Comparing ESC and iPSC—Based Models for Human Genetic Disorders

Abstract
:1. Introduction
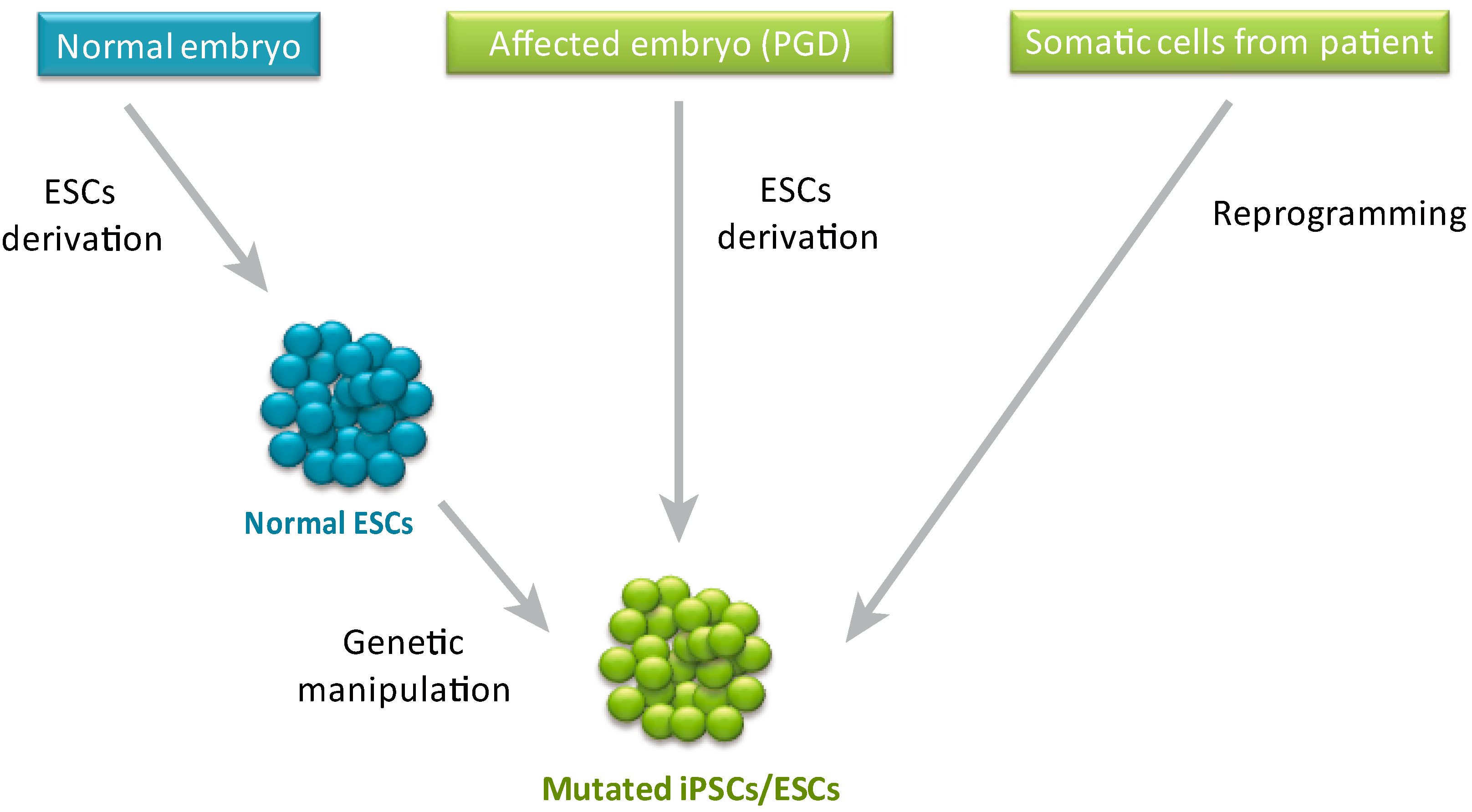
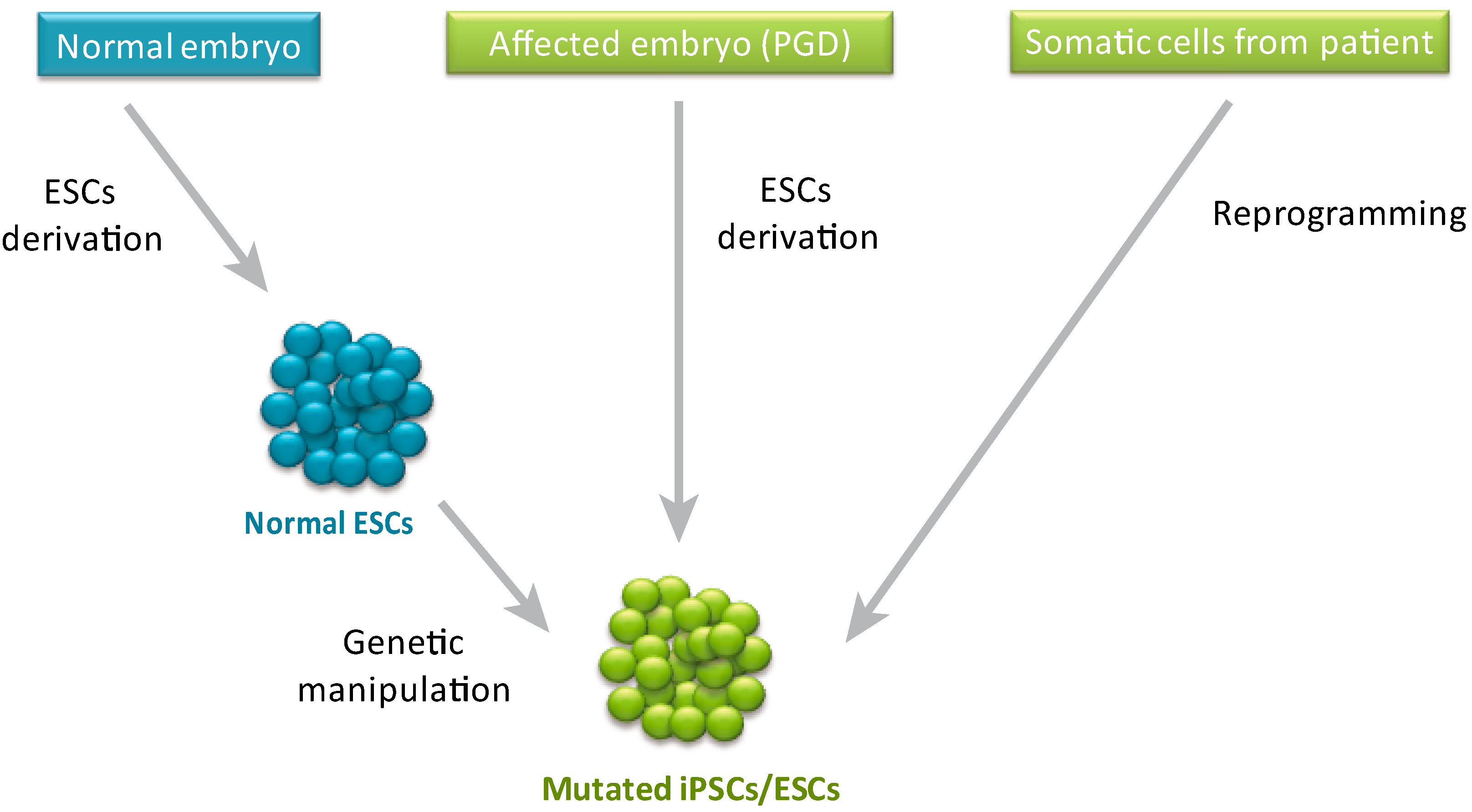
2. ESCs vs. iPSCs in Disease Modeling
- (1)
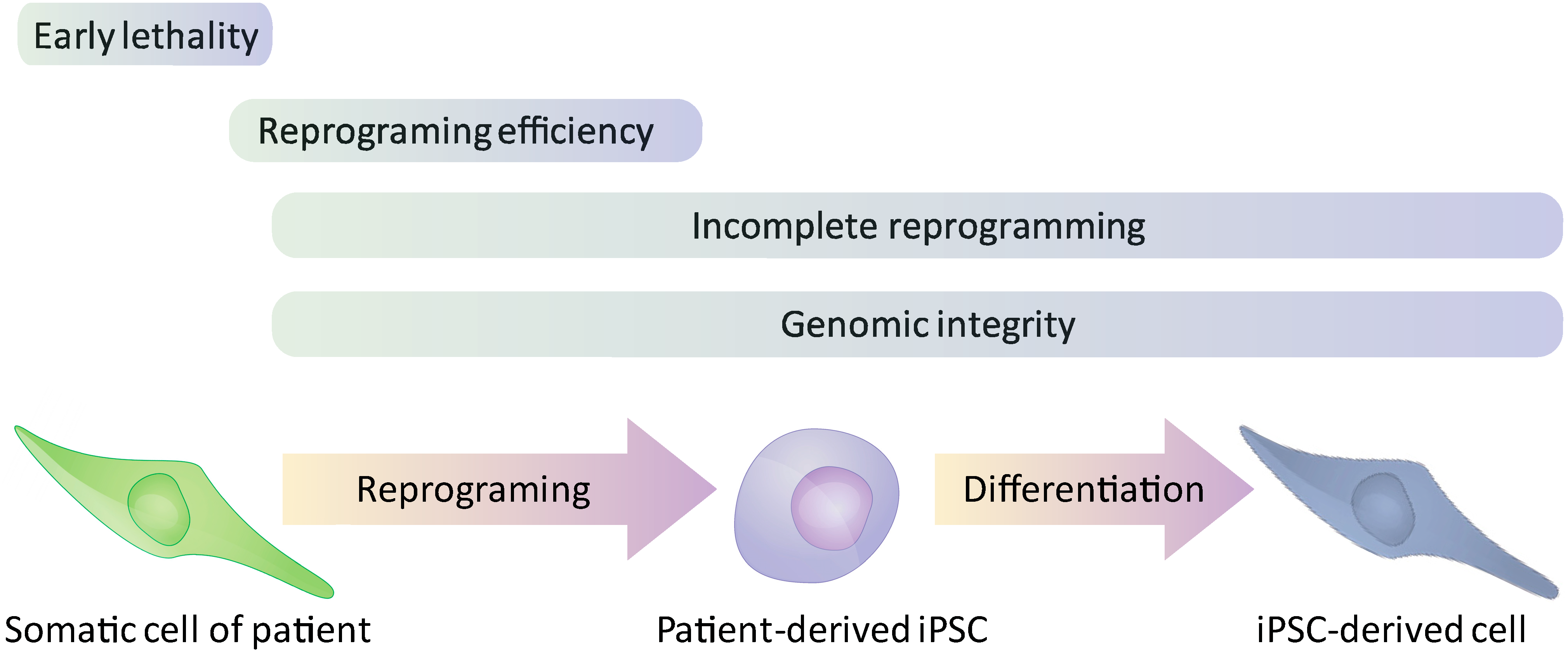
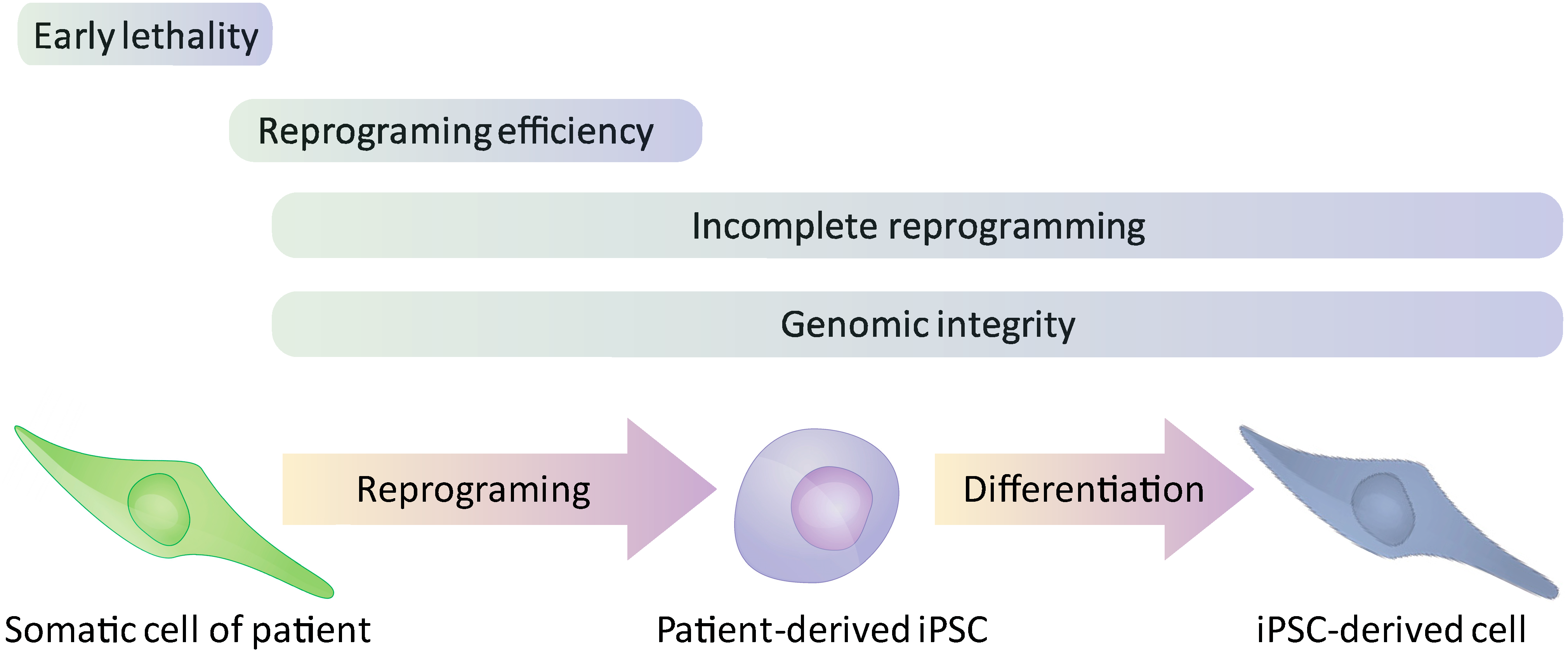
- The use of normal human ESCs to model a genetic disorder requires genetic manipulation to induce the specific mutation that one would like to study. The way to obtain a mutation that will be identical to the natural occurring mutation, seen in patients, is by genome editing. However, the efficiency of genome editing in human ESCs, before the establishment of gene targeting technologies as discussed below, was extremely low (especially in cases where a homozygous mutation was required) [17] and derivation of iPSCs that already contain the specific mutation obviates the needs for this inefficient process.
- (2)
- While the above mentioned limitation can be overcome by derivation of mutant ESCs from affected embryos identified by Preimplantation Genetic Diagnosis (PGD), this procedure is limited to a small number of diseases in which PGD is normally preformed, and can be done only in labs that are associated with in vitro fertilization (IVF) units.
