
The Role of NLRP3 Inflammasome in Type 2 Diabetes Mellitus and Its Macrovascular Complications


 ,
,  ,
,  ,
,  , ,
, , 

Abstract
1. Introduction
2. Search Methodology
3. The NLRP3 Inflammasome; Structure and Activation
4. Regulation of T2DM by NLRP3 Inflammasome
5. NLRP3 Inflammasome and Diabetic Macrovascular Disease
6. NLRP3 Inflammasome-Targeted Pharmacotherapy
6.1. MCC950
6.2. Glyburide Derivatives
6.3. Bay 11-7082
6.4. OLT1177
6.5. Colchicine
6.6. CY-09
6.7. Tranilast
6.8. INF4E
6.9. Hydrogen Sulfide Donors
7. Off-Target Modulation of the NLRP3 Inflammasome by Conventional Drugs and Natural Compounds
7.1. Diabetic Medications and NLRP3 Modulation
7.2. Other Pharmaceutical Agents
7.3. Natural Compounds and Derivatives
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Cole, J.B.; Florez, J.C. Genetics of Diabetes Mellitus and Diabetes Complications. Nat. Rev. Nephrol. 2020, 16, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Menini, S.; Iacobini, C.; Vitale, M.; Pugliese, G. The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders. Cells 2020, 9, 1812. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Theofilis, P.; Patoulias, D.; Vlachakis, P.K.; Antoniadis, A.P.; Fragakis, N. Diabetes-Driven Atherosclerosis: Updated Mechanistic Insights and Novel Therapeutic Strategies. Int. J. Mol. Sci. 2025, 26, 2196. [Google Scholar] [CrossRef]
- Dal Canto, E.; Ceriello, A.; Rydén, L.; Ferrini, M.; Hansen, T.B.; Schnell, O.; Standl, E.; Beulens, J.W.J. Diabetes as a Cardiovascular Risk Factor: An Overview of Global Trends of Macro and Micro Vascular Complications. Eur. J. Prev. Cardiol. 2019, 26, 25–32. [Google Scholar] [CrossRef]
- Lontchi-Yimagou, E.; Sobngwi, E.; Matsha, T.E.; Kengne, A.P. Diabetes Mellitus and Inflammation. Curr. Diab Rep. 2013, 13, 435–444. [Google Scholar] [CrossRef]
- Huang, D.; Refaat, M.; Mohammedi, K.; Jayyousi, A.; Al Suwaidi, J.; Abi Khalil, C. Macrovascular Complications in Patients with Diabetes and Prediabetes. Biomed. Res. Int. 2017, 2017, 7839101. [Google Scholar] [CrossRef]
- Henning, R.J. Type-2 Diabetes Mellitus and Cardiovascular Disease. Future Cardiol. 2018, 14, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Nițulescu, I.M.; Ciulei, G.; Cozma, A.; Procopciuc, L.M.; Orășan, O.H. From Innate Immunity to Metabolic Disorder: A Review of the NLRP3 Inflammasome in Diabetes Mellitus. J. Clin. Med. 2023, 12, 6022. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- de Zoete, M.R.; Palm, N.W.; Zhu, S.; Flave, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef]
- Sepehri, Z.; Kiani, Z.; Afshari, M.; Kohan, F.; Dalvand, A.; Ghavami, S. Inflammasomes and Type 2 Diabetes: An Updated Systematic Review. Immunol. Lett. 2017, 192, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Li, Q.; Xu, G.; Xiao, X.; Bai, Z. The Mechanism of NLRP3 Inflammasome Activation and Its Pharmacological Inhibitors. Front. Immunol. 2023, 13, 1109938. [Google Scholar] [CrossRef]
- Bulté, D.; Rigamonti, C.; Romano, A.; Mortellaro, A. Inflammasomes: Mechanisms of Action and Involvement in Human Diseases. Cells 2023, 12, 1766. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mezzaroma, E.; Buckley, L.F.; Potere, N.; Di Nisio, M.; Biondi-Zoccai, G.; Van Tassell, B.W.; Abbate, A. Targeting the NLRP3 Inflammasome in Cardiovascular Diseases. Pharmacol. Ther. 2022, 236, 108053. [Google Scholar] [CrossRef]
