
A Novel Screening Approach for Familial Hypercholesterolemia: A Genetic Study on Patients Detected Using Preexisting Centralized Analytics

Abstract
1. Introduction
2. Materials and Methods
Patient Selection
3. Genetic Analysis
4. Statistical Analysis
5. Results
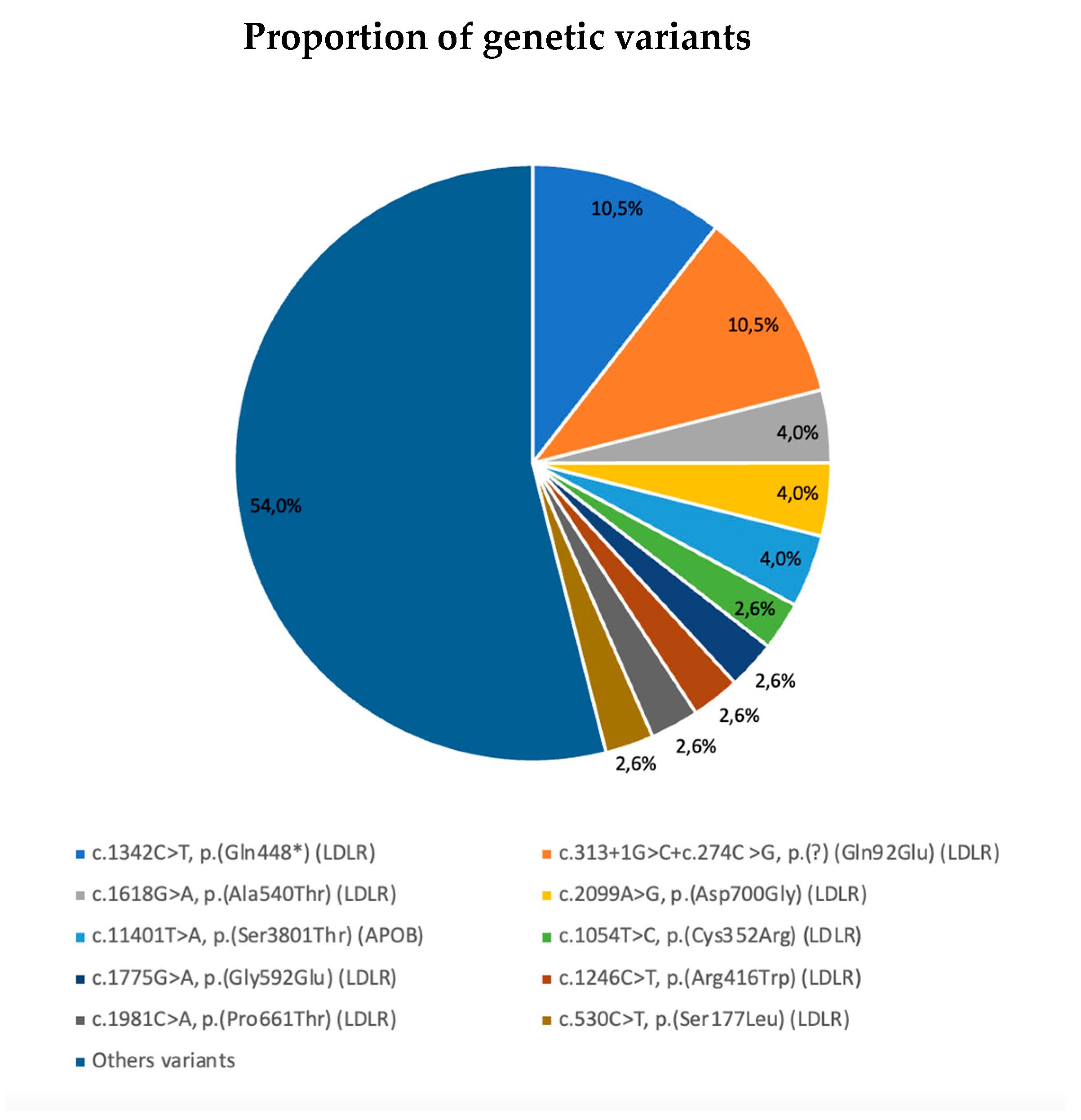
5.1. Mutational Analysis
5.2. Phenotype Versus Genotype
6. Discussion
7. What Is New?
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, e34–e47. [Google Scholar] [CrossRef] [PubMed]
- Austin, M.A.; Hutter, C.M.; Zimmern, R.L.; Humphries, S.E. Familial hypercholesterolemia and coronary heart disease: A HuGE association review. Am. J. Epidemiol. 2004, 160, e421–e429. [Google Scholar] [CrossRef] [PubMed]
- Pérez de Isla, L.; Alonso, R.; Mata, N.; Saltijeral, A.; Muñiz, O.; Rubio-Marin, P.; Diaz-Diaz, J.L.; Fuentes, F.; de Andrés, R.; Zambón, D.; et al. Coronary heart disease, peripheral arterial disease, and stroke in familial hypercholesterolaemia: Insights from the SAFEHEART registry (Spanish familial hypercholesterolaemia cohort study). Arterioscler. Thromb. Vasc. Biol. 2016, 36, e2004–e2010. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.; Wonderling, D.; Thorogood, M.; Lambert, H.; Humphries, S.E.; Neil, H.A. Screening for hypercholesterolaemia versus case finding for familial hypercholesterolaemia: A systematic review and cost-effectiveness analysis. Health Technol. Assess. 2000, 4, e1–e123. [Google Scholar] [CrossRef]
- De Isla, L.P.; Arroyo-Olivares, R.; Alonso, R.; Muñiz-Grijalvo, O.; Díaz-Díaz, J.L.; Zambón, D.; Fuentes, F.; Mata, N.; Piedecausa, M.; Mañas, M.D.; et al. Incidence of Cardiovascular Events and Changes in the Estimated Risk and Treatment of Familial Hypercholesterolemia: The SAFEHEART Registry. Rev. Española Cardiol. (Engl. Ed.) 2020, 73, 828–834. [Google Scholar] [CrossRef]
- Singh, A.; Gupta, A.; Collins, B.L.; Qamar, A.; Monda, K.L.; Biery, D.; Lopez, J.A.G.; de Ferranti, S.D.; Plutzky, J.; Cannon, C.P.; et al. Familial Hypercholesterolemia Among Young Adults With Myocardial Infarction. J. Am. Coll. Cardiol. 2019, 73, 2439–2450. [Google Scholar] [CrossRef]
- Mata, P.; Alonso, R.; González-Juanatey, J.R.; Badimón, L.; Díaz-Díaz, J.L.; Muñoz, M.T. Diagnóstico y tratamiento de la hiper-colesterolemia familiar en España. Documento de consenso. Aten Primaria. 2015, 47, 56–65. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef]