- (3)
- By contrast to iPSCs from affected individuals, in the case of ESCs based models, the correlation between the genotype and the phenotype is not obvious, and the penetrance of the mutation might be low as a results of specific “protective” genetic background [18].
- (4)
3. Turner Syndrome

{kind=link}
{kind=link}
{kind=link}
| ESCs | iPSCs | ||||
|---|---|---|---|---|---|
| Reference (ref #) | Method of Derivation of Mutate ESCs | Reference (ref #) | Reprogramming Method | ||
| Fragile X | Eiges 2007 [7] | PGD | Urbach 2009 [36] | Retroviruses | |
| Rett syndrome | Cheung 2011 [37] | Retroviruses | |||
| Li 2013 [38] | Gene targeting by TALEN | Marchetto 2010 [39] | Retroviruses | ||
| Turner syndrome | Urbach 2011 [6] | Screen for XO colonies | Li 2012 [35] | Retroviruses and Lentiviruses | |
| Fanconi Anemia | Yung 2013 [25]; Tulpule 2010 [40] | Knockdown of FANCA and FANCD2 | Yung 2013 [25] | Lentiviruses | |
| Liu 2014 [41] | Gene targeting by TALEN | Liu 2014 [41] | Reprogramming | ||
| Spinal muscular atrophy | Wang 2013 [27] | Knockdown of SMN | Ebert 2009 [28] | Lentiviruses | |
| Shwachman-Diamond syndrome | Tulpule 2013 [29] | Knockdown of SBDS | Tulpule 2013 [29] | Retroviruses | |
| Long QT | Bellin 2013 [30] | Gene targeting | Bellin 2013 [30] | Retroviruses | |
| Huntington’s disease | Lu 2013 [42] | Over-expression of HTTexon1 with 23, 73 or 145 glutamine repeats in HESCs | HD iPSC Consortium 2012 [43] | Lentiviruses (OSKM + Nanog + Lin28) | ESCs > iPSCs (Based on Lu et al.) mHTT aggregated appeared only in ESCs based model |
| Myotonic Dystrophy | Seriola 2011 [31] | Du 2013 [32] | Retroviruses | ESCs ~ iPSCs TNR becomes stable upon differentiation in ES and iPS. Down-regulation of MMR genes upon differentiation was observed only in ESCs | |
4. Fanconi Anemia
5. Fragile X Syndrome
6. Huntington’s Disease
7. Disease Modeling by Gene Targeting of hPSCs
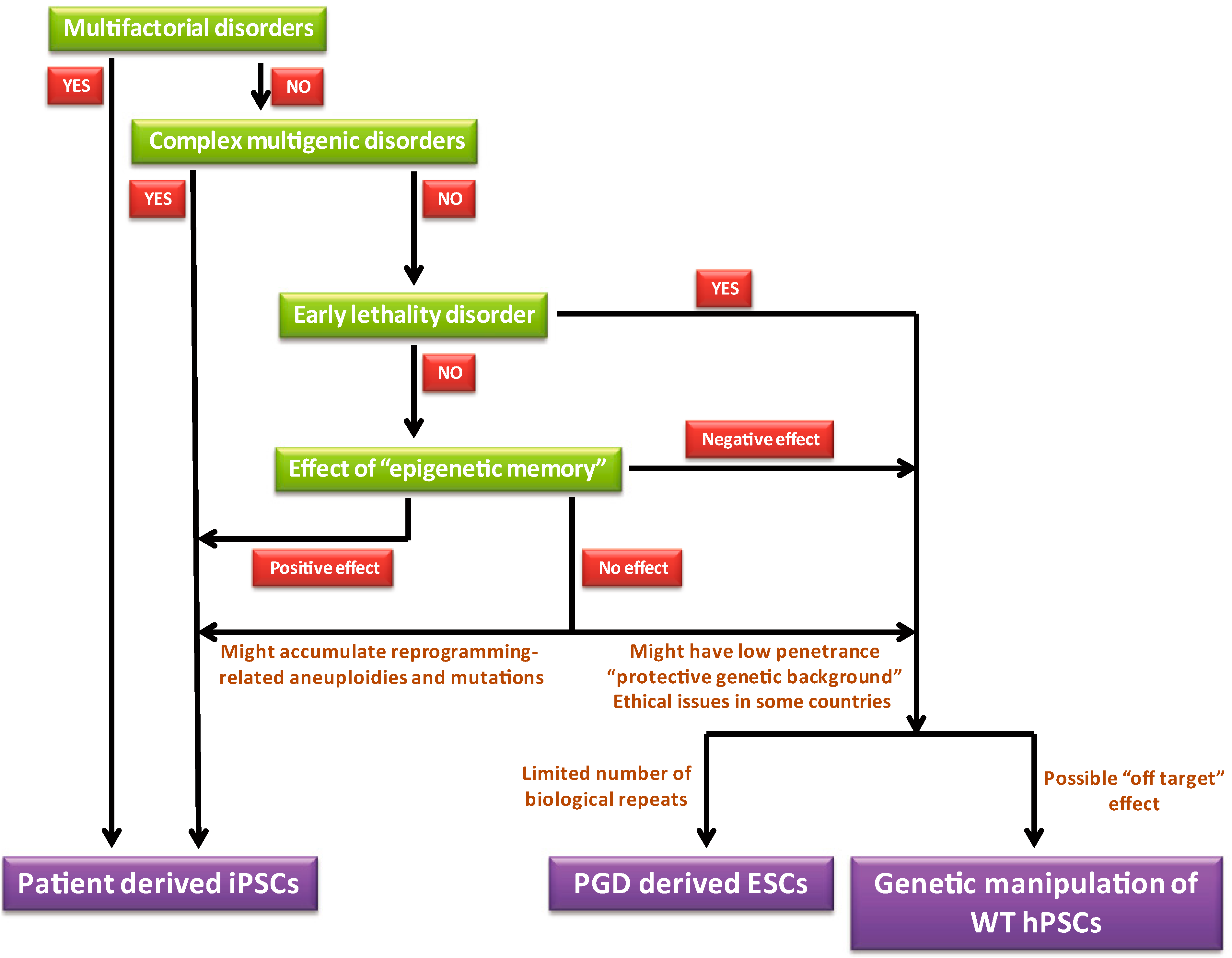
8. “Guidelines” for Choosing the Optimal Model System for a Given Disease
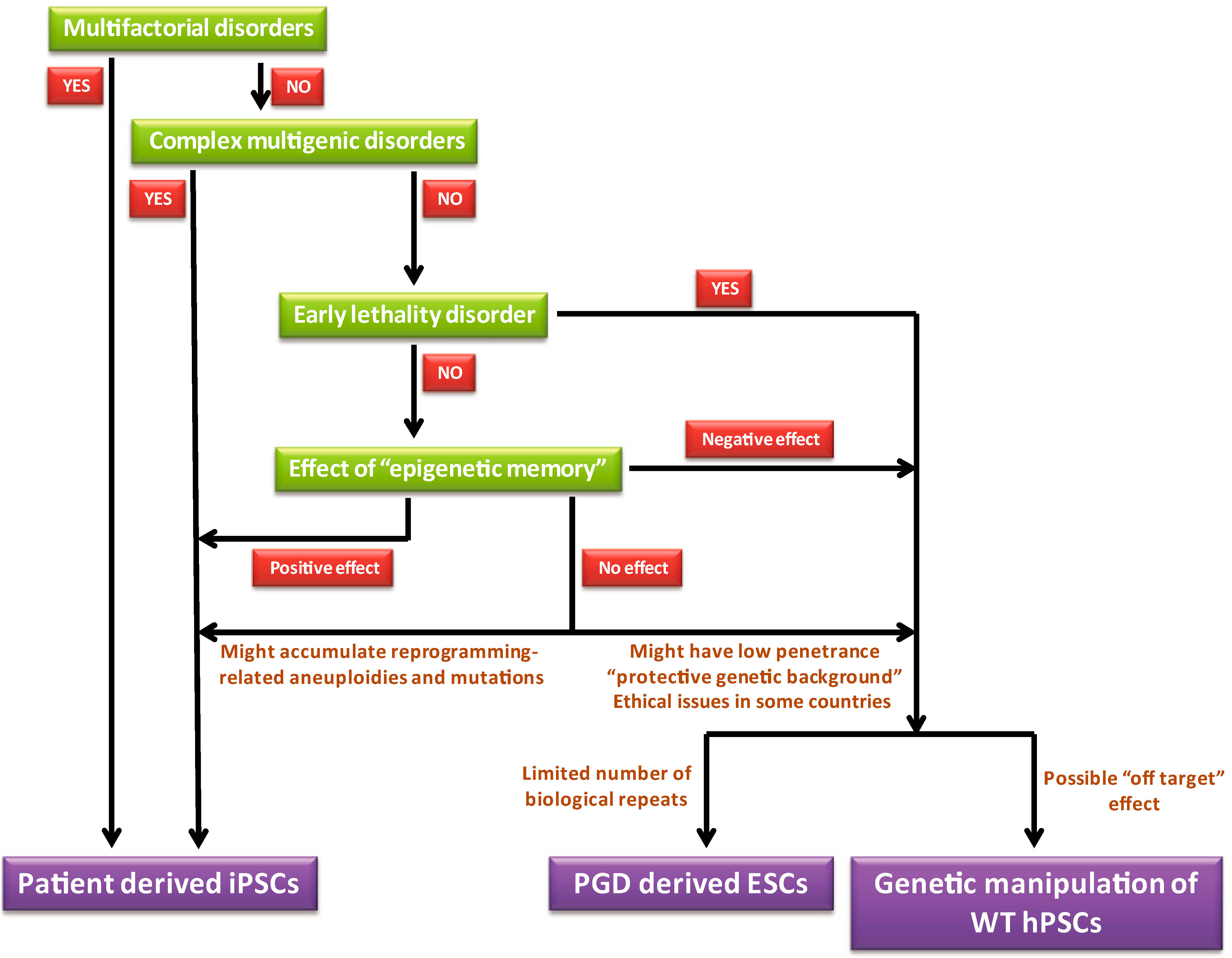
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Robinton, D.A.; Daley, G.Q. The promise of induced pluripotent stem cells in research and therapy. Nature 2012, 481, 295–305. [Google Scholar]
- Roberts, R.M.; Loh, K.M.; Amita, M.; Bernardo, A.S.; Adachi, K.; Alexenko, A.P.; Schust, D.J.; Schulz, L.C.; Telugu, B.P.V.L.; Ezashi, T.; et al. Differentiation of trophoblast cells from human embryonic stem cells: To be or not to be? Reproduction 2014, 147. [Google Scholar] [CrossRef]
- Xu, R.-H.; Chen, X.; Li, D.S.; Li, R.; Addicks, G.C.; Glennon, C.; Zwaka, T.P.; Thomson, J.A. BMP4 Initiates Human Embryonic Stem Cell Differentiation to Trophoblast. Nat. Biotechnol. 2002, 20, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, T.F. Use of rodents as models of human diseases. J. Pharm. Bioallied Sci. 2014, 6, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Urbach, A.; Schuldiner, M.; Benvenisty, N. Modeling for Lesch-Nyhan disease by gene targeting in human embryonic stem cells. Stem Cells 2004, 22, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Urbach, A.; Benvenisty, N. Studying early lethality of 45,XO (Turner’s syndrome) embryos using human embryonic stem cells. PLoS One 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Eiges, R.; Urbach, A.; Malcov, M.; Frumkin, T.; Schwartz, T.; Amit, A.; Yaron, Y.; Eden, A.; Yanuka, O.; Benvenisty, N.; et al. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 2007, 1, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Maury, Y.; Gauthier, M.; Peschanski, M.; Martinat, C. Human pluripotent stem cells for disease modelling and drug screening. Bioessays 2012, 34, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Verlinsky, Y.; Strelchenko, N.; Kukharenko, V.; Rechitsky, S.; Verlinsky, O.; Galat, V.; Kuliev, A. Human embryonic stem cell lines with genetic disorders. Reprod. Biomed. Online 2005, 10, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Mateizel, I.; Spits, C.; de Rycke, M.; Liebaers, I.; Sermon, K. Derivation, culture, and characterization of VUB hESC lines. Vitr. Cell. Dev. Biol. Anim. 2010, 46, 300–308. [Google Scholar] [CrossRef]
- Park, I.-H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Kiskinis, E.; Sandoe, J.; Williams, L.; Boulting, G.; Moccia, R.; Wainger, B.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways Disrupted in Human ALS Motor Neurons Identified through Genetic Correction of Mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [PubMed]
- Puri, M.C.; Nagy, A. Concise review: Embryonic stem cells versus induced pluripotent stem cells: The game is on. Stem Cells 2012, 30, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Bilic, J.; Izpisua Belmonte, J.C. Concise review: Induced pluripotent stem cells versus embryonic stem cells: Close enough or yet too far apart? Stem Cells 2012, 30, 33–41. [Google Scholar] [CrossRef]
- Cherry, A.B.C.; Daley, G.Q. Reprogrammed cells for disease modeling and regenerative medicine. Annu. Rev. Med. 2013, 64, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, J.-S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Sandoe, J.; Eggan, K. Opportunities and challenges of pluripotent stem cell neurodegenerative disease models. Nat. Neurosci. 2013, 16, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.A. Human embryonic stem cell research: Ethical and legal issues. Nat. Rev. Genet. 2001, 2, 74–78. [Google Scholar] [CrossRef] [PubMed]
- De Wert, G.; Mummery, C. Human embryonic stem cells: Research, ethics and policy. Hum. Reprod. 2003, 18, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nur, O.; Russ, H.A.; Efrat, S.; Benvenisty, N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 2011, 9, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Morey, R.; O’Neil, R.C.; He, Y.; Daughtry, B.; Schultz, M.D.; Hariharan, M.; Nery, J.R.; Castanon, R.; Sabatini, K.; et al. Abnormalities in human pluripotent cells due to reprogramming mechanisms. Nature 2014, 511, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Li, Z.; Fung, H.-L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Yung, S.K.; Tilgner, K.; Ledran, M.H.; Habibollah, S.; Neganova, I.; Singhapol, C.; Saretzki, G.; Stojkovic, M.; Armstrong, L.; Przyborski, S.; et al. Brief report: Human pluripotent stem cell models of fanconi anemia deficiency reveal an important role for fanconi anemia proteins in cellular reprogramming and survival of hematopoietic progenitors. Stem Cells 2013, 31, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.U.W.; Milsom, M.D.; Harris, C.E.; Vyas, R.; Brumme, K.M.; Parmar, K.; Moreau, L.A.; Schambach, A.; Park, I.H.; London, W.B.; et al. Overcoming reprogramming resistance of Fanconi anemia cells. Blood 2012, 119, 5449–5457. [Google Scholar] [PubMed]