- Platnich, J.M.; Muruve, D.A. NOD-like Receptors and Inflammasomes: A Review of Their Canonical and Non-Canonical Signaling Pathways. Arch. Biochem. Biophys. 2019, 670, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 Inflammasome in Cancer and Metabolic Diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Ma, Q. Pharmacological Inhibition of the NLRP3 Inflammasome: Structure, Molecular Activation, and Inhibitor-NLRP3 Interaction. Pharmacol. Rev. 2023, 75, 487–520. [Google Scholar] [CrossRef]
- Yu, Z.W.; Zhang, J.; Li, X.; Wang, Y.; Fu, Y.H.; Gao, X.Y. A New Research Hot Spot: The Role of NLRP3 Inflammasome Activation, a Key Step in Pyroptosis, in Diabetes and Diabetic Complications. Life Sci 2020, 240, 117138. [Google Scholar] [CrossRef]
- Gora, I.M.; Ciechanowska, A.; Ladyzynski, P. Nlrp3 Inflammasome at the Interface of Inflammation, Endothelial Dysfunction, and Type 2 Diabetes. Cells 2021, 10, 314. [Google Scholar] [CrossRef]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.; Yu, T.; Chu, X. NLRP3 Inflammasome in Endothelial Dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef]
- Rovira-Llopis, S.; Apostolova, N.; Bañuls, C.; Muntané, J.; Rocha, M.; Victor, V.M. Mitochondria, the NLRP3 Inflammasome, and Sirtuins in Type 2 Diabetes: New Therapeutic Targets. Antioxid. Redox Signal. 2018, 29, 749–791. [Google Scholar] [CrossRef]
- Shi, C.; Cao, P.; Wang, Y.; Zhang, Q.; Zhang, D.; Wang, Y.; Wang, L.; Gong, Z. PANoptosis: A Cell Death Characterized by Pyroptosis, Apoptosis, and Necroptosis. J. Inflamm. Res. 2023, 16, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhu, Y.; Zhang, L.; Guo, L.; Wang, X.; Pan, Z.; Jiang, X.; Wu, F.; He, G. Mechanisms of PANoptosis and Relevant Small-Molecule Compounds for Fighting Diseases. Cell Death Dis. 2023, 14, 851. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Chen, S.; Sun, R.; Zhang, X.; Wang, D. The NLRP3 Inflammasome: Role in Metabolic Disorders and Regulation by Metabolic Pathways. Cancer Lett. 2018, 419, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; L’Homme, L.; De Roover, A.; Kohnen, L.; Scheen, A.J.; Moutschen, M.; Piette, J.; Legrand-Poels, S.; Paquot, N. Obesity Phenotype Is Related to NLRP3 Inflammasome Activity and Immunological Profile of Visceral Adipose Tissue. Diabetologia 2013, 56, 2487–2497. [Google Scholar] [CrossRef]
- Yin, Z.; Deng, T.; Peterson, L.E.; Yu, R.; Lin, J.; Hamilton, D.J.; Reardon, P.R.; Sherman, V.; Winnier, G.E.; Zhan, M.; et al. Transcriptome Analysis of Human Adipocytes Implicates the NOD-like Receptor Pathway in Obesity-Induced Adipose Inflammation. Mol. Cell. Endocrinol. 2014, 394, 80–87. [Google Scholar] [CrossRef]
- Wang, X.; He, G.; Peng, Y.; Zhong, W.; Wang, Y.; Zhang, B. Sodium Butyrate Alleviates Adipocyte Inflammation by Inhibiting NLRP3 Pathway. Sci. Rep. 2015, 5, 12676. [Google Scholar] [CrossRef]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; Mcallan, L.; Nilaweera, K.N.; et al. Monounsaturated Fatty Acid-Enriched High-Fat Diets Impede Adipose NLRP3 Inflammasome-Mediated IL-1β Secretion and Insulin Resistance despite Obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef]
