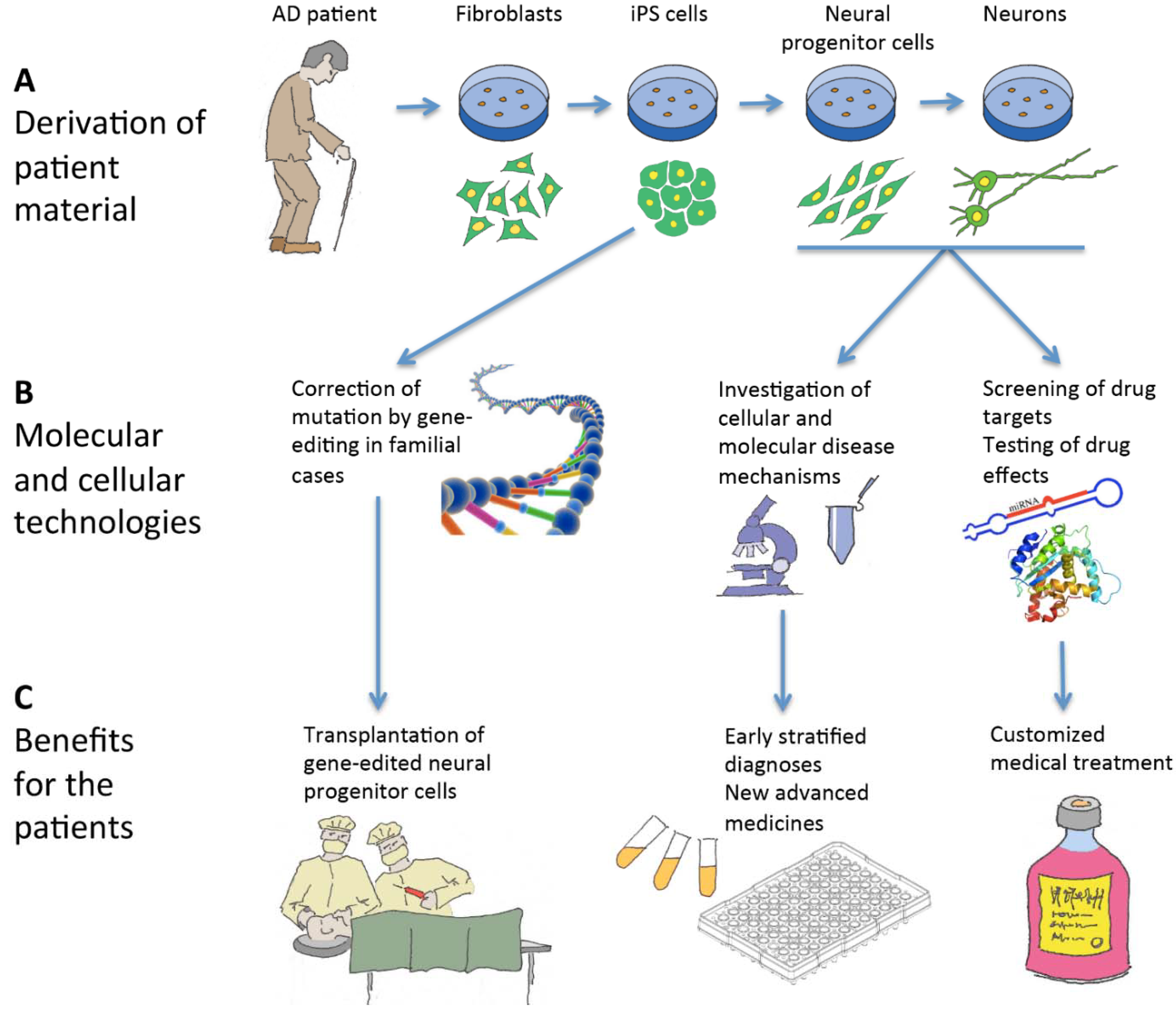
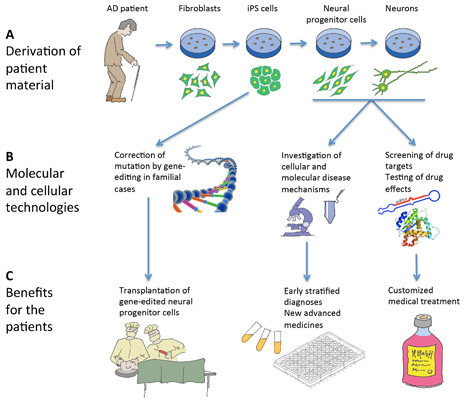
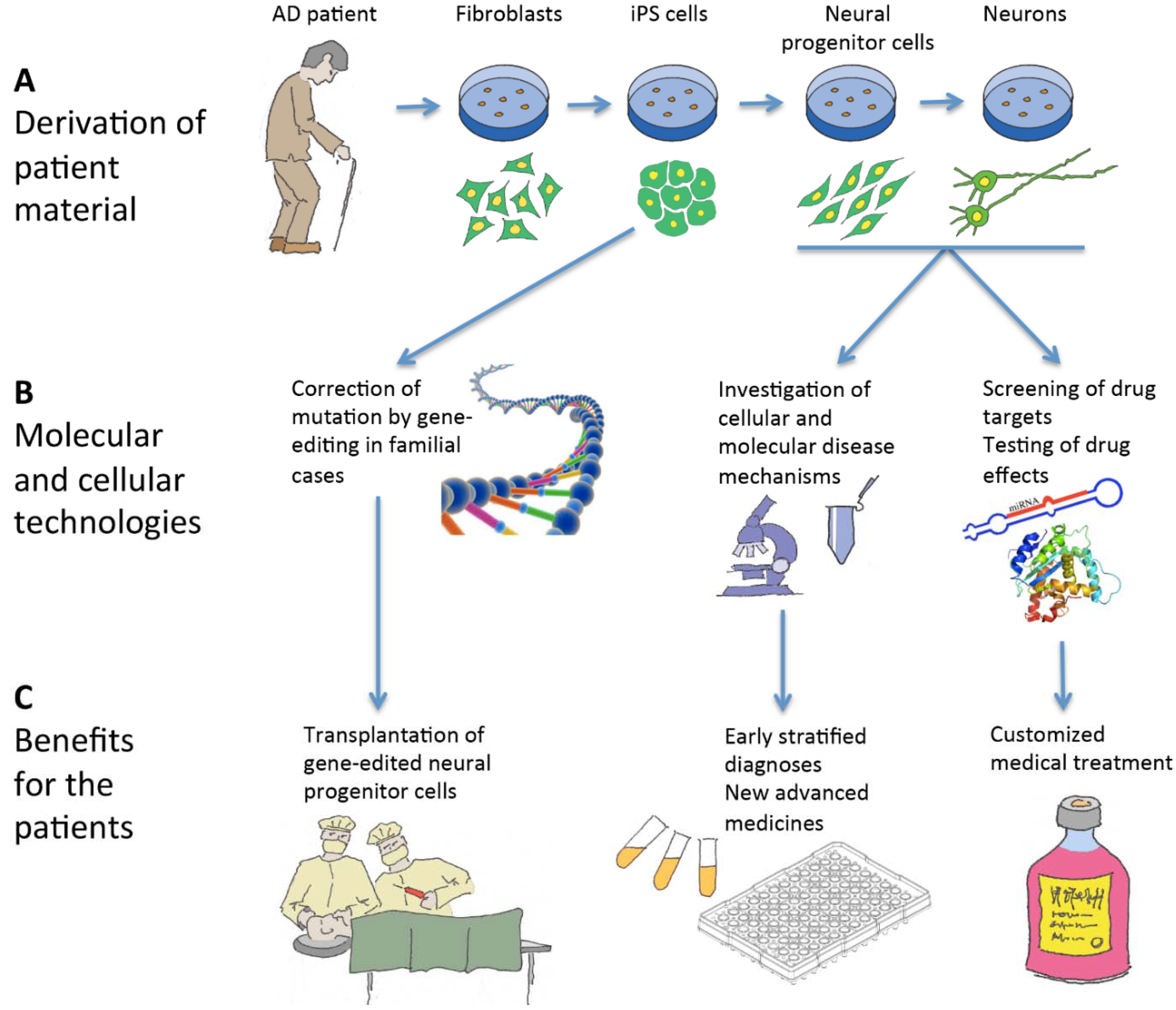
6. Induced Pluripotent Stem Cells and Neural Cell Derivatives Have Been Produced from Several AD Patients

{kind=link}
{kind=link}
| Cell Type Analyzed | Analyses Performed | Phenotype |
|---|---|---|
| Neurons (βIIITubulin+ MAP2+) | Extracellular Aβ Tau accumulation (HT7 antibody) Tangle formation Treatment with γ-secretase inhibitor and modulator of γ-secretase-mediated APP cleavage | Increased Aβ42:Aβ40 No Tau accumulation No tangle formation Decreased Aβ40+Aβ42 with γ-secretase inhibitor and modulator of APP cleavage |
| Neurons (βIII-Tubulin+ MAP2+) >90% | Genome-wide mRNA seq. Extracellular Aβ p-tau (Thr231) GSK-3β activity Treatment with γ-secretase and β-secretase inhibitors Endosome markers Synaptic markers | Increased Aβ40, Increased p-tau (Thr231), Increased aGSK-3β γ-/β-secretase inhibitors reduced Aβ40 |
| Increased Aβ40 Increased p-tau (Thr231), Increased aGSK-3β γ-/β-secretase inhibitors reduced Aβ40 β-secretase inhibitor reduced aGSK-3β+p-tau had large/very large Rab5 + early endosomes No change in synapsin I + puncta on dendrites | ||
| No change in Aβ40, No increase of p-tau (Thr231), No increase aGSK-3β γ-/β-secretase inhibitors reduced Ab40 | ||
| No change in Aβ40 Increased p-tau (Thr231) and aGSK-3β γ-/β-secretase inhibitors reduced Aβ40 β-secretase inhibitor reduced aGSK-3β+p-tau had large/very large Rab5 + early endosomes No change in synapsin I + puncta on dendrites | ||
| It-NES progenitor cells (NESTIN+SOX2+) | Expression APP+ γ-secretase components Extracellular Aβ Aβ length qPCR | Dominant-negative effect on S3 cleavage of Notch in progenitors, decreased HES5 |
| Neurons (βIII-Tubulin+MAP2ab+ GABA+) + <10% Astrocyte (GFAP+) | Increased full-length APP Decreased Aβ40 | |
| It-NES cells (NESTIN+SOX2+) | Dominant-negative effect on S3 cleavage of Notch in progenitors, decreased HES5 | |
| Neurons (βIII-Tubulin+MAP2ab+ GABA+) <10% Astrocyte (GFAP+) | Decreased Aβ40 | |
| Cortical neurons; Early born, (TBR1+ βIII-Tubulin+ /CTIP2+ βIII-Tubulin+) 30% Late born, (BM2+ βIII-Tubulin+/SATB2+ βIII-Tubulin+) 20%–25% Functional synapses Glutamatergic (PSD95+) | Extracellular Aβ Aggregation of Aβ Treatment with γ-secretase inhibitor p-tau expression Cell death | Increased Aβ40 Increased Aβ42 (>70 days cultures) Increased Aβ42:Aβ40 Intracellular and extracellular Aβ42 aggregates Decreased Aβ40+Aβ42 with γ-secretase inhibitor p-tau localized in cell bodies and dendrites Increased secretion of total tau and p-tau Increased cell death (2 fold) |
| Cortical neurons (SATB2+TBR1+) | Extracellular Aβ Intracellular Aβ Aβ Oligomers Gene expression profiling ROS expression Aβ Oligomers ROS expression | Decreased Aβ40 and Aβ42 Elevated Aβ oligomers in neural cells Elevated levels of oxidative stress-related genes Elevated ROS |
| Astrocytes | Elevated Aβ oligomers Elevated ROS | |
| Cortical neurons (SATB2+TBR1+) | Increased Aβ42, increased Aβ42:Aβ40 Elevated levels of oxidative stress-related genes | |
| Cortical neurons (SATB2+TBR1+) | No change in Aβ40 or Aβ42 Elevated levels of oxidative stress-related genes | |
| Cortical neurons (SATB2+TBR1+) | No change in Aβ40 or Aβ42 Elevated Aβ oligomers in neural cells Elevated levels of oxidative stress-related genes and ROS | |
| Astrocytes | Elevated Aβ oligomers, Elevated ROS | |
| Basal forebrain cholinergic neurons (MAP2+ChAT+ VaChT+P75R+NKX2.1+HB9−) Expressed tetrodotoxin-sensitive voltage-activated currents and voltage-gated calcium channels | Extracellular Aβ Treatment with γ-secretase inhibitors Treatment with ionomycin + glutamate Fura-2 calcium imaging | Elevated Aβ42, Increased Aβ40 with γ-secretase inhibitor Increased susceptibility to glutamate-induced excitotoxic death Increased calcium transient |
| Elevated Aβ42 Increased Aβ40 with γ-secretase inhibitor Susceptibility to cell death following calcium influx | ||
| No elevated Aβ42 Increased susceptibility to glutamate-induced excitotoxic death Increased calcium transient | ||
| Elevated Aβ42 Reduced Ab40 with γ-secretase inhibitor | ||
| No elevated Aβ42 Reduced Aβ40 with γ-secretase inhibitor | ||
| Forebrain neurons (MAP2+Tau+ βIII-Tubulin+Cux1+ TBR1+PSD95+ VGLUT1+) | Extracellular Aβ APP cleavage product expression Treatment with γ-secretase inhibitor Expression of tau Treatment with Aβ antibodies | APP holoprotein 1.4× increased Increased Aβ42:Aβ40 Increased Aβ42 Increased Aβ38 Decreased APPsα:APPsβ (Increased APPsβ) γ-secretase inhibitor blocked APPsβ cleavage Increased total tau Increased p-tau (Ser262) d100 Aβ antibodies blocked increased total tau (early differentiated neurons only) |
| Neurons (βIII-Tubulin+MAP2+) | Extracellular Aβ Treatment with γ-secretase inhibitors | Increased Aβ42:Aβ40 Increased Aβ42 γ-secretase inhibitor lowered total Aβ, Aβ40, Aβ42, Aβ38 |