- Wang, Z.-B.; Zhang, X.; Li, X.-J. Recapitulation of spinal motor neuron-specific disease phenotypes in a human cell model of spinal muscular atrophy. Cell Res. 2013, 23, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Yu, J.; Rose, F.F.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Tulpule, A.; Kelley, J.M.; Lensch, M.W.; McPherson, J.; Park, I.H.; Hartung, O.; Nakamura, T.; Schlaeger, T.M.; Shimamura, A.; Daley, G.Q. Pluripotent stem cell models of shwachman-diamond syndrome reveal a common mechanism for pancreatic and hematopoietic dysfunction. Cell Stem Cell 2013, 12, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Bellin, M.; Casini, S.; Davis, R.P.; D’Aniello, C.; Haas, J.; Ward-van Oostwaard, D.; Tertoolen, L.G.J.; Jung, C.B.; Elliott, D.A.; Welling, A.; et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J. 2013, 32, 3161–3175. [Google Scholar] [CrossRef] [PubMed]
- Seriola, A.; Spits, C.; Simard, J.P.; Hilven, P.; Haentjens, P.; Pearson, C.E.; Sermon, K. Huntington’s and myotonic dystrophy hESCs: Down-regulated trinucleotide repeat instability and mismatch repair machinery expression bupon differentiation. Hum. Mol. Genet. 2011, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Campau, E.; Soragni, E.; Jespersen, C.; Gottesfeld, J.M. Length-dependent CTG.CAG triplet-repeat expansion in myotonic dystrophy patient-derived induced pluripotent stem cells. Hum. Mol. Genet. 2013, 22, 5276–5287. [Google Scholar] [PubMed]
- Saenger, P. Turner’s syndrome. N. Engl. J. Med. 1996, 335, 1749–1754. [Google Scholar] [CrossRef] [PubMed]
- Ranke, M.B.; Saenger, P. Turner’s syndrome. Lancet 2001, 358, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, X.; Fan, W.; Zhao, P.; Chan, Y.C.; Chen, S.; Zhang, S.; Guo, X.; Zhang, Y.; Li, Y.; et al. Modeling abnormal early development with induced pluripotent stem cells from aneuploid syndromes. Hum. Mol. Genet. 2012, 21, 32–45. [Google Scholar] [PubMed]
- Urbach, A.; Bar-Nur, O.; Daley, G.Q.; Benvenisty, N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 2010, 6, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.Y.L.; Horvath, L.M.; Grafodatskaya, D.; Pasceri, P.; Weksberg, R.; Hotta, A.; Carrel, L.; Ellis, J. Isolation of MECP2-null Rett Syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum. Mol. Genet. 2011, 20, 2103–2115. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Muffat, J.; Cheng, A.W.; Orlando, D.A.; Lovén, J.; Kwok, S.M.; Feldman, D.A.; Bateup, H.S.; Gao, Q.; et al. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell 2013, 13, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef]
- Tulpule, A.; William Lensch, M.; Miller, J.D.; Austin, K.; D’Andrea, A.; Schlaeger, T.M.; Shimamura, A.; Daley, G.Q. Knockdown of Fanconi anemia genes in human embryonic stem cells reveals early developmental defects in the hematopoietic lineage. Blood 2010, 115, 3453–3462. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-H.; Suzuki, K.; Li, M.; Qu, J.; Montserrat, N.; Tarantino, C.; Gu, Y.; Yi, F.; Xu, X.; Zhang, W.; et al. Modelling Fanconi anemia pathogenesis and therapeutics using integration-free patient-derived iPSCs. Nat. Commun. 2014, 5, 4330. [Google Scholar] [PubMed]
- Lu, B.; Palacino, J. A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. 2013, 27, 1820–1829. [Google Scholar] [CrossRef] [PubMed]
- HD iPSC Consortium. Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 2012, 11, 264–278. [Google Scholar]
- D’Andrea, A.D.; Grompe, M. Molecular biology of Fanconi anemia: Implications for diagnosis and therapy. Blood 1997, 90, 1725–1736. [Google Scholar]
- Raya, A.; Rodríguez-Pizà, I.; Guenechea, G.; Vassena, R.; Navarro, S.; Barrero, M.J.; Consiglio, A.; Castellà, M.; Río, P.; Sleep, E.; et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature 2009, 460, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Boyle, L.; Kaufmann, W.E. The behavioral phenotype of FMR1 mutations. Am. J. Med. Genet. Part C Semin. Med. Genet. 2010, 154C, 469–476. [Google Scholar] [CrossRef]
- Penagarikano, O.; Mulle, J.G.; Warren, S.T. The pathophysiology of fragile X syndrome. Annu. Rev. Genomics Hum. Genet. 2007, 8, 109–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Bray, S.M.; Warren, S.T. New perspectives on the biology of fragile X syndrome. Curr. Opin. Genet. Dev. 2012, 22, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Callan, M.A.; Zarnescu, D.C. Heads-up: New roles for the fragile X mental retardation protein in neural stem and progenitor cells. Genesis 2011, 49, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.R.; Bray, S.M.; Warren, S.T. Molecular mechanisms of fragile X syndrome: A twenty-year perspective. Annu. Rev. Pathol. 2012, 7, 219–245. [Google Scholar] [CrossRef] [PubMed]
- Devys, D.; Lutz, Y.; Rouyer, N.; Bellocq, J.P.; Mandel, J.L. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 1993, 4, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Turetsky, T.; Aizenman, E.; Gil, Y.; Weinberg, N.; Shufaro, Y.; Revel, A.; Laufer, N.; Simon, A.; Abeliovich, D.; Reubinoff, B.E. Laser-assisted derivation of human embryonic stem cell lines from IVF embryos after preimplantation genetic diagnosis. Hum. Reprod. 2008, 23, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Telias, M.; Segal, M.; Ben-Yosef, D. Neural differentiation of Fragile X human Embryonic Stem Cells reveals abnormal patterns of development despite successful neurogenesis. Dev. Biol. 2013, 374, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Hatton, D.D.; Sideris, J.; Skinner, M.; Mankowski, J.; Bailey, D.B.; Roberts, J.; Mirrett, P. Autistic behavior in children with fragile X syndrome: Prevalence, stability, and the impact of FMRP. Am. J. Med. Genet. Part A 2006, 140, 1804–1813. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Cortell, R.; Kau, A.S.M.; Bukelis, I.; Tierney, E.; Gray, R.M.; Cox, C.; Capone, G.T.; Stanard, P. Autism spectrum disorder in fragile X syndrome: Communication, social interaction, and specific behaviors. Am. J. Med. Genet. A 2004, 129A, 225–234. [Google Scholar] [CrossRef]
- Pietrobono, R.; Tabolacci, E.; Zalfa, F.; Zito, I.; Terracciano, A.; Moscato, U.; Bagni, C.; Oostra, B.; Chiurazzi, P.; Neri, G. Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum. Mol. Genet. 2005, 14, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Smeets, H.J.; Smits, A.P.; Verheij, C.E.; Theelen, J.P.; Willemsen, R.; van de Burgt, I.; Hoogeveen, A.T.; Oosterwijk, J.C.; Oostra, B.A. Normal phenotype in two brothers with a full FMR1 mutation. Hum. Mol. Genet. 1995, 4, 2103–2108. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, S.D.; Theriault, K.M.; Reis, S.A.; Zhou, F.; Madison, J.M.; Daheron, L.; Loring, J.F.; Haggarty, S.J. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One 2011, 6, e26203. [Google Scholar] [CrossRef] [PubMed]
- Alisch, R.S.; Wang, T.; Chopra, P.; Visootsak, J.; Conneely, K.N.; Warren, S.T. Genome-wide analysis validates aberrant methylation in fragile X syndrome is specific to the FMR1 locus. BMC Med. Genet. 2013, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nur, O.; Caspi, I.; Benvenisty, N. Molecular analysis of FMR1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell Biol. 2012, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.B.; Gusella, J.F. Huntington’s disease. Pathogenesis and management. N. Engl. J. Med. 1986, 315, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Huntington, T.; Macdonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groat, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Landles, C.; Sathasivam, K.; Weiss, A.; Woodman, B.; Moffitt, H.; Finkbeiner, S.; Sun, B.; Gafni, J.; Ellerby, L.M.; Trottier, Y.; et al. Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J. Biol. Chem. 2010, 285, 8808–8823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; An, M.C.; Montoro, D.; Ellerby, L.M. Characterization of human Huntington’s disease cell model from induced pluripotent stem cells. PLoS Curr. 2010, 2, 1–11. [Google Scholar] [CrossRef]
- Camnasio, S.; Carri, A.D.; Lombardo, A.; Grad, I.; Mariotti, C.; Castucci, A.; Rozell, B.; Riso, P.L.; Castiglioni, V.; Zuccato, C.; et al. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol. Dis. 2012, 46, 41–51. [Google Scholar] [CrossRef]
- Bradley, C.K.; Scott, H.A.; Chami, O.; Peura, T.T.; Dumevska, B.; Schmidt, U.; Stojanov, T. Derivation of Huntington’s disease-affected human embryonic stem cell lines. Stem Cells Dev. 2011, 20, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.Y.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halevy, T.; Urbach, A. Comparing ESC and iPSC—Based Models for Human Genetic Disorders. J. Clin. Med. 2014, 3, 1146-1162. https://doi.org/10.3390/jcm3041146
Halevy T, Urbach A. Comparing ESC and iPSC—Based Models for Human Genetic Disorders. Journal of Clinical Medicine. 2014; 3(4):1146-1162. https://doi.org/10.3390/jcm3041146
Chicago/Turabian StyleHalevy, Tomer, and Achia Urbach. 2014. "Comparing ESC and iPSC—Based Models for Human Genetic Disorders" Journal of Clinical Medicine 3, no. 4: 1146-1162. https://doi.org/10.3390/jcm3041146
APA StyleHalevy, T., & Urbach, A. (2014). Comparing ESC and iPSC—Based Models for Human Genetic Disorders. Journal of Clinical Medicine, 3(4), 1146-1162. https://doi.org/10.3390/jcm3041146