Opportunities and Limitations of Modelling Alzheimer’s Disease with Induced Pluripotent Stem Cells


Abstract
:
1. Opportunities and Limitations of Modelling Alzheimer’s Disease with Induced Pluripotent Stem Cells
2. iPSCs as a Model System

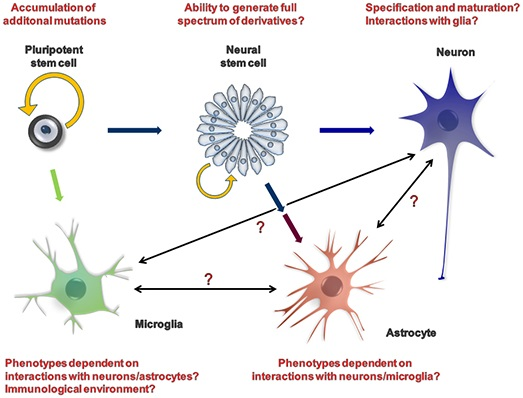{kind=link}
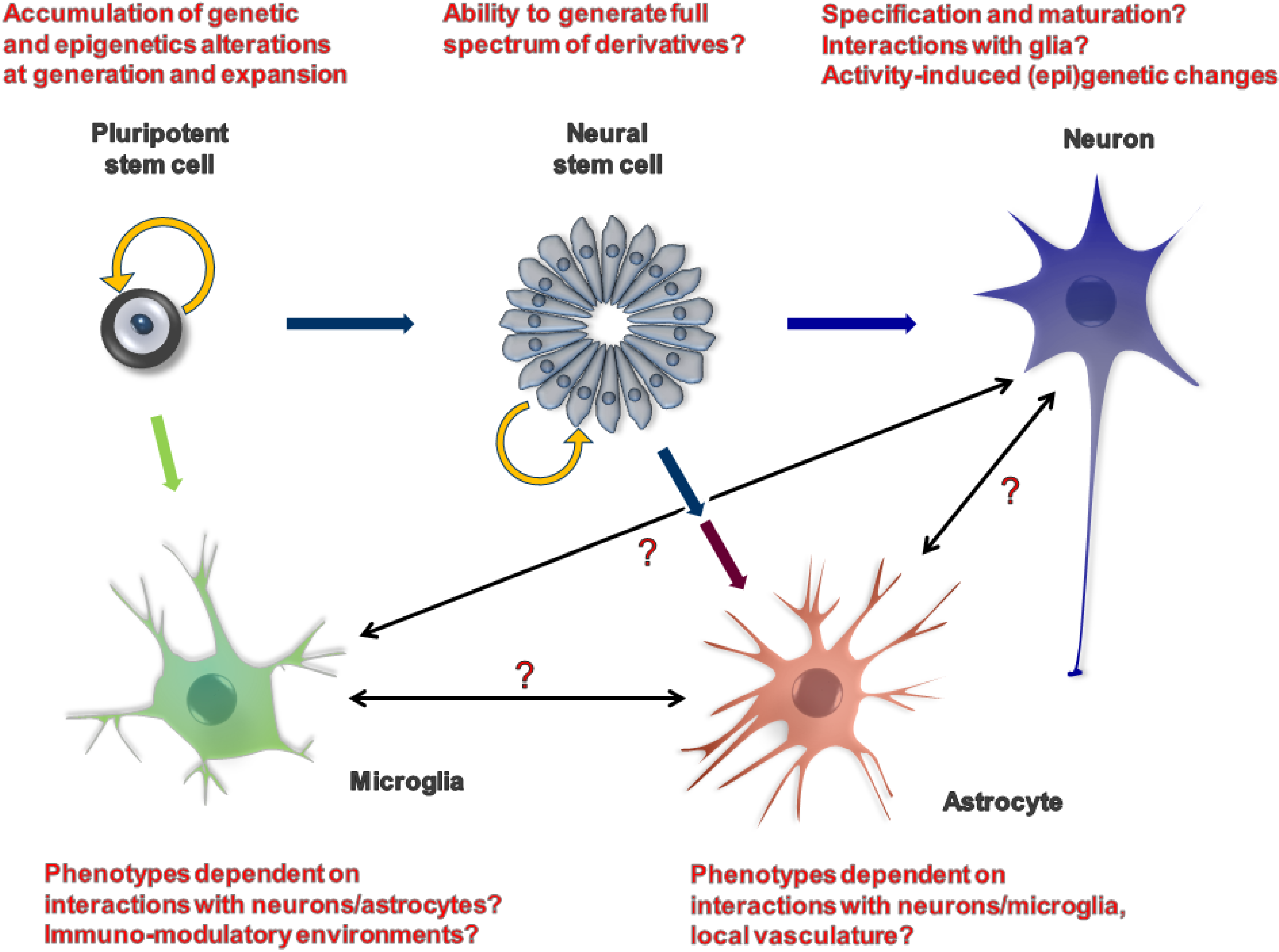{kind=link}
{kind=link}
| Genetic Defect | Affected Process(es) | Disease Type, iPS/hES N and n * | Transgene-Free? | Investigated Cell Type(s) | Reference(s) |
|---|---|---|---|---|---|
| APP | Aβ production and aggregation, MAPT | Familial early-onset (N = 2, father + daughter; n = 2 pre-selected) | N | Neurons | [31] |
| APP | Aβ production, ER stress | Familial early-onset (N = 2, n = 2 and 3) and sporadic (N = 2; n = 2) | Y | Cortical neurons, astrocytes | [32] |
| PSEN1 | β-amyloid processing | Early-onset AD, OE model in N = 1 hES and N = 1 iPS | Y/N | Neurons | [33] |
| PSEN1, PSEN2 | β-amyloid processing | Early-onset AD, N = 2 PSEN1&2; n = 2 | N | Neurons | [34] |
| ApoE(4) | Aβ levels | Early and late-onset DA, familial (N = 2) and sporadic (N = 3) | N | Basal forebrain cholinergic neuron | [35] |
| PSEN1 | Aβ production and aggregation, MAPT | Familial AD, N = 4 | N | Neural stem cells, neurons | [36] |
| APP and PSEN1 OE | Aβ production and processing | OE models of familial AD mutations | N | Neural precursor cells, neurons | [37] |
3. Making the Right Cell Type
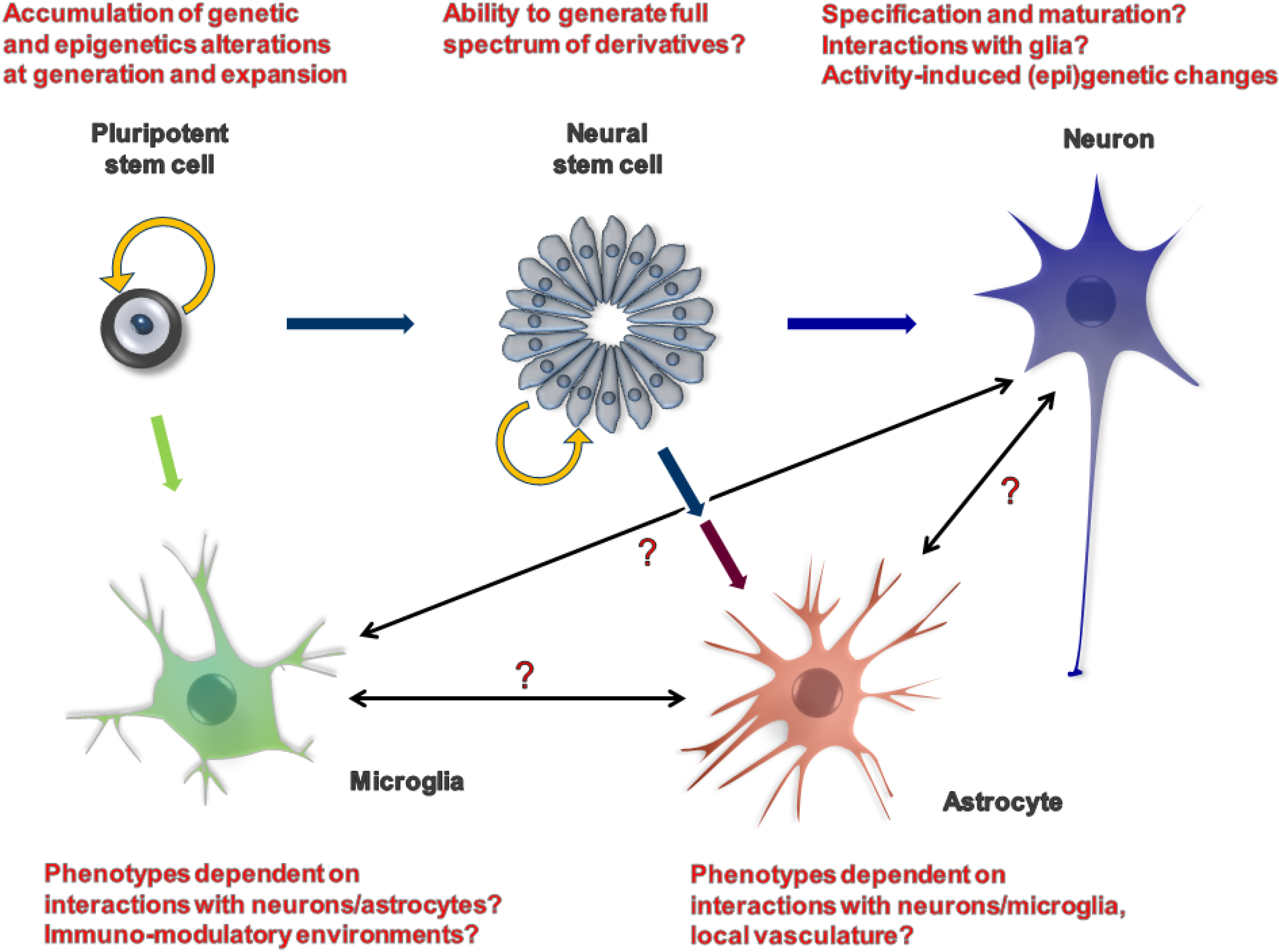
4. AD Phenotypes Which Can be Reliably Modelled in Vitro
5. Down Syndrome iPSC as a Model for AD
6. Drug Screening Utilizing AD iPSC-Derived Cell Types
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.Y.; Kim, D.S.; Kim, D.W. Human ES and iPS cells as cell sources for the treatment of parkinson’s disease: Current state and problems. J. Cell. Biochem. 2010, 109, 292–301. [Google Scholar] [PubMed]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M.; et al. Isogenic human iPSC parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schondorf, D.C.; Wagner, L.; Glatza, M.; Hoing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Schondorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. IPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Li, Z.; Fung, H.L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.C.; Ulitsky, I.; Slavin, I.; Tran, H.; Schork, A.; Morey, R.; Lynch, C.; Harness, J.V.; Lee, S.; Barrero, M.J.; et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 2011, 8, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.; Yeo, G.W.; Kainohana, O.; Marsala, M.; Gage, F.H.; Muotri, A.R. Transcriptional signature and memory retention of human-induced pluripotent stem cells. PLoS One 2009, 4, e7076. [Google Scholar] [CrossRef] [PubMed]