| Increased Aβ42:Aβ40 Increased Aβ42 γ-secretase inhibitor lowered Aβ42 | ||
| Increased Aβ42:Aβ40 Increased Aβ42 γ-secretase inhibitor lowered Aβ42 | ||
| D14 immature neurons (79% NESTIN+ small pop’n TUJ1+) | Extracellular Aβ Total Aβ | Increased Aβ42:Aβ40 Increased NLRP2, ASB9, NDP |
| Neurons Electrical signaling properties | Increased Aβ42:Aβ40 | |
| D14 immature neurons (79% NESTIN+ small pop’n TUJ1+) | Increased Aβ42:Aβ40 Increased NLRP2, ASB9, NDP | |
| Neurons Electrical signaling properties | Increased Aβ42:Aβ40 |
| Differentiation Protocol | Cell Type Formed |
|---|---|
| EB induction w/o bFGF 8 days EBs plated gelatin w/o bFGF 8 days Neuron induction w/o growth factors 2 weeks Added compound E or compound W 48 h | Neurons βIII-Tubulin+MAP2+ |
| Neuronal rosette induction on PA6 stromal cells 11 days NPCs isolated by FACS CD184+CD15+CD44−CD271− NPC cultured 4 weeks Neuron induction-BDNF/GDNF/cAMP 3 weeks CD24+CD184−CD44− neurons selected by FACS Cultured in BDNF/GDNF/cAMP 5 days | Neurons βIII-Tubulin+MAP2+ >90% VGluT1+ 15% GABA+ 8% Expressed tetrodotoxin-sensitive voltage-activated currents GABA+AMPA receptors Spontaneous inhibitory/excitatory synaptic currents |
| It-NES induction with bFGF+EGF+B27 Neuron induction—Matrigel w/o factors, +N2+B27+cAMP 4 weeks | Neurons βIII-Tubulin+ 80% Astrocytes 6% |
| Matrigel+N2+B27+Noggin+SB431542 Dissociated and cultured with 3N+bFGF 100 days | Cortical neurons; Early born, TBR1+βIII-Tubulin+ CTIP2+βIII-Tubulin+ 30% Late born, BM2+βIII-Tubulin+ SATB2+βIII-Tubulin+ 20%–25% Functional synapses Glutamatergic+ PSD95+ |
| EB induction DMEM/HamsF12+ 5% KSR+SB431542 8 days Neural induction—plated on Matrigel+N2+SB431542 16 days Cortical neuron induction—dissociated and cultured in NB media+B27+BDNF+GDNF+NT3 48 days As above, but on day 58 cortical neuron induction, cells passaged Repeated passages on day 96, 126, 156, 176 | Cortical neurons SATB2+TBR1+ Astrocytes |
| RA+bFGF 7 days Neurosphere formation w/o bFGF 7 days Neurospheres cultured with bFGF+EGF 4 days Neurospheres cultured with SHH+FGF8 3 days Dissociated and transfected with Lhx8/Gbx1-IRES-EGFP 2 days Lhx8+/Gbx1+ cells selected by FACS and cultured in NB media+ bFGF+NGF 2 weeks (+arabinoside from day 5–10 of NB culture step) | Basal forebrain cholinergic neurons 95% MAP2 66% ChAT VaChT+P75R+, NKX2.1+HB9− Expessed tetrodotoxin-sensitive voltage-activated currents, voltage-gated calcium channels |
| Aggregates with iPS cell media 4 days + neural media+N2 2 days Aggregates plated on matrigel, Neural media + N2 10 days Suspension culture, neural media+B27+N2+cAMP+IGF1 7 days Neural rosettes selected manually or Neural Rosette selection agent Dissociated+plated on Matrigel+NBmedia+N2+B27+ cAMP+BDNF+GDNF+IGF1 35 days–76 days | Neurons 90% MAP2 Tau+, βIII-Tubulin+Cux1+Tbr1+PSD95+VGLUT1+ Spontaneous activity from microelectrode array |
| Neuronal rosette induction on PA6 stromal cells +Noggin and SB431542 6 days −Noggin and SB431542 8 days CD24+/CD184+/CD271−/CD44− cells selected by FACS Cultured in neural media (DMEM:F12+N2+B27+BDNF+GDNF+dcAMP) for 3 weeks w/o bFGF CD24+/CD184−/CD44− neurons selected by FACS | Neurons βIII-Tubulin+MAP2+ |
| Neuronal progenitor induction using dual-SMAD inhibition 9 days NB media 26 days–46 days | Neural progenitors 79% NESTIN+, small pop’n βIII-Tubulin+ Neurons Active Na+ channels K+ channels Produce action potentials 40% neurons Ca2+ spikes |
7. Modeling Impaired APP Processing from Patient-Specific Induced Pluripotent Stem Cells Reveals Considerable Variability
8. Tau Processing, Cell Death and Oxidative Stress in iPS Cell Lines Modeling AD
9. Hunting for New Genes of Interest in AD
10. Current Use of AD-Modeling Stem Cells for Compound Screening and Drug Testing
11. Production of AD Isogenic Controls for Potential Gene/Cell Therapy
12. Current General Limitations of Use of iPS Cells for Disease Modeling
13. Hurdles Needed to be Overcome in Order to Recapitulate AD Faithfully in a Dish
14. Future Induction of an AD Phenotype Using Components that Introduce Cellular Stress
15. Induction of an AD Phenotype by Manipulating the Gene Expression of Age Inducing genes
16. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Abbreviations
| B27 | (B27 supplement) |
| BDNF | (brain-derived neurotrophic factor) |
| bFGF | (basic fibroblast growth factor) |
| cAMP | (cyclic AMP) |
| dcAMP | (dibutryl cyclic AMP) |
| EB | (embryoid body) |
| EGF | (epidermal growth factor) |
| EGFP | (enhanced green fluorescent protein) |
| FACS | (fluorescence activated cell sorting) |
| GDNF | (glial cell-derived neurotrophic factor) |
| IGF | (insulin growth factor) |
| IPS cell | (induced pluripotent stem cells) |
| It-NES | (neuroepithelial stem cells) |
| KSR | (knockout serum replacement) |
| N2 | (N2 supplement) |
| NB | (neural basal media) |
| NGF | (nerve growth factor) |
| NPC | (neural progenitor cell) |
| RA | (retinoic acid) |
| seq. | (sequencing) |
| SHH | (sonic hedgehog) |
| wks | (weeks) |
| w/o | (without) |
| 3N | (modified bold 3N medium) |
Conflicts of Interest
References
- World Health Organization and Alzheimer’s Disease International. Dementia: A Public Health Priority; WHO Press: Geneva, Switzerland, 2012; p. 103. [Google Scholar]
- Aalten, P.; Verhey, F.R.; Boziki, M.; Brugnolo, A.; Bullock, R.; Byrne, E.J.; Camus, V.; Caputo, M.; Collins, D.; de Deyn, P.P.; et al. Consistency of neuropsychiatric syndromes across dementias: Results from the European Alzheimer Disease Consortium. Part II. Dement. Geriatr. Cogn. Disord. 2008, 25, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kamboh, M.I. Molecular genetics of late-onset Alzheimer’s disease. Ann. Hum. Genet. 2004, 68, 381–404. [Google Scholar] [CrossRef] [PubMed]
- Roses, A.D.; Saunders, A.M. Perspective on a pathogenesis and treatment of Alzheimer’s disease. Alzheimer’s Dement. 2006, 2, 59–70. [Google Scholar] [CrossRef]
- Bertram, L.; McQueen, M.B.; Mullin, K.; Blacker, D.; Tanzi, R.E. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 2007, 39, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Cruchaga, C.; Kauwe, J.S.; Harari, O.; Jin, S.C.; Cai, Y.; Karch, C.M.; Benitez, B.A.; Jeng, A.T.; Skorupa, T.; Carrell, D.; et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron 2013, 78, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Rao, A.T.; Degnan, A.J.; Levy, L.M. Genetics of Alzheimer disease. Am. J. Neuroradiol. 2014, 35, 457–458. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induced pluripotent stem cells in medicine and biology. Development 2013, 140, 2457–2461. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.D. Genetic aspects of Alzheimer disease. Genet. Med. 2008, 10, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: Evidence that an initially deposited species is A beta 42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.C.; Li, B.; Grundke-Iqbal, I.; Iqbal, K. Mechanism of tau-induced neurodegeneration in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2008, 5, 375–384. [Google Scholar] [CrossRef]
- Braak, H.; Rub, U.; Schultz, C.; del Tredici, K. Vulnerability of cortical neurons to Alzheimer’s and Parkinson’s diseases. J. Alzheimer’s Dis. 2006, 9, 35–44. [Google Scholar]
- Mann, D.M. Pyramidal nerve cell loss in Alzheimer’s disease. Neurodegeneration 1996, 5, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Baglietto-Vargas, D.; Moreno-Gonzalez, I.; Sanchez-Varo, R.; Jimenez, S.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Torres, M.; Romero-Acebal, M.; Ruano, D.; Vizuete, M.; et al. Calretinin interneurons are early targets of extracellular amyloid-beta pathology in PS1/AbetaPP Alzheimer mice hippocampus. J. Alzheimer’s Dis. 2010, 21, 119–132. [Google Scholar]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- West, M.J.; Coleman, P.D.; Flood, D.G.; Troncoso, J.C. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet 1994, 344, 769–772. [Google Scholar] [CrossRef] [PubMed]
- Schliebs, R.; Arendt, T. The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 2011, 221, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Lee, S.C.; Mattiace, L.A.; Yen, S.H.; Brosnan, C. Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia 1993, 7, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.; Sheng, J.G.; Roberts, G.W.; Mrak, R.E. Interleukin-1 expression in different plaque types in Alzheimer’s disease: Significance in plaque evolution. J. Neuropathol. Exp. Neurol. 1995, 54, 276–281. [Google Scholar] [CrossRef] [PubMed]
- White, J.A.; Manelli, A.M.; Holmberg, K.H.; van Eldik, L.J.; Ladu, M.J. Differential effects of oligomeric and fibrillar amyloid-beta 1–42 on astrocyte-mediated inflammation. Neurobiol. Dis. 2005, 18, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Munch, G.; Thome, J.; Foley, P.; Schinzel, R.; Riederer, P. Advanced glycation endproducts in ageing and Alzheimer’s disease. Brain Res. Rev. 1997, 23, 134–143. [Google Scholar] [CrossRef] [PubMed]
- McShea, A.; Harris, P.L.; Webster, K.R.; Wahl, A.F.; Smith, M.A. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am. J. Pathol. 1997, 150, 1933–1939. [Google Scholar] [PubMed]
- McShea, A.; Lee, H.G.; Petersen, R.B.; Casadesus, G.; Vincent, I.; Linford, N.J.; Funk, J.O.; Shapiro, R.A.; Smith, M.A. Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim. Biophys. Acta 2007, 1772, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Castellani, R.J.; Hirai, K.; Smith, M.A. Reactive Oxygen Species Mediate Cellular Damage in Alzheimer Disease. J. Alzheimer’s Dis. 1998, 1, 45–55. [Google Scholar]