- Bitto, A.; Altavilla, D.; Pizzino, G.; Irrera, N.; Pallio, G.; Colonna, M.R.; Squadrito, F. Inhibition of Inflammasome Activation Improves the Impaired Pattern of Healing in Genetically Diabetic Mice. Br. J. Pharmacol. 2014, 171, 2300–2307. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 Directly Targets the NLRP3 ATP-Hydrolysis Motif for Inflammasome Inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Henriksbo, B.D.; Lau, T.C.; Cavallari, J.F.; Denou, E.; Chi, W.; Lally, J.S.; Crane, J.D.; Duggan, B.M.; Foley, K.P.; Fullerton, M.D.; et al. Fluvastatin Causes NLRP3 Inflammasome-Mediated Adipose Insulin Resistance. Diabetes 2014, 63, 3742–3747. [Google Scholar] [CrossRef]
- Kim, Y.; Wang, W.; Okla, M.; Kang, I.; Moreau, R.; Chung, S. Suppression of NLRP3 Inflammasome by γ -Tocotrienol Ameliorates Type 2 Diabetes. J. Lipid Res. 2016, 57, 66–76. [Google Scholar] [CrossRef]
- Tanti, J.F.; Ceppo, F.; Jager, J.; Berthou, F. Implication of Inflammatory Signaling Pathways in Obesity-Induced Insulin Resistance. Front. Endocrinol. 2013, 3, 181. [Google Scholar] [CrossRef]
- Ringling, R.E.; Gastecki, M.L.; Woodford, M.L.; Lum-Naihe, K.J.; Grant, R.W.; Pulakat, L.; Vieira-Potter, V.J.; Padilla, J. Loss of Nlrp3 Does Not Protect Mice from Western Diet-Induced Adipose Tissue Inflammation and Glucose Intolerance. PLoS ONE 2016, 11, e0161939. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat. Med. 2011, 17, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Chan, K.L.; Zhang, S.; Mejdani, M.; Jacobson, M.R.; Ducos, A.; Bilan, P.J.; Niu, W.; Klip, A. Saturated Fatty Acids Activate Caspase-4/5 in Human Monocytes, Triggering IL-1 and IL-18 Release. Am. J. Physiol. Endocrinol. Metab. 2016, 311, 825–835. [Google Scholar] [CrossRef]
- Sokolowska, E.; Blachnio-Zabielska, A. The Role of Ceramides in Insulin Resistance. Front. Endocrinol. 2019, 10, 577. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lim, W.-G.; Jung, H.; Jeong, Y.C.; Park, C.-Y.; Yang, S.B.; Lee, C.H.; Wang, D.; Sohn, K.; Han, J.W.; et al. Protective Catalytic Layer Powering Activity and Stability of Electrocatalyst for High-Energy Lithium-Sulfur Pouch Cell. Nat. Commun. 2025, 16, 1649. [Google Scholar] [CrossRef]
- Feng, H.; Gu, J.; Gou, F.; Huang, W.; Gao, C.; Chen, G.; Long, Y.; Zhou, X.; Yang, M.; Liu, S.; et al. High Glucose and Lipopolysaccharide Prime NLRP3 Inflammasome via ROS/TXNIP Pathway in Mesangial Cells. J. Diabetes Res. 2016, 2016, 6973175. [Google Scholar] [CrossRef]
- Kong, X.; Lu, A.L.; Yao, X.M.; Hua, Q.; Li, X.Y.; Qin, L.; Zhang, H.M.; Meng, G.X.; Su, Q. Activation of NLRP3 Inflammasome by Advanced Glycation End Products Promotes Pancreatic Islet Damage. Oxid. Med. Cell. Longev. 2017, 2017, 9692546. [Google Scholar] [CrossRef]
- Chutkow, W.A.; Birkenfeld, A.L.; Brown, J.D.; Lee, H.Y.; Frederick, D.W.; Yoshioka, J.; Patwari, P.; Kursawe, R.; Cushman, S.W.; Plutzky, J.; et al. Deletion of the α-Arrestin Protein Txnip in Mice Promotes Adiposity and Adipogenesis While Preserving Insulin Sensitivity. Diabetes 2010, 59, 1424–1434. [Google Scholar] [CrossRef]