- Benn, M.; Watts, G.F.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Corrigendum to Familial Hypercholesterolemia in the Danish General Population: Prevalence, Coronary Artery Disease, and Cholesterol-Lowering Medication. J. Clin. Endocrinol. Metab. 2014, 99, 4758–4759. [Google Scholar]
- Wierzbicki, A.S.; Humphries, S.E.; Minhas, R. Guideline Development Group. Familial hypercholesterolemia: Summary of NICE guidance. BMJ 2008, 337, a1095. [Google Scholar] [CrossRef]
- World Health Organization. Familial Hypercholesterolemia: Report of a Second WHO Consultation; WHO Publication No. WHO7HGN/FH/CONS/99.2; World Health Organisation, Human Genetics programme, Division of Non-communicable Diseases: Geneva, Switzerland, 1999. [Google Scholar]
- Sturm, A.C.; Knowles, J.W.; Gidding, S.S.; Ahmad, Z.S.; Ahmed, C.D.; Ballantyne, C.M.; Baum, S.J.; Bourbon, M.; Carrié, A.; Cuchel, M.; et al. Clinical Genetic Testing for Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2018, 72, 662. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; de Ferranti, S.D.; Durrington, M.P.H.P. Inherited Disorders of LDL-Cholesterol Metabolism; UpToDate: Waltham, MA, USA, 2014. [Google Scholar]
- Sabatel-Pérez, F.; Sánchez-Prieto, J.; Becerra-Muñoz, V.M.; Alonso-Briales, J.H.; Mata, P.; Rodríguez-Padial, L. Improving Familial Hypercholesterolemia Index Case Detection: Sequential Active Screening from Centralized Analytical Data. J. Clin. Med. 2021, 10, 749. [Google Scholar] [CrossRef] [PubMed]
- Amor-Salamanca, A.; Castillo, S.; Gonzalez-Vioque, E.; Dominguez, F.; Quintana, L.; Lluís-Ganella, C.; Escudier, J.M.; Ortega, J.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Genetically Confirmed Familial Hypercholesterolemia in Patients with Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2017, 70, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Watts, G.F.; Gidding, S.; Wierzbicki, A.S.; Toth, P.P.; Alonso, R.; Brown, W.V.; Bruckert, E.; Defesche, J.; Lin, K.K.; Livingston, M.; et al. Integrated guidance on the care of familial hypercholesterolaemia from the International FH Foundation. Int. J. Cardiol. 2014, 171, 309–325. [Google Scholar] [CrossRef]
- Stock, J. New EAS Consensus Statement on FH: Improving the care of FH patients. Atherosclerosis 2013, 231, 69–71. [Google Scholar] [CrossRef]
- Wintjens, R.; Bozon, D.; Belabbas, K.; Mbou, F.; Girardet, J.-P.; Tounian, P.; Jolly, M.; Boccara, F.; Cohen, A.; Karsenty, A.; et al. Global molecular analysis and APOE mutations in a cohort of autosomal dominant hypercholesterolemia patients in France. J. Lipid Res. 2016, 57, 482–491. [Google Scholar] [CrossRef]