- Hussein, S.M.; Batada, N.N.; Vuoristo, S.; Ching, R.W.; Autio, R.; Narva, E.; Ng, S.; Sourour, M.; Hamalainen, R.; Olsson, C.; et al. Copy number variation and selection during reprogramming to pluripotency. Nature 2011, 471, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Shoemaker, R.; Xie, B.; Gore, A.; LeProust, E.M.; Antosiewicz-Bourget, J.; Egli, D.; Maherali, N.; Park, I.H.; Yu, J.; et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat. Biotechnol. 2009, 27, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Doi, A.; Park, I.H.; Wen, B.; Murakami, P.; Aryee, M.J.; Irizarry, R.; Herb, B.; Ladd-Acosta, C.; Rho, J.; Loewer, S.; et al. Differential methylation of tissue- and cancer-specific CPG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009, 41, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H.; et al. Reference maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Nazor, K.L.; Altun, G.; Lynch, C.; Tran, H.; Harness, J.V.; Slavin, I.; Garitaonandia, I.; Muller, F.J.; Wang, Y.C.; Boscolo, F.S.; et al. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell 2012, 10, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, G.; Ganat, Y.; Papapetrou, E.P.; Lipchina, I.; Socci, N.D.; Sadelain, M.; Studer, L. Mir-371-3 expression predicts neural differentiation propensity in human pluripotent stem cells. Cell Stem Cell 2011, 8, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Guenther, M.G.; Frampton, G.M.; Soldner, F.; Hockemeyer, D.; Mitalipova, M.; Jaenisch, R.; Young, R.A. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell Stem Cell 2010, 7, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Mallon, B.S.; Chenoweth, J.G.; Johnson, K.R.; Hamilton, R.S.; Tesar, P.J.; Yavatkar, A.S.; Tyson, L.J.; Park, K.; Chen, K.G.; Fann, Y.C.; et al. Stemcelldb: The human pluripotent stem cell database at the national institutes of health. Stem Cell Res. 2013, 10, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Hartjes, K.A.; Li, X.; Martinez-Fernandez, A.; Roemmich, A.J.; Larsen, B.T.; Terzic, A.; Nelson, T.J. Selection via pluripotency-related transcriptional screen minimizes the influence of somatic origin on iPSC differentiation propensity. Stem Cells 2014, 32, 2350–2359. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C.A.; Christodoulou, C.; Gianotti-Sommer, A.; Shen, S.S.; Sailaja, B.S.; Hezroni, H.; Spira, A.; Meshorer, E.; Kotton, D.N.; Mostoslavsky, G. Residual expression of reprogramming factors affects the transcriptional program and epigenetic signatures of induced pluripotent stem cells. PLoS One 2012, 7, e51711. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Morey, R.; O’Neil, R.C.; He, Y.; Daughtry, B.; Schultz, M.D.; Hariharan, M.; Nery, J.R.; Castanon, R.; Sabatini, K.; et al. Abnormalities in human pluripotent cells due to reprogramming mechanisms. Nature 2014, 511, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.S.; Wang, Z.; Hu, Y.; Yang, H.H.; Gere, S.; Buetow, K.H.; Lee, M.P. Allelic variation in gene expression is common in the human genome. Genome Res. 2003, 13, 1855–1862. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Yuan, W.; Velculescu, V.E.; Vogelstein, B.; Kinzler, K.W. Allelic variation in human gene expression. Science 2002, 297, 1143. [Google Scholar] [CrossRef] [PubMed]
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Wutz, A. Epigenetic alterations in human pluripotent stem cells: A tale of two cultures. Cell Stem Cell 2012, 11, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Mekhoubad, S.; Bock, C.; de Boer, A.S.; Kiskinis, E.; Meissner, A.; Eggan, K. Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 2012, 10, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Matsuno, Y.; Fouse, S.D.; Rao, N.; Root, S.; Xu, R.; Pellegrini, M.; Riggs, A.D.; Fan, G. X-inactivation in female human embryonic stem cells is in a nonrandom pattern and prone to epigenetic alterations. Proc. Natl. Acad. Sci. USA 2008, 105, 4709–4714. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Bernitz, J.M.; Lee, D.F.; Lemischka, I.R. Genomic editing tools to model human diseases with isogenic pluripotent stem cells. Stem Cells Dev. 2014, 23, 2673–2686. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Suzuki, K.; Kim, N.Y.; Liu, G.H.; Izpisua Belmonte, J.C. A cut above the rest: Targeted genome editing technologies in human pluripotent stem cells. J. Biol. Chem. 2014, 289, 4594–4599. [Google Scholar] [CrossRef] [PubMed]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The familial Alzheimer’s disease APPV717I mutation alters APP processing and tau expression in iPSC-derived neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular abeta and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Koch, P.; Tamboli, I.Y.; Mertens, J.; Wunderlich, P.; Ladewig, J.; Stuber, K.; Esselmann, H.; Wiltfang, J.; Brustle, O.; Walter, J. Presenilin-L l166P mutant human pluripotent stem cell-derived neurons exhibit partial loss of γ-secretase activity in endogenous amyloid-β generation. Am. J. Pathol. 2012, 180, 2404–2416. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Bhattacharyya, B.J.; Belmadani, A.; Pan, L.; Miller, R.J.; Kessler, J.A. Stem cell derived basal forebrain cholinergic neurons from Alzheimer’s disease patients are more susceptible to cell death. Mol. Neurodegener. 2014, 9. [Google Scholar] [CrossRef]
- Liu, Q.; Waltz, S.; Woodruff, G.; Ouyang, J.; Israel, M.A.; Herrera, C.; Sarsoza, F.; Tanzi, R.E.; Koo, E.H.; Ringman, J.M.; et al. Effect of potent γ-secretase modulator in human neurons derived from multiple presenilin 1-induced pluripotent stem cell mutant carriers. JAMA Neurol. 2014. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Holtzman, D.M. Origins of Alzheimer’s disease: Reconciling cerebrospinal fluid biomarker and neuropathology data regarding the temporal sequence of amyloid-β and tau involvement. Curr. Opin. Neurol. 2012, 25, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Morphological criteria for the recognition of Alzheimer’s disease and the distribution pattern of cortical changes related to this disorder. Neurobiol. Aging 1994, 15, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [PubMed]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.L.; Herrera, C.; Hefferan, M.P.; van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [PubMed]
- Shi, Y.; Kirwan, P.; Smith, J.; MacLean, G.; Orkin, S.H.; Livesey, F.J. A human stem cell model of early Alzheimer’s disease pathology in down syndrome. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Yahata, N.; Asai, M.; Kitaoka, S.; Takahashi, K.; Asaka, I.; Hioki, H.; Kaneko, T.; Maruyama, K.; Saido, T.C.; Nakahata, T.; et al. Anti-abeta drug screening platform using human iPS cell-derived neurons for the treatment of Alzheimer’s disease. PLoS One 2011, 6, e25788. [Google Scholar] [PubMed]
- Sanchez-Danes, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jimenez-Delgado, S.; Caig, C.; Mora, S.; di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.W.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Batista, L.F.; Pech, M.F.; Zhong, F.L.; Nguyen, H.N.; Xie, K.T.; Zaug, A.J.; Crary, S.M.; Choi, J.; Sebastiano, V.; Cherry, A.; et al. Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature 2011, 474, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Moskalev, A.A.; Shaposhnikov, M.V.; Plyusnina, E.N.; Zhavoronkov, A.; Budovsky, A.; Yanai, H.; Fraifeld, V.E. The role of DNA damage and repair in aging through the prism of koch-like criteria. Ageing Res. Rev. 2013, 12, 661–684. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Gee, J.; Ding, Q. The proteasome in brain aging. Ageing Res. Rev. 2002, 1, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Romer, I.; Nebrich, G.; Klein, O.; Koppelstatter, A.; Hin, S.C.; Hartl, D.; Zabel, C. Aging in mouse brain is a cell / tissue-level phenomenon exacerbated by proteasome loss. J. Proteome Res. 2010, 9, 3551–3560. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease—A brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Steele, M.L.; Robinson, S.R. Reactive astrocytes give neurons less support: Implications for Alzheimer’s disease. Neurobiol. Aging 2012, 33. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, F.P.; Carrasco, M.A.; Siao, M.C.; Maniatis, T.; Eggan, K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based als model. Nat. Neurosci. 2007, 10, 608–614. [Google Scholar]
- Marchetto, M.C.; Muotri, A.R.; Mu, Y.; Smith, A.M.; Cezar, G.G.; Gage, F.H. Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 2008, 3, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Jiang, P.; Xue, H.; Peterson, S.E.; Tran, H.T.; McCann, A.E.; Parast, M.M.; Li, S.; Pleasure, D.E.; Laurent, L.C.; et al. Role of astroglia in down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Beutner, C.; Roy, K.; Linnartz, B.; Napoli, I.; Neumann, H. Generation of microglial cells from mouse embryonic stem cells. Nat. Protoc. 2010, 5, 1481–1494. [Google Scholar] [CrossRef] [PubMed]
- Napoli, I.; Kierdorf, K.; Neumann, H. Microglial precursors derived from mouse embryonic stem cells. Glia 2009, 57, 1660–1671. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, T.; Park, K.C.; Toyonaga, S.; Yamada, S.M.; Nakabayashi, H.; Nakai, E. Characterization of microglia induced from mouse embryonic stem cells and their migration into the brain parenchyma. J. Neuroimmunol. 2005, 160, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E.; Gold, G. Alzheimer disease therapy—Moving from amyloid-β to tau. Nat. Rev. Neurol. 2013, 9, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Gotz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Fox, N.C.; Jack, C.R., Jr.; Scheltens, P.; Thompson, P.M. The clinical use of structural MRI in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Putcha, D.; Brickhouse, M.; O’Keefe, K.; Sullivan, C.; Rentz, D.; Marshall, G.; Dickerson, B.; Sperling, R. Hippocampal hyperactivation associated with cortical thinning in Alzheimer’s disease signature regions in non-demented elderly adults. J. Neurosci.: Off. J. Soc. Neurosci. 2011, 31, 17680–17688. [Google Scholar] [CrossRef]
- Briggs, J.A.; Sun, J.; Shepherd, J.; Ovchinnikov, D.A.; Chung, T.L.; Nayler, S.P.; Kao, L.P.; Morrow, C.A.; Thakar, N.Y.; Soo, S.Y.; et al. Integration-free induced pluripotent stem cells model genetic and neural developmental features of down syndrome etiology. Stem Cells 2013, 31, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, P.; Herrmann, F.R.; Bussiere, T.; Bouras, C.; Kovari, E.; Perl, D.P.; Morrison, J.H.; Gold, G.; Hof, P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 2003, 60, 1495–1500. [Google Scholar] [CrossRef]
- Cho, J.H.; Johnson, G.V. Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3β (GSK3β) plays a critical role in regulating tau’s ability to bind and stabilize microtubules. J. Neurochem. 2004, 88, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.A.; Goldstein, L.S. Capturing Alzheimer’s disease genomes with induced pluripotent stem cells: Prospects and challenges. Genome Med. 2011, 3. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Mucke, L. A network dysfunction perspective on neurodegenerative diseases. Nature 2006, 443, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Marie, H. Hippocampal synaptic plasticity in Alzheimer’s disease: What have we learned so far from transgenic models? Rev. Neurosci. 2011, 22, 373–402. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Brennand, K.J.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, D.; DiFiglia, M. Neurobiotin, a useful neuroanatomical tracer for in vivo anterograde, retrograde and transneuronal tract-tracing and for in vitro labeling of neurons. J. Neurosci. Methods 1992, 41, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Muotri, A.R.; Marchetto, M.C.; Coufal, N.G.; Oefner, R.; Yeo, G.; Nakashima, K.; Gage, F.H. L1 retrotransposition in neurons is modulated by MECP2. Nature 2010, 468, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Ma, D.K.; Mo, H.; Ball, M.P.; Jang, M.H.; Bonaguidi, M.A.; Balazer, J.A.; Eaves, H.L.; Xie, B.; Ford, E.; et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011, 14, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Sweatt, J.D. Covalent modification of DNA regulates memory formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Schor, I.E.; Rascovan, N.; Pelisch, F.; Allo, M.; Kornblihtt, A.R. Neuronal cell depolarization induces intragenic chromatin modifications affecting ncam alternative splicing. Proc. Natl. Acad. Sci. USA 2009, 106, 4325–4330. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.P.; Tun, N.; Grayson, D.R. Depolarization induces downregulation of DNMT1 and DNMT3a in primary cortical cultures. Epigenetics 2008, 3, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. DNMT1 and DNMT3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.; Woldemichael, B.T.; Berchtold, D.; Dewarrat, G.; Mansuy, I.M. Dynamic histone marks in the hippocampus and cortex facilitate memory consolidation. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef]
- Barry, G.; Briggs, J.A.; Vanichkina, D.P.; Poth, E.M.; Beveridge, N.J.; Ratnu, V.S.; Nayler, S.P.; Nones, K.; Hu, J.; Bredy, T.W.; et al. The long non-coding RNA gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol. Psychiatry 2014, 19, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Lipovich, L.; Dachet, F.; Cai, J.; Bagla, S.; Balan, K.; Jia, H.; Loeb, J.A. Activity-dependent human brain coding / noncoding gene regulatory networks. Genetics 2012, 192, 1133–1148. [Google Scholar] [CrossRef] [PubMed]
- Sleegers, K.; Brouwers, N.; Gijselinck, I.; Theuns, J.; Goossens, D.; Wauters, J.; del-Favero, J.; Cruts, M.; van Duijn, C.M.; van Broeckhoven, C. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain: A J. Neurol. 2006, 129, 2977–2983. [Google Scholar] [CrossRef]
- Rovelet-Lecrux, A.; Frebourg, T.; Tuominen, H.; Majamaa, K.; Campion, D.; Remes, A.M. APP locus duplication in a finnish family with dementia and intracerebral haemorrhage. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1158–1159. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, K.; Shimohata, T.; Nishimura, A.; Shiga, A.; Mizuguchi, T.; Tokunaga, J.; Ohno, T.; Miyashita, A.; Kuwano, R.; Matsumoto, N.; et al. Identification of independent APP locus duplication in Japanese patients with early-onset Alzheimer disease. J. Neurol. Neurosurg. Psychiatry 2009, 80, 1050–1052. [Google Scholar] [CrossRef] [PubMed]
- Korbel, J.O.; Tirosh-Wagner, T.; Urban, A.E.; Chen, X.N.; Kasowski, M.; Dai, L.; Grubert, F.; Erdman, C.; Gao, M.C.; Lange, K.; et al. The genetic architecture of down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc. Natl. Acad. Sci. USA 2009, 106, 12031–12036. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, S.R.; Cho, H.J.; Lee, H.W.; Jeong, H.K.; Radnaabazar, C.; Kim, Y.S.; Kim, M.J.; Son, M.Y.; Seo, H.; Chung, S.H.; et al. Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: Evidence for a functional link between down syndrome and Alzheimer’s disease. J. Neurochem. 2008, 104, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liang, Z.; Wegiel, J.; Hwang, Y.W.; Iqbal, K.; Grundke-Iqbal, I.; Ramakrishna, N.; Gong, C.X. Overexpression of DYRK1A contributes to neurofibrillary degeneration in down syndrome. FASEB J. 2008, 22, 3224–3233. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.S.; Park, J.H.; Ryu, Y.S.; Choi, S.H.; Yoon, S.H.; Kwen, M.Y.; Oh, J.Y.; Song, W.J.; Chung, S.H. Regulation of rcan1 protein activity by DYRK1A protein-mediated phosphorylation. J. Biol. Chem. 2011, 286, 40401–40412. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.R.; Corlett, A.; Dubach, D.; Mustafa, T.; Coleman, H.A.; Parkington, H.C.; Merson, T.D.; Bourne, J.A.; Porta, S.; Arbones, M.L.; et al. Over-expression of RCAN1 causes down syndrome-like hippocampal deficits that alter learning and memory. Hum. Mol. Genet. 2012, 21, 3025–3041. [Google Scholar] [CrossRef] [PubMed]
- Lloret, A.; Badia, M.C.; Giraldo, E.; Ermak, G.; Alonso, M.D.; Pallardo, F.V.; Davies, K.J.; Vina, J. Amyloid-β toxicity and tau hyperphosphorylation are linked via RCAN1 in Alzheimer’s disease. JAD 2011, 27, 701–709. [Google Scholar] [PubMed]
- Wolvetang, E.J.; Bradfield, O.M.; Hatzistavrou, T.; Crack, P.J.; Busciglio, J.; Kola, I.; Hertzog, P.J. Overexpression of the chromosome 21 transcription factor ETS2 induces neuronal apoptosis. Neurobiol. Dis. 2003, 14, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Wolvetang, E.W.; Bradfield, O.M.; Tymms, M.; Zavarsek, S.; Hatzistavrou, T.; Kola, I.; Hertzog, P.J. The chromosome 21 transcription factor ETS2 transactivates the beta-APP promoter: Implications for down syndrome. BBA 2003, 1628, 105–110. [Google Scholar] [PubMed]
- Helguera, P.; Pelsman, A.; Pigino, G.; Wolvetang, E.; Head, E.; Busciglio, J. ETS-2 promotes the activation of a mitochondrial death pathway in down’s syndrome neurons. J. Neurosci. 2005, 25, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.R.; Holler, C.J.; Webb, R.L.; Li, F.; Beckett, T.L.; Murphy, M.P. BACE1 and BACE2 enzymatic activities in Alzheimer’s disease. J. Neurochem. 2010, 112, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Holler, C.J.; Webb, R.L.; Laux, A.L.; Beckett, T.L.; Niedowicz, D.M.; Ahmed, R.R.; Liu, Y.; Simmons, C.R.; Dowling, A.L.; Spinelli, A.; et al. BACE2 expression increases in human neurodegenerative disease. Am. J. Pathol. 2012, 180, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; He, G.; Song, W. BACE2, as a novel APP theta-secretase, is not responsible for the pathogenesis of Alzheimer’s disease in down syndrome. FASEB J. 2006, 20, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Jing, Y.; Cost, G.J.; Chiang, J.C.; Kolpa, H.J.; Cotton, A.M.; Carone, D.M.; Carone, B.R.; Shivak, D.A.; Guschin, D.Y.; et al. Translating dosage compensation to trisomy 21. Nature 2013, 500, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Medway, C.; Morgan, K. Review: The genetics of Alzheimer’s disease; putting flesh on the bones. Neuropathol. Appl. Neurobiol. 2014, 40, 97–105. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ovchinnikov, D.A.; Wolvetang, E.J. Opportunities and Limitations of Modelling Alzheimer’s Disease with Induced Pluripotent Stem Cells. J. Clin. Med. 2014, 3, 1357-1372. https://doi.org/10.3390/jcm3041357
Ovchinnikov DA, Wolvetang EJ. Opportunities and Limitations of Modelling Alzheimer’s Disease with Induced Pluripotent Stem Cells. Journal of Clinical Medicine. 2014; 3(4):1357-1372. https://doi.org/10.3390/jcm3041357
Chicago/Turabian StyleOvchinnikov, Dmitry A., and Ernst J. Wolvetang. 2014. "Opportunities and Limitations of Modelling Alzheimer’s Disease with Induced Pluripotent Stem Cells" Journal of Clinical Medicine 3, no. 4: 1357-1372. https://doi.org/10.3390/jcm3041357
APA StyleOvchinnikov, D. A., & Wolvetang, E. J. (2014). Opportunities and Limitations of Modelling Alzheimer’s Disease with Induced Pluripotent Stem Cells. Journal of Clinical Medicine, 3(4), 1357-1372. https://doi.org/10.3390/jcm3041357