- Wang, M.; Lin, X.; Yang, X.; Yang, Y. Research Progress on Related Mechanisms of Uric Acid Activating NLRP3 Inflammasome in Chronic Kidney Disease. Ren. Fail. 2022, 44, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Chang, H.W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of Diet on the Gut Microbiome and Implications for Human Health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chang, Y.; Zhang, K.; Chen, H.; Tao, S.; Zhang, Z. Implication of the Gut Microbiome Composition of Type 2 Diabetic Patients from Northern China. Sci. Rep. 2020, 10, 5450. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Jin, Y.; Liu, Y.; Xu, L.; Xu, J.; Xiong, Y.; Peng, Y.; Ding, K.; Zheng, S.; Yang, N.; Zhang, Z.; et al. Novel Role for Caspase 1 Inhibitor VX765 in Suppressing NLRP3 Inflammasome Assembly and Atherosclerosis via Promoting Mitophagy and Efferocytosis. Cell Death Dis. 2022, 13, 512. [Google Scholar] [CrossRef]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 Inflammasome Activation in Patients with Type 2 Diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. NLRP3 Gene Silencing Ameliorates Diabetic Cardiomyopathy in a Type 2 Diabetes Rat Model. PLoS ONE 2014, 9, e104771. [Google Scholar] [CrossRef]
- Wan, Z.; Fan, Y.; Liu, X.; Xue, J.; Han, Z.; Zhu, C.; Wang, X. NLRP3 Inflammasome Promotes Diabetes-Induced Endothelial Inflammation and Atherosclerosis. Diabetes Metab. Syndr. Obes. 2019, 12, 1931–1942. [Google Scholar] [CrossRef]
- Li, X.X.; Ling, S.K.; Hu, M.Y.; Ma, Y.; Li, Y.; Huang, P.L. Protective Effects of Acarbose against Vascular Endothelial Dysfunction through Inhibiting Nox4/NLRP3 Inflammasome Pathway in Diabetic Rats. Free Radic. Biol. Med. 2019, 145, 175–186. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, C.; Yan, B.; Shi, X.; Shi, Y.; Qu, L.; Liang, X. Jinmaitong Ameliorates Diabetic Peripheral Neuropathy through Suppressing TXNIP/NLRP3 Inflammasome Activation in the Streptozotocin-Induced Diabetic Rat Model. Diabetes Metab. Syndr. Obes. 2019, 12, 2145–2155. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Jiang, B.; Cohen, R.A.; Zang, M. Activation of Sterol Regulatory Element Binding Protein and NLRP3 Inflammasome in Atherosclerotic Lesion Development in Diabetic Pigs. PLoS ONE 2013, 8, e67532. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nũez, G.; Schnurr, M.; et al. NLRP3 Inflammasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wada, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of Interleukin-1ß Decreases the Severity of Atherosclerosis in ApoE-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 656–660. [Google Scholar] [CrossRef]
- An, Q.; Hu, Q.; Wang, B.; Cui, W.; Wu, F.; Ding, Y. Oleanolic Acid Alleviates Diabetic Rat Carotid Artery Injury through the Inhibition of NLRP3 Inflammasome Signaling Pathways. Mol. Med. Rep. 2017, 16, 8413–8419. [Google Scholar] [CrossRef]
- Song, J.; Li, J.; Hou, F.; Wang, X.; Liu, B. Mangiferin Inhibits Endoplasmic Reticulum Stress-Associated Thioredoxin-Interacting Protein/NLRP3 Inflammasome Activation with Regulation of AMPK in Endothelial Cells. Metabolism 2015, 64, 428–437. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef]
- Adams, V.; Linke, A. Impact of Exercise Training on Cardiovascular Disease and Risk. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 728–734. [Google Scholar] [CrossRef]