- Fouchier, S.W.; Dallinga-Thie, G.M.; Meijers, J.C.; Zelcer, N.; Kastelein, J.J.; Defesche, J.C.; Hovingh, G.K. Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ. Res. 2014, 115, 552–555. [Google Scholar] [CrossRef]
- Roberts, R.; Chang, C.; Hadley, T. Genetic Risk Stratification: A Paradigm Shift in Prevention of Coronary Artery Disease. J. Am. Coll. Cardiol. Basic Trans. Sci. 2021, 6, 287–304. [Google Scholar]
- Iribarren, C.; Lu, M.; Jorgenson, E.; Martínez, M.; Lluis-Ganella, C.; Subirana, I.; Salas, E.; Elosua, R. Clinical Utility of Multimarker Genetic Risk Scores for Prediction of Incident Coronary Heart Disease: A Cohort Study Among Over 51 Thousand Individuals of European Ancestry. Circ. Cardiovasc. Genet. 2016, 9, 531–540. [Google Scholar] [CrossRef]
- Ascaso, J.F.; Civeira, F.; Guijarro, C.; López Miranda, J.; Masana, L.; Mostaza, J.M.; Pedro-Botet, J.; Pintó, X.; Valdivielso, P. Indications of PCSK9 inhibitors in clinical practice. Recommendations of the Spanish Sociey of Arteriosclerosis (SEA). Clin. Investig. Arterioscler. 2019, 31, 128–139. [Google Scholar] [PubMed]
- Bourbon, M.; Alves, A.C.; Alonso, R.; Mata, N.; Aguiar, P.; Padró, T.; Mata, P. Mutational Analysis and Genotype-Phenotype Relation in Familial Hypercholesterolemia: The SAFEHEART Registry. Atherosclerosis 2017, 262, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Diakou, M.; Miltiadous, G.; Xenophontos, S.L.; Manoli, P.; Cariolou, M.A.; Elisaf, M. Spectrum of LDLR Gene Mutations, Including a Novel Mutation Causing Familial Hypercholesterolaemia, in North-Western Greece. Eur. J. Intern. Med. 2011, 22, e55–e59. [Google Scholar] [CrossRef]
- Esser, V.; Russell, D.W. Transport-deficient mutations in the low density lipoprotein receptor: Alterations in the cysteine rich and cysteine-poor regions of the protein block intracellular transport. J. Biol. Chem. 1988, 263, 13276–13281. [Google Scholar] [CrossRef]
- Jensen, H.; Jensen, L.; Meinertz, H.; Hansen, P.; Gregersen, N.; Færgeman, O. Spectrum of LDL Receptor Gene Mutations in Denmark: Implications for Molecular Diagnostic Strategy in Heterozygous Familial Hypercholesterolemia. Atherosclerosis 1999, 146, 337–344. [Google Scholar] [CrossRef]
- Medeiros, A.M.; Alves, A.C.; Bourbon, M. Mutational analysis of a cohort with clinical diagnosis of familial hypercholesterolemia: Considerations for genetic diagnosis improvement. Genet. Med. 2016, 18, e316–e324. [Google Scholar] [CrossRef]
- Usifo, E.; Leigh, S.E.A.; Whittall, R.A.; Lench, N.; Taylor, A.; Yeats, C.; Orengo, C.A.; Martin, A.C.R.; Celli, J.; Humphries, S.E. Low density lipoprotein receptor gene familial hypercholesterolemia variant database: Update and pathological assessment. Ann. Hum. Genet. 2012, 76, e387–e401. [Google Scholar] [CrossRef]
- Ferrieres, J.; Lambert, J.; Lussier-Cacan, S.; Davignon, J. Coronary artery disease in heterozygous familial hypercholesterolaemia patients with the same LDL receptor gene mutation. Circulation 1995, 92, 290–295. [Google Scholar] [CrossRef]
- Jansen, A.C.M.; van Aalst-Cohen, E.S.; Tanck, M.W.; Trip, M.D.; Lansberg, P.J.; Liem, A.H.; van Lennep, H.W.O.R.; Sijbrands, E.J.G.; Kastelein, J.J.P. The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: Data in 2400 patients. J. Intern. Med. 2004, 256, 482–490. [Google Scholar] [CrossRef]
- Alonso, R.; Mata, N.; Castillo, S.; Fuentes, F.; Saenz, P.; Muñiz, O.; Galiana, J.; Figueras, R.; Diaz, J.; Gomez-Enterría, P.; et al. Cardiovascular Disease in Familial Hypercholesterolaemia: Influence of Low-Density Lipoprotein Receptor Mutation Type and Classic Risk Factors. Atherosclerosis 2008, 200, 315–321. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the Management of Dyslipidaemias: Lipid Modification to Reduce Cardiovascular Risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Variables | Global (n = 469) | DLCN 3–5 (n = 385) | DLCN ≥ 6 (n = 84) | Standardized Effect Size (CI 95%) (a) |
|---|---|---|---|---|
| Men/Women | 191 (40.7)/278 (59.3) | 162 (42.1)/223 (57.9) | 29 (34.5)/55 (65.5) | 0.73 (0.44–1.19) |
| Age | 53.2 ± 12.8 | 54.6 ± 12.3 | 47.1 ± 12.9 | 0.59 (0.36–0.82) |
| HTA | 148 (31.6) | 130 (33.8) | 18 (21.4) | 0.54 (0.31–0.94) |
| DM2 | 47 (10.0) | 43 (11.2) | 4 (4.8) | 0.39 (0.14–1.14) |
| History of Smoking | 233 (49.7) | 181 (47.0) | 52 (61.9) | 1.83 (1.13–2.97) |
| Average BMI | 27.7 ± 4.4 | 27.8 ± 4.3 | 27.0 ± 4.7 | 0.19 (−0.04–0.43) |
| Normal weight | 135 (28.8) | 104 (27.1) | 31 (36.9) | 1.57 (0.96–2.59) |
| Overweight | 209 (44.6) | 173 (45.1) | 36 (42.9) | 0.92 (0.57–1.47) |
| Obesity | 124 (26.4) | 107 (27.9) | 17 (20.2) | 0.66 (0.37–1.17) |
| Average DLCN score | 4.25 ± 1.9 | 3.52 ± 0.8 | 7.56 ± 2.2 | −2.07 (−2.21–−1.92) |
| Hypothyroidism | 37 (7.9) | 29 (7.5) | 8 (9.5) | 1.29 (0.57–2.94) |
| Chronic kidney disease | 14 (3) | 13 (3.4) | 1 (1.2) | 0.35 (0.04–2.67) |
| Cardiovascular disease | 26 (5.5) | 13 (3.4) | 13 (15.5) | 5.24 (2.33–11.77) |
| Family history of early ischemic heart disease | 34 (7.2) | 15 (3.9) | 19 (22.6) | 7.21 (3.49–14.91) |
| Family history of hypercholesterolemia | 131 (27.9) | 61 (15.9) | 70 (83.3) | 26.48 (14.02–49.99) |
| Corneal arcus (≤45 years) | 7 (6.2) | 0 | 7 (20.6) | - |
| Xanthomas | 1 (0.2) | 0 | 1 (1.1) | - |
| Total Cholesterol (mg/dL) | 320.4 ± 25.3 | 316.1 ± 18.6 | 340.4 ± 38.9 | −0.96 (−1.18–−0.74) |
| LDL-C (mg/dL) | 238.3 ± 21.9 | 233.6 ± 13.6 | 259.8 ± 35.9 | −1.19 (−1.40–−0.98) |
| HDL-C (mg/dL) | 56.2 ± 14.0 | 56.4 ± 13.6 | 55.2 ± 15.9 | 0.08 (−0.15–0.33) |
| Triglycerides (mg/dL) | 128.3 ± 36.3 | 129.5 ± 35.8 | 122.8 ± 38.4 | 0.18 (−0.05–0.42) |
| TSH (mlU/L) | 2.1 ± 2.12 | 2.1 ± 2.3 | 2.2 ± 1.30 | −0.08 (−0.32–0.17) |
| Hypolipidemic | 344 (73.4) | 269 (69.9) | 75 (89.3) | 3.59 (1.74–7.42) |
| High potency hypolipidemic | 109 (23.2) | 64 (16.6) | 45 (53.6) | 5.79 (3.49–9.59) |
| cDNA | Protein | Gene | Exon | Effect | Pathogenic Class | Allele Type | N (%) |
|---|---|---|---|---|---|---|---|
| c.1342C>T, | p.(Gln448*) | LDLR (NM_000527.4) | 9 | Exonic-NonSense | Class I | Null | 8/76 (10.5%) |
| c.313+1G>C+c.274C>G | p.(?) (Gln92Glu) | LDLR (NM_000527.4) | 3i | Intronic-Splicing | Class I | Not available | 8/76 (10.5%) |
| c.1618G>A | p.(Ala540Thr) | LDLR (NM_000527.4) | 11 | Exonic-Missense | Class II | Defective | 3/76 (3.9%) |
| c.2099A>G | p.(Asp700Gly) | LDLR (NM_000527.4) | 14 | Exonic-Missense | Class II | Not available | 3/76 (3.9%) |
| c.11401T>A | p.(Ser3801Thr) | APOB (NM_000384.2) | 26 | Exonic_Missense | Class III | Not available | 3/76 (3.9%) |
| c.1054T>C | p.(Cys352Arg) | LDLR (NM_000527.4) | 7 | Exonic-Missense | Class II | Not available | 2/76 (2.6%) |
| c.1775G>A | p.(Gly592Glu) | LDLR (NM_000527.4) | 12 | Exonic-Missense | Class I | Defective | 2/76 (2.6%) |
| c.1246C>T | p.(Arg416Trp) | LDLR (NM_000527.4) | 9 | Exonic-Missense | Class I | Defective | 2/76 (2.6%) |
| c.1981C>A | p.(Pro661Thr) | LDLR (NM_000527.4) | 13 | Exonic-Missense | Class III | Not available | 2/76 (2.6%) |
| c.530C>T | p.(Ser177Leu) | LDLR (NM_000527.4) | 4 | Exonic-Missense | Class I | Null | 2/76 (2.6%) |
| Allele Type | N | Total Cholest (mg/dL)/IQR | HDLC (mg/dL)/IQR | Triglycerides (mg/dL)/IQR | LDLC (mg/dL)/IQR | DLCN (points)/IQR |
|---|---|---|---|---|---|---|
| Defective | 19 | 339 (291.1–386.9) | 53.1 (42.9–63.2) | 110.9 (71.5–150.3) | 287.6 (244.2–330.9) | 7.3 (5.0–9.6) |
| Not available | 18 | 337.4 (302.1–372.6) | 53.2 (38.2–68.3) | 128.7 (90.4–167.0) | 305.6 (253.1–358.1) | 7.3 (6.1–8.6) |
| Null | 22 | 355.7 (312.0–399.4) | 52.9 (36.2–69.7) | 126.8 (86.1–167.5) | 321.4 (257.8–385.0) | 8.2 (6.1–10.3) |
| Effect | ||||||
| Exonic-Del | 3 | 400.0 (377.3–422.7) | 46.7 (42.1–51.3) | 147.3 (95.3–199.4) | 363.3 (288.8–437.9) | 8 (6.3–9.7) |
| Exonic-Deletion-InFrame | 1 | 442 | 99 | 118 | 319 | 6 |
| Exonic-Synonymous | 1 | 382 | 49 | 190 | 295 | 7 |
| Intronic-Splicing | 9 | 347.1 (295.0–399.2) | 48.1 (41.2–55.0) | 112.0 (70.0–153.9) | 340.8 (292.3–389.2) | 9.5 (7.5–11.6) |
| Missense | 32 | 334.6 (297.6–371.6) | 53.4 (42.4–64.3) | 117.1 (83.1–151.2) | 290.3 (240.8–339.9) | 7.0 (5.5–8.6) |
| Nonsense | 12 | 345.8 (305.3–386.3) | 53.2 (32.2–74.3) | 126.0 (80.9–171.1) | 306.2 (244.1–368.2) | 8.1 (5.7–10.6) |
| Promoter | 1 | 333 | 63 | 195 | 300 | 7 |
| Pathogenic class | ||||||
| Class I | 32 | 351.1 (300.0–402.1) | 53.0 (37.1–69.0) | 117.7 (79.2–156.2) | 319.7 (260.7–378.7) | 8.2 (5.9–10.6) |
| Class II | 17 | 335.5 (307.5–363.5) | 53.9 (43.5–64.3) | 107.9 (73.8–141.9) | 289.6 (238.3–340.9) | 7.0 (5.9–8.1) |
| Class III | 10 | 340.0 (306.0–374.0) | 51.8 (37.2–66.3) | 161.2 (132.1–190.3) | 288.2 (249.5–236.9) | 6.9 (5.9–7.9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Prieto, J.; Sabatel, F.; Moreno, F.; Arias, M.A.; Rodríguez-Padial, L. A Novel Screening Approach for Familial Hypercholesterolemia: A Genetic Study on Patients Detected Using Preexisting Centralized Analytics. J. Clin. Med. 2025, 14, 2780. https://doi.org/10.3390/jcm14082780
Sánchez-Prieto J, Sabatel F, Moreno F, Arias MA, Rodríguez-Padial L. A Novel Screening Approach for Familial Hypercholesterolemia: A Genetic Study on Patients Detected Using Preexisting Centralized Analytics. Journal of Clinical Medicine. 2025; 14(8):2780. https://doi.org/10.3390/jcm14082780
Chicago/Turabian StyleSánchez-Prieto, Joaquín, Fernando Sabatel, Fátima Moreno, Miguel A. Arias, and Luis Rodríguez-Padial. 2025. "A Novel Screening Approach for Familial Hypercholesterolemia: A Genetic Study on Patients Detected Using Preexisting Centralized Analytics" Journal of Clinical Medicine 14, no. 8: 2780. https://doi.org/10.3390/jcm14082780
APA StyleSánchez-Prieto, J., Sabatel, F., Moreno, F., Arias, M. A., & Rodríguez-Padial, L. (2025). A Novel Screening Approach for Familial Hypercholesterolemia: A Genetic Study on Patients Detected Using Preexisting Centralized Analytics. Journal of Clinical Medicine, 14(8), 2780. https://doi.org/10.3390/jcm14082780