- Unterberger, U.; Hoftberger, R.; Gelpi, E.; Flicker, H.; Budka, H.; Voigtlander, T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. J. Neuropathol. Exp. Neurol. 2006, 65, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Sturchler-Pierrat, C.; Abramowski, D.; Duke, M.; Wiederhold, K.H.; Mistl, C.; Rothacher, S.; Ledermann, B.; Burki, K.; Frey, P.; Paganetti, P.A.; et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA 1997, 94, 13287–13292. [Google Scholar] [CrossRef]
- Duff, K.; Eckman, C.; Zehr, C.; Yu, X.; Prada, C.M.; Perez-tur, J.; Hutton, M.; Buee, L.; Harigaya, Y.; Yager, D.; et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 1996, 383, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, M.C.; McNamara, M.; Fedorchak, K.; Hsiao, K.; Hyman, B.T. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J. Neuropathol. Exp. Neurol. 1997, 56, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, L.; Gordon, M.N.; McGowan, E.; Yu, X.; Benkovic, S.; Jantzen, P.; Wright, K.; Saad, I.; Mueller, R.; Morgan, D.; et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 1998, 4, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Chishti, M.A.; Yang, D.S.; Janus, C.; Phinney, A.L.; Horne, P.; Pearson, J.; Strome, R.; Zuker, N.; Loukides, J.; French, J.; et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 2001, 276, 21562–21570. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; van Nostrand, W.E. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Konietzko, U.; Krebs, D.C.; Nitsch, R.M. Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol. Aging 2007, 28, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Casas, C.; Sergeant, N.; Itier, J.M.; Blanchard, V.; Wirths, O.; van der Kolk, N.; Vingtdeux, V.; van de Steeg, E.; Ret, G.; Canton, T.; et al. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am. J. Pathol. 2004, 165, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Lim, F.; Hernandez, F.; Lucas, J.J.; Gomez-Ramos, P.; Moran, M.A.; Avila, J. FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and Tau filaments in forebrain. Mol. Cell. Neurosci. 2001, 18, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef] [PubMed]
- Shineman, D.W.; Basi, G.S.; Bizon, J.L.; Colton, C.A.; Greenberg, B.D.; Hollister, B.A.; Lincecum, J.; Leblanc, G.G.; Lee, L.B.; Luo, F.; et al. Accelerating drug discovery for Alzheimer’s disease: Best practices for preclinical animal studies. Alzheimer’s Res. Ther. 2011, 3. [Google Scholar] [CrossRef]
- Franco, R.; Cedazo-Minguez, A. Successful therapies for Alzheimer’s disease: Why so many in animal models and none in humans? Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef]
- Becker, R.E.; Greig, N.H. Increasing the success rate for Alzheimer’s disease drug discovery and development. Exp. Opin. Drug Discov. 2012, 7, 367–370. [Google Scholar] [CrossRef]
- Sperling, R.A.; Karlawish, J.; Johnson, K.A. Preclinical Alzheimer disease-the challenges ahead. Nat. Rev. Neurol. 2013, 9, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Lerou, P.H.; Zhao, R.; Huo, H.; Daley, G.Q. Generation of human-induced pluripotent stem cells. Nat. Protoc. 2008, 3, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Malchenko, S.; Galat, V.; Seftor, E.A.; Vanin, E.F.; Costa, F.F.; Seftor, R.E.; Soares, M.B.; Hendrix, M.J. Cancer hallmarks in induced pluripotent cells: New insights. J. Cell. Physiol. 2010, 225, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H.; et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Nishino, K.; Toyoda, M.; Yamazaki-Inoue, M.; Fukawatase, Y.; Chikazawa, E.; Sakaguchi, H.; Akutsu, H.; Umezawa, A. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 2011, 7, e1002085. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Huang, K.; Shen, Y.; Xue, Z.; Cai, C.; Horvath, S.; Fan, G. Functional modules distinguish human induced pluripotent stem cells from embryonic stem cells. Stem Cells Dev. 2011, 20, 1937–1950. [Google Scholar] [CrossRef] [PubMed]
- Bellin, M.; Marchetto, M.C.; Gage, F.H.; Mummery, C.L. Induced pluripotent stem cells: The new patient? Nat. Rev. Mol. Cell Biol. 2012, 13, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced pluripotent stem cells generated without viral integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, C.H.; Moon, J.I.; Chung, Y.G.; Chang, M.Y.; Han, B.S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Amit, M.; Itskovitz-Eldor, J. Feeder-free culture of human embryonic stem cells. Methods Enzymol. 2006, 420, 37–49. [Google Scholar] [PubMed]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; Propson, N.E.; et al. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.H.; Hartung, O.; Li, H.; Guo, C.; Sahalie, J.M.; Manos, P.D.; Urbach, A.; Heffner, G.C.; Grskovic, M.; Vigneault, F.; et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell 2010, 7, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Taniguchi, Y.; Senda, S.; Takizawa, N.; Ichisaka, T.; Asano, K.; Morizane, A.; Doi, D.; Takahashi, J.; Nishizawa, M.; et al. A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Hyun, I. The bioethics of stem cell research and therapy. J. Clin. Investig. 2010, 120, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Charron, D.; Suberbielle-Boissel, C.; Al-Daccak, R. Immunogenicity and allogenicity: A challenge of stem cell therapy. J. Cardiovasc. Transl. Res. 2009, 2, 130–138. [Google Scholar] [CrossRef]
- Noggle, S.; Fung, H.L.; Gore, A.; Martinez, H.; Satriani, K.C.; Prosser, R.; Oum, K.; Paull, D.; Druckenmiller, S.; Freeby, M.; et al. Human oocytes reprogram somatic cells to a pluripotent state. Nature 2011, 478, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R. Human cloning—The science and ethics of nuclear transplantation. N. Engl. J. Med. 2004, 351, 2787–2791. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.; Dottori, M.; Sourris, K.; Jamshidi, P.; Hatzistavrou, T.; Davis, R.; Azzola, L.; Jackson, S.; Lim, S.M.; Pera, M.; et al. A method for genetic modification of human embryonic stem cells using electroporation. Nat. Protoc. 2007, 2, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Maeder, M.L.; Mali, P.; Pruett-Miller, S.M.; Thibodeau-Beganny, S.; Chou, B.K.; Chen, G.; Ye, Z.; Park, I.H.; Daley, G.Q.; et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell 2009, 5, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, J.L.; Bryne, S.; Pan, J.; Church, G.M. CRISPR/Cas9-directed genome editing of cultured cells. In Current Protocols in Molecular Biology; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2014; Volume 107, pp. 1–17. [Google Scholar]
- Cherry, A.B.; Daley, G.Q. Reprogrammed cells for disease modeling and regenerative medicine. Ann. Rev. Med. 2013, 64, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Matsumoto, Y.; Shindo, T.; Miyake, K.; Shindo, A.; Kawanishi, M.; Kawai, N.; Tamiya, T.; Nagao, S. Neural stem cells transplantation in cortex in a mouse model of Alzheimer’s disease. J. Med. Investig. 2006, 53, 61–69. [Google Scholar] [CrossRef]
- Yamasaki, T.R.; Blurton-Jones, M.; Morrissette, D.A.; Kitazawa, M.; Oddo, S.; LaFerla, F.M. Neural stem cells improve memory in an inducible mouse model of neuronal loss. J. Neurosci. 2007, 27, 11925–11933. [Google Scholar] [CrossRef] [PubMed]
- Blurton-Jones, M.; Kitazawa, M.; Martinez-Coria, H.; Castello, N.A.; Muller, F.J.; Loring, J.F.; Yamasaki, T.R.; Poon, W.W.; Green, K.N.; LaFerla, F.M. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc. Natl. Acad. Sci. USA 2009, 106, 13594–13599. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Lee, H.J.; Joo, S.S.; Bae, D.K.; Yang, G.; Yang, Y.H.; Lim, I.; Matsuo, A.; Tooyama, I.; Kim, Y.B.; et al. Human neural stem cells over-expressing choline acetyltransferase restore cognition in rat model of cognitive dysfunction. Exp. Neurol. 2012, 234, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.M.; Djukic, B.; Arnold, C.; Gillespie, A.K.; Yoon, S.Y.; Wang, M.M.; Zhang, O.; Knoferle, J.; Rubenstein, J.L.; Alvarez-Buylla, A.; et al. Inhibitory Interneuron Progenitor Transplantation Restores Normal Learning and Memory in ApoE4 Knock-In Mice without or with Abeta Accumulation. J. Neurosci. 2014, 34, 9506–9515. [Google Scholar] [CrossRef] [PubMed]
- Xuan, A.G.; Luo, M.; Ji, W.D.; Long, D.H. Effects of engrafted neural stem cells in Alzheimer’s disease rats. Neurosci. Lett. 2009, 450, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.S.; Maclean, K.N.; Jiang, H.; Synder, E.Y.; Sladek, J.R., Jr.; Bjugstad, K.B. Neural stem cells reduce hippocampal tau and reelin accumulation in aged Ts65Dn Down syndrome mice. Cell Transplant. 2011, 20, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Babaei, P.; Soltani Tehrani, B.; Alizadeh, A. Transplanted bone marrow mesenchymal stem cells improve memory in rat models of Alzheimer’s disease. Stem Cells Int. 2012, 2012. [Google Scholar] [CrossRef]
- Kim, S.; Chang, K.A.; Kim, J.; Park, H.G.; Ra, J.C.; Kim, H.S.; Suh, Y.H. The preventive and therapeutic effects of intravenous human adipose-derived stem cells in Alzheimer’s disease mice. PLoS One 2012, 7, e45757. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lee, J.K.; Lee, H.; Carter, J.E.; Chang, J.W.; Oh, W.; Yang, Y.S.; Suh, J.G.; Lee, B.H.; Jin, H.K.; et al. Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer’s disease mouse model through modulation of neuroinflammation. Neurobiol. Aging 2012, 33, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, F.H.; Alaie, H.; Karbalaie, K.; Tanhaei, S.; Nasr Esfahani, M.H.; Baharvand, H. Transplantation of primed or unprimed mouse embryonic stem cell-derived neural precursor cells improves cognitive function in Alzheimerian rats. Differ. Res. Biol. Divers. 2009, 78, 59–68. [Google Scholar] [CrossRef]
- Yahata, N.; Asai, M.; Kitaoka, S.; Takahashi, K.; Asaka, I.; Hioki, H.; Kaneko, T.; Maruyama, K.; Saido, T.C.; Nakahata, T.; et al. Anti-Abeta drug screening platform using human iPS cell-derived neurons for the treatment of Alzheimer’s disease. PLoS One 2011, 6, e25788. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Liang, P.; Wu, J.C. Induced pluripotent stem cells as a disease modeling and drug screening platform. J. Cardiovasc. Pharmacol. 2012, 60, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Noetzli, M.; Eap, C.B. Pharmacodynamic, pharmacokinetic and pharmacogenetic aspects of drugs used in the treatment of Alzheimer’s disease. Clin. Pharmacokinet. 2013, 52, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Yu, J.T.; Tan, L. Biomarkers for preclinical Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 42, 1051–1069. [Google Scholar]
- Duan, L.; Bhattacharyya, B.J.; Belmadani, A.; Pan, L.; Miller, R.J.; Kessler, J.A. Stem cell derived basal forebrain cholinergic neurons from Alzheimer’s disease patients are more susceptible to cell death. Mol. Neurodegener. 2014, 9. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [PubMed]
- Koch, P.; Tamboli, I.Y.; Mertens, J.; Wunderlich, P.; Ladewig, J.; Stuber, K.; Esselmann, H.; Wiltfang, J.; Brustle, O.; Walter, J. Presenilin-1 L166P mutant human pluripotent stem cell-derived neurons exhibit partial loss of gamma-secretase activity in endogenous amyloid-beta generation. Am. J. Pathol. 2012, 180, 2404–2416. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kirwan, P.; Smith, J.; MacLean, G.; Orkin, S.H.; Livesey, F.J. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Liu, Q.; Waltz, S.; Woodruff, G.; Ouyang, J.; Israel, M.A.; Herrera, C.; Sarsoza, F.; Tanzi, R.E.; Koo, E.H.; Ringman, J.M.; et al. Effect of Potent gamma-Secretase Modulator in Human Neurons Derived From Multiple Presenilin 1-Induced Pluripotent Stem Cell Mutant Carriers. JAMA Neurol. 2014. [Google Scholar] [CrossRef]
- Sproul, A.A.; Jacob, S.; Pre, D.; Kim, S.H.; Nestor, M.W.; Navarro-Sobrino, M.; Santa-Maria, I.; Zimmer, M.; Aubry, S.; Steele, J.W.; et al. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS One 2014, 9, e84547. [Google Scholar] [CrossRef] [PubMed]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 2008, 63, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Quiroz, Y.T.; Fleisher, A.S.; Chen, K.; Velez-Pardo, C.; Jimenez-Del-Rio, M.; Fagan, A.M.; Shah, A.R.; Alvarez, S.; Arbelaez, A.; et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: A case-control study. Lancet. Neurol. 2012, 11, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Boss, M.A. Diagnostic approaches to Alzheimer’s disease. Biochim. Biophys. Acta 2000, 1502, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, B.; Siepi, D.; Sabalich, I.; Tranfaglia, C.; Parnetti, L. Cerebrospinal fluid neuron-specific enolase: A further marker of Alzheimer’s disease? Funct. Neurol. 2008, 23, 93–96. [Google Scholar] [PubMed]
- Kumar-Singh, S.; Theuns, J.; van Broeck, B.; Pirici, D.; Vennekens, K.; Corsmit, E.; Cruts, M.; Dermaut, B.; Wang, R.; van Broeckhoven, C. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum. Mutat. 2006, 27, 686–695. [Google Scholar] [CrossRef] [PubMed]
- McIntire, L.B.; Landman, N.; Kang, M.S.; Finan, G.M.; Hwang, J.C.; Moore, A.Z.; Park, L.S.; Lin, C.S.; Kim, T.W. Phenotypic assays for beta-amyloid in mouse embryonic stem cell-derived neurons. Chem. Biol. 2013, 20, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lei, Y.; Luo, J.; Wang, J.; Zhang, S.; Yang, X.J.; Sun, M.; Nuwaysir, E.; Fan, G.; Zhao, J.; et al. Prevention of beta-amyloid induced toxicity in human iPS cell-derived neurons by inhibition of Cyclin-dependent kinases and associated cell cycle events. Stem Cell Res. 2013, 10, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xue, W.; Chen, S.; Bogorad, R.L.; Benedetti, E.; Grompe, M.; Koteliansky, V.; Sharp, P.A.; Jacks, T.; Anderson, D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014, 32, 551–553. [Google Scholar] [PubMed]
- Wu, Y.; Liang, D.; Wang, Y.; Bai, M.; Tang, W.; Bao, S.; Yan, Z.; Li, D.; Li, J. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 2013, 13, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Low, B.E.; Krebs, M.P.; Joung, J.K.; Tsai, S.Q.; Nishina, P.M.; Wiles, M.V. Correction of the Crb1rd8 allele and retinal phenotype in C57BL/6N mice via TALEN-mediated homology-directed repair. Investig. Ophthalmol. Visual Sci. 2014, 55, 387–395. [Google Scholar] [CrossRef]
- Ma, N.; Liao, B.; Zhang, H.; Wang, L.; Shan, Y.; Xue, Y.; Huang, K.; Chen, S.; Zhou, X.; Chen, Y.; et al. Transcription activator-like effector nuclease (TALEN)-mediated gene correction in integration-free beta-thalassemia induced pluripotent stem cells. J. Biol. Chem. 2013, 288, 34671–34679. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Zhao, H. Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnol. Bioeng. 2014, 111, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Perez-Pinera, P.; Thakore, P.I.; Kabadi, A.M.; Brown, M.T.; Qin, X.; Fedrigo, O.; Mouly, V.; Tremblay, J.P.; Gersbach, C.A. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol. Ther. 2013, 21, 1718–1726. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.J.; Starker, C.G.; McElroy, A.N.; Webber, B.R.; Riddle, M.J.; Xia, L.; DeFeo, A.P.; Gabriel, R.; Schmidt, M.; von Kalle, C.; et al. TALEN-based gene correction for epidermolysis bullosa. Mol. Ther. 2013, 21, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhang, Y. Embryonic stem cell and induced pluripotent stem cell: An epigenetic perspective. Cell Res. 2013, 23, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Loh, Y.H.; McLoughlin, E.M.; Huang, J.; Park, I.H.; Miller, J.D.; Huo, H.; Okuka, M.; dos Reis, R.M.; Loewer, S.; et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature 2010, 464, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Prigione, A.; Hossini, A.M.; Lichtner, B.; Serin, A.; Fauler, B.; Megges, M.; Lurz, R.; Lehrach, H.; Makrantonaki, E.; Zouboulis, C.C.; et al. Mitochondrial-associated cell death mechanisms are reset to an embryonic-like state in aged donor-derived iPS cells harboring chromosomal aberrations. PLoS One 2011, 6, e27352. [Google Scholar] [CrossRef] [PubMed]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Ait-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.M.; de Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef] [PubMed]
- Suhr, S.T.; Chang, E.A.; Rodriguez, R.M.; Wang, K.; Ross, P.J.; Beyhan, Z.; Murthy, S.; Cibelli, J.B. Telomere dynamics in human cells reprogrammed to pluripotency. PLoS One 2009, 4, e8124. [Google Scholar] [PubMed]
- Yagi, T.; Kosakai, A.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Nabetani, A.; Ishikawa, F.; Arai, Y.; Hirose, N.; et al. Establishment of induced pluripotent stem cells from centenarians for neurodegenerative disease research. PLoS One 2012, 7, e41572. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [PubMed]
- Cohen, A.D.; Klunk, W.E. Early detection of Alzheimer’s disease using PiB and FDG PET. Neurobiol. Dis. 2014. [Google Scholar]
- Lista, S.; Garaci, F.G.; Ewers, M.; Teipel, S.; Zetterberg, H.; Blennow, K.; Hampel, H. CSF Abeta 1–42 combined with neuroimaging biomarkers in the early detection, diagnosis and prediction of Alzheimer’s disease. Alzheimer’s Dement. 2014, 10, 381–392. [Google Scholar] [CrossRef]
- Caselli, R.J.; Reiman, E.M. Characterizing the preclinical stages of Alzheimer’s disease and the prospect of presymptomatic intervention. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1), 405–416. [Google Scholar]
- Koronyo, Y.; Salumbides, B.C.; Black, K.L.; Koronyo-Hamaoui, M. Alzheimer’s disease in the retina: Imaging retinal abeta plaques for early diagnosis and therapy assessment. Neurodegener. Dis. 2012, 10, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, T.; Orban-Kis, K.; Horvath, E.; Metz, J.; Pap, Z.; Pavai, Z. Morphological identification of neuron types in the rat hippocampus. Rom. J. Morphol. Embryol. 2011, 52, 15–20. [Google Scholar] [PubMed]
- Molyneaux, B.J.; Arlotta, P.; Menezes, J.R.; Macklis, J.D. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 2007, 8, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Espuny-Camacho, I.; Michelsen, K.A.; Gall, D.; Linaro, D.; Hasche, A.; Bonnefont, J.; Bali, C.; Orduz, D.; Bilheu, A.; Herpoel, A.; et al. Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron 2013, 77, 440–456. [Google Scholar] [CrossRef] [PubMed]
- De la Torre-Ubieta, L.; Bonni, A. Transcriptional regulation of neuronal polarity and morphogenesis in the mammalian brain. Neuron 2011, 72, 22–40. [Google Scholar] [CrossRef] [PubMed]
- Badger, J.L.; Cordero-Llana, O.; Hartfield, E.M.; Wade-Martins, R. Parkinson’s disease in a dish—Using stem cells as a molecular tool. Neuropharmacology 2014, 76, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Roybon, L.; Lamas, N.J.; Garcia-Diaz, A.; Yang, E.J.; Sattler, R.; Jackson-Lewis, V.; Kim, Y.A.; Kachel, C.A.; Rothstein, J.D.; Przedborski, S.; et al. Human stem cell-derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Rep. 2013, 4, 1035–1048. [Google Scholar] [CrossRef] [PubMed]
- Juopperi, T.A.; Kim, W.R.; Chiang, C.H.; Yu, H.; Margolis, R.L.; Ross, C.A.; Ming, G.L.; Song, H. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol. Brain 2012, 5. [Google Scholar] [CrossRef]
- Hu, B.Y.; Weick, J.P.; Yu, J.; Ma, L.X.; Zhang, X.Q.; Thomson, J.A.; Zhang, S.C. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. USA 2010, 107, 4335–4340. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Yan, P.; Ma, X.; Liu, H.; Perez, R.; Zhu, A.; Gonzales, E.; Burchett, J.M.; Schuler, D.R.; Cirrito, J.R.; et al. Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J. Neurosci. 2014, 34, 9607–9620. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.W.; Hu, X.; Yoon, H.; Yan, P.; Xiao, Q.; Wang, Y.; Gil, S.C.; Brown, J.; Wilhelmsson, U.; Restivo, J.L.; et al. Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J. 2013, 27, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wu, S.; Bu, G.; Onifade, M.K.; Patel, S.N.; LaDu, M.J.; Fagan, A.M.; Holtzman, D.M. Glial fibrillary acidic protein-apolipoprotein E (apoE) transgenic mice: Astrocyte-specific expression and differing biological effects of astrocyte-secreted apoE3 and apoE4 lipoproteins. J. Neurosci. 1998, 18, 3261–3272. [Google Scholar] [PubMed]
- Doens, D.; Fernandez, P.L. Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Beal, M.F. Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic. Biol. Med. 2011, 51, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Perez Estrada, C.; Covacu, R.; Sankavaram, S.R.; Svensson, M.; Brundin, L. Oxidative Stress Increases Neurogenesis and Oligodendrogenesis in Adult Neural Progenitor Cells. Stem Cells Dev. 2014, 23, 2311–2327. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Wang, X.; Nunomura, A.; Moreira, P.I.; Lee, H.G.; Perry, G.; Smith, M.A.; Zhu, X. Oxidative stress signaling in Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Young, J.E.; Martinez, R.A.; la Spada, A.R. Nutrient deprivation induces neuronal autophagy and implicates reduced insulin signaling in neuroprotective autophagy activation. J. Biol. Chem. 2009, 284, 2363–2373. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.W.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Hamamichi, S.; Rivas, R.N.; Knight, A.L.; Cao, S.; Caldwell, K.A.; Caldwell, G.A. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. USA 2008, 105, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Herranz, D.; Munoz-Martin, M.; Canamero, M.; Mulero, F.; Martinez-Pastor, B.; Fernandez-Capetillo, O.; Serrano, M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef]
- Giblin, W.; Skinner, M.E.; Lombard, D.B. Sirtuins: Guardians of mammalian healthspan. Trends Genet. 2014, 30, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Jayasena, T.; Poljak, A.; Sachdev, P.S. Sirtuins in cognitive ageing and Alzheimer’s disease. Curr. Opin. Psychiatry 2012, 25, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Aron, L.; Zullo, J.; Pan, Y.; Kim, H.; Chen, Y.; Yang, T.H.; Kim, H.M.; Drake, D.; Liu, X.S.; et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 2014, 507, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.J.; Yacoubian, T.A.; Slone, S.R.; Caldwell, K.A.; Caldwell, G.A. Functional analysis of VPS41-mediated neuroprotection in Caenorhabditis elegans and mammalian models of Parkinson’s disease. J. Neurosci. 2012, 32, 2142–2153. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A.; Ojala, J.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Impaired autophagy and APP processing in Alzheimer’s disease: The potential role of Beclin 1 interactome. Prog. Neurobiol. 2013, 106–107, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Morizane, A.; Doi, D.; Kikuchi, T.; Okita, K.; Hotta, A.; Kawasaki, T.; Hayashi, T.; Onoe, H.; Shiina, T.; Yamanaka, S.; et al. Direct Comparison of Autologous and Allogeneic Transplantation of iPSC-Derived Neural Cells in the Brain of a Nonhuman Primate. Stem Cell Rep. 2013, 1, 283–292. [Google Scholar] [CrossRef]
- Doi, D.; Samata, B.; Katsukawa, M.; Kikuchi, T.; Morizane, A.; Ono, Y.; Sekiguchi, K.; Nakagawa, M.; Parmar, M.; Takahashi, J. Isolation of human induced pluripotent stem cell-derived dopaminergic progenitors by cell sorting for successful transplantation. Stem Cell Rep. 2014, 2, 337–350. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freude, K.; Pires, C.; Hyttel, P.; Hall, V.J. Induced Pluripotent Stem Cells Derived from Alzheimer’s Disease Patients: The Promise, the Hope and the Path Ahead. J. Clin. Med. 2014, 3, 1402-1436. https://doi.org/10.3390/jcm3041402
Freude K, Pires C, Hyttel P, Hall VJ. Induced Pluripotent Stem Cells Derived from Alzheimer’s Disease Patients: The Promise, the Hope and the Path Ahead. Journal of Clinical Medicine. 2014; 3(4):1402-1436. https://doi.org/10.3390/jcm3041402
Chicago/Turabian StyleFreude, Kristine, Carlota Pires, Poul Hyttel, and Vanessa Jane Hall. 2014. "Induced Pluripotent Stem Cells Derived from Alzheimer’s Disease Patients: The Promise, the Hope and the Path Ahead" Journal of Clinical Medicine 3, no. 4: 1402-1436. https://doi.org/10.3390/jcm3041402
APA StyleFreude, K., Pires, C., Hyttel, P., & Hall, V. J. (2014). Induced Pluripotent Stem Cells Derived from Alzheimer’s Disease Patients: The Promise, the Hope and the Path Ahead. Journal of Clinical Medicine, 3(4), 1402-1436. https://doi.org/10.3390/jcm3041402