- Wang, F.; Guo, X.; Shen, X.; Kream, R.M.; Mantione, K.J.; Stefano, G.B. Vascular Dysfunction Associated with Type 2 Diabetes and Alzheimer’s Disease: A Potential Etiological Linkage. Med. Sci. Monit. Basic. Res. 2014, 20, 118–129. [Google Scholar] [CrossRef]
- Theofilis, P.; Oikonomou, E.; Chasikidis, C.; Tsioufis, K.; Tousoulis, D. Inflammasomes in Atherosclerosis—From Pathophysiology to Treatment. Pharmaceuticals 2023, 16, 1211. [Google Scholar] [CrossRef]
- Shi, X.; Xie, W.L.; Kong, W.W.; Chen, D.; Qu, P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2015, 24, 2455–2466. [Google Scholar] [CrossRef]
- Pereira, C.A.; Carlos, D.; Ferreira, N.S.; Silva, J.F.; Zanotto, C.Z.; Zamboni, D.S.; Garcia, V.D.; Ventura, D.F.; Silva, J.S.; Tostes, R.C. Mitochondrial DNA Promotes NLRP3 Inflammasome Activation and Contributes to Endothelial Dysfunction and Inflammation in Type 1 Diabetes. Front. Physiol. 2020, 10, 1557. [Google Scholar] [CrossRef]
- Zhou, X.; Kang, C.; Hu, Y.; Wang, X. Study on insulin resistance and ischemic cerebrovascular disease: A biblio-metric analysis via CiteSpace. Front Public Health 2023, 11, 1021378. [Google Scholar] [CrossRef]
- Cai, H.; Wang, P.; Zhang, B.; Dong, X. Expression of the NEK7/NLRP3 Inflammasome Pathway in Patients with Diabetic Lower Extremity Arterial Disease. BMJ Open Diabetes Res. Care 2020, 8, e001808. [Google Scholar] [CrossRef]
- Vlachakis, P.K.; Theofilis, P.; Kachrimanidis, I.; Giannakopoulos, K.; Drakopoulou, M.; Apostolos, A.; Kordalis, A.; Leontsinis, I.; Tsioufis, K.; Tousoulis, D. The Role of Inflammasomes in Heart Failure. Int. J. Mol. Sci. 2024, 25, 5372. [Google Scholar] [CrossRef]
- Bogdanova, D.; Samsonov, M.Y.; Lebedeva, S.; Bukhanova, D.; Materenchuk, M.; Mutig, K. Targeting Interleukin-1 Signaling for Renoprotection. Front. Immunol. 2025, 16, 1591197. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A Small-Molecule Inhibitor of the NLRP3 Inflammasome for the Treatment of Inflammatory Diseases. Nat. Med. 2015, 21, 248–257. [Google Scholar] [CrossRef]
- Takahashi, M. NLRP3 Inflammasome as a Key Driver of Vascular Disease. Cardiovasc. Res. 2022, 118, 372–385. [Google Scholar] [CrossRef]
- Ye, J.; Li, L.; Wang, M.; Ma, Q.; Tian, Y.; Zhang, Q.; Liu, J.; Li, B.; Zhang, B.; Liu, H.; et al. Diabetes Mellitus Promotes the Development of Atherosclerosis: The Role of NLRP3. Front. Immunol. 2022, 13, 900254. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide Inhibits the Cryopyrin/Nalp3 Inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the Nlrp3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef]
- Luzi, L.; Pozza, G. Glibenclamide: An Old Drug with a Novel Mechanism of Action? Acta Diabetol. 1997, 34, 239–244. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-Inflammatory Compounds Parthenolide and Bay 11-7082 Are Direct Inhibitors of the Inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Lei, S.; Zhao, B.; Wu, Y.; Su, W.; Liu, M.; Meng, Q.; Zhou, B.; Leng, Y.; Xia, Z.Y. NLRP3 Inflammasome Activation-Mediated Pyroptosis Aggravates Myocardial Ischemia/Reperfusion Injury in Diabetic Rats. Oxid. Med. Cell. Longev. 2017, 2017, 9743280. [Google Scholar] [CrossRef]
- Marchetti, C.; Swartzwelter, B.; Koenders, M.I.; Azam, T.; Tengesdal, I.W.; Powers, N.; de Graaf, D.M.; Dinarello, C.A.; Joosten, L.A.B. NLRP3 Inflammasome Inhibitor OLT1177 Suppresses Joint Inflammation in Murine Models of Acute Arthritis. Arthritis Res. Ther. 2018, 20, 169. [Google Scholar] [CrossRef]
- Klück, V.; Jansen, T.L.T.A.; Janssen, M.; Comarniceanu, A.; Efdé, M.; Tengesdal, I.W.; Schraa, K.; Cleophas, M.C.P.; Scribner, C.L.; Skouras, D.B.; et al. Dapansutrile, an Oral Selective NLRP3 Inflammasome Inhibitor, for Treatment of Gout Flares: An Open-Label, Dose-Adaptive, Proof-of-Concept, Phase 2a Trial. Lancet Rheumatol. 2020, 2, e270–e280. [Google Scholar] [CrossRef]
- Cocco, M.; Garella, D.; Di Stilo, A.; Borretto, E.; Stevanato, L.; Giorgis, M.; Marini, E.; Fantozzi, R.; Miglio, G.; Bertinaria, M. Electrophilic Warhead-Based Design of Compounds Preventing NLRP3 Inflammasome-Dependent Pyroptosis. J. Med. Chem. 2014, 57, 10366–10382. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Yao Hui, L.L.; Kraus, V.B. Colchicine-Update on Mechanisms of Action and Therapeutic Uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef]
- Weng, J.H.; Koch, P.D.; Luan, H.; Tu, H.C.; Shimada, K.; Ngan, I.; Ventura, R.; Jiang, R.; Mitchison, T.J. Colchicine Acts Selectively in the Liver to Induce Hepatokines That Inhibit Myeloid Cell Activation. Nat. Metab. 2021, 3, 513–522. [Google Scholar] [CrossRef]
- Fujisue, K.; Sugamura, K.; Kurokawa, H.; Matsubara, J.; Ishii, M.; Izumiya, Y.; Kaikita, K.; Sugiyama, S. Colchicine Improves Survival, Left Ventricular Remodeling, and Chronic Cardiac Function after Acute Myocardial Infarction. Circ. J. 2017, 81, 1174–1182. [Google Scholar] [CrossRef]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Imazio, M.; Brucato, A.; Cemin, R.; Ferrua, S.; Belli, R.; Maestroni, S.; Trinchero, R.; Spodick, D.H.; Adler, Y. Colchicine for Recurrent Pericarditis (CORP) A Randomized Trial. Ann. Intern. Med. 2011, 155, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroma, E.; Abbate, A.; Toldo, S. NLRP3 Inflammasome Inhibitors in Cardiovascular Diseases. Molecules 2021, 26, 976. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, Y.; Pan, Y.; Liu, Y.; Zheng, S.; Ding, K.; Mu, K.; Yuan, Y.; Li, Z.; Song, H.; et al. Novel Role for Tranilast in Regulating Nlrp3 Ubiquitination, Vascular Inflammation, and Atherosclerosis. J. Am. Heart Assoc. 2020, 9, e015513. [Google Scholar] [CrossRef]
- Mastrocola, R.; Penna, C.; Tullio, F.; Femminò, S.; Nigro, D.; Chiazza, F.; Serpe, L.; Collotta, D.; Alloatti, G.; Cocco, M.; et al. Pharmacological Inhibition of NLRP3 Inflammasome Attenuates Myocardial Ischemia/Reperfusion Injury by Activation of RISK and Mitochondrial Pathways. Oxid. Med. Cell. Longev. 2016, 2016, 5271251. [Google Scholar] [CrossRef]
- Cocco, M.; Miglio, G.; Giorgis, M.; Garella, D.; Marini, E.; Costale, A.; Regazzoni, L.; Vistoli, G.; Orioli, M.; Massulaha-Ahmed, R.; et al. Design, Synthesis, and Evaluation of Acrylamide Derivatives as Direct NLRP3 Inflammasome Inhibitors. ChemMedChem 2016, 11, 1790–1803. [Google Scholar] [CrossRef]
- Toldo, S.; Das, A.; Mezzaroma, E.; Chau, V.Q.; Marchetti, C.; Durrant, D.; Samidurai, A.; Van Tassell, B.W.; Yin, C.; Ockaili, R.A.; et al. Induction of Microrna-21 with Exogenous Hydrogen Sulfide Attenuates Myocardial Ischemic and Inflammatory Injury in Mice. Circ. Cardiovasc. Genet. 2014, 7, 311–320. [Google Scholar] [CrossRef]
- Byrne, N.J.; Matsumura, N.; Maayah, Z.H.; Ferdaoussi, M.; Takahara, S.; Darwesh, A.M.; Levasseur, J.L.; Jahng, J.W.S.; Vos, D.; Parajuli, N.; et al. Empagliflozin Blunts Worsening Cardiac Dysfunction Associated with Reduced NLRP3 (Nucleotide-Binding Domain-Like Receptor Protein 3) Inflammasome Activation in Heart Failure. Circ. Heart Fail. 2020, 13, E006277. [Google Scholar] [CrossRef]
- Chen, H.; Tran, D.; Yang, H.C.; Nylander, S.; Birnbaum, Y.; Ye, Y. Dapagliflozin and Ticagrelor Have Additive Effects on the Attenuation of the Activation of the NLRP3 Inflammasome and the Progression of Diabetic Cardiomyopathy: An AMPK–MTOR Interplay. Cardiovasc. Drugs Ther. 2020, 34, 443–461. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, B.; Wang, L.; Yang, M.; Xia, Z.; Wei, W.; Zhang, F.; Yuan, X. Pioglitazone Ameliorates Glomerular NLRP3 Inflammasome Activation in Apolipoprotein E Knockout Mice with Diabetes Mellitus. PLoS ONE 2017, 12, e0181248. [Google Scholar] [CrossRef]
- Dwivedi, D.K.; Jena, G.B. NLRP3 Inhibitor Glibenclamide Attenuates High-Fat Diet and Streptozotocin-Induced Non-Alcoholic Fatty Liver Disease in Rat: Studies on Oxidative Stress, Inflammation, DNA Damage and Insulin Signalling Pathway. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Ishikawa, A.; Watanabe, E.; Nakamura, Y.; Aruga, Y.; Hasegawa, H.; Onogi, Y.; Honda, H.; Nagai, Y.; Takatsu, K.; et al. Eplerenone Prevented Obesity-Induced Inflammasome Activation and Glucose Intolerance. J. Endocrinol. 2017, 235, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Keech, A.C.; Mitchell, P.; Summanen, P.A.; O’Day, J.; Davis, T.M.; Moffitt, M.S.; Taskinen, M.-R.; Simes, R.J.; Tse, D.; Williamson, E.; et al. Effect of Fenofibrate on the Need for Laser Treatment for Diabetic Retinopathy (FIELD Study): A Randomised Controlled Trial. Lancet 2007, 370, 1687–1697. [Google Scholar] [CrossRef]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, Y.; Zhang, M.; An, F. Rosuvastatin Alleviates Diabetic Cardiomyopathy by Inhibiting NLRP3 Inflammasome and MAPK Pathways in a Type 2 Diabetes Rat Model. Cardiovasc. Drugs Ther. 2014, 28, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The Ketone Metabolite β-Hydroxybutyrate Blocks NLRP3 Inflammasome-Mediated Inflammatory Disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Wu, K.K.L.; Cheung, S.W.M.; Cheng, K.K.Y. NLRP3 Inflammasome Activation in Adipose Tissues and Its Implications on Metabolic Diseases. Int. J. Mol. Sci. 2020, 21, 4184. [Google Scholar] [CrossRef]
- Li, A.; Zhang, S.; Li, J.; Liu, K.; Huang, F.; Liu, B. Metformin and Resveratrol Inhibit Drp1-Mediated Mitochondrial Fission and Prevent ER Stress-Associated NLRP3 Inflammasome Activation in the Adipose Tissue of Diabetic Mice. Mol. Cell. Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef]
- Zhao, W.; Zhou, L.; Novák, P.; Shi, X.; Lin, C.B.; Zhu, X.; Yin, K. Metabolic Dysfunction in the Regulation of the NLRP3 Inflammasome Activation: A Potential Target for Diabetic Nephropathy. J. Diabetes Res. 2022, 2022, 2193768. [Google Scholar] [CrossRef]
- Zhang, X.H.; Peng, L.; Zhang, J.; Dong, Y.P.; Wang, C.J.; Liu, C.; Xia, D.Y.; Zhang, X.S. Berberine Ameliorates Subarachnoid Hemorrhage Injury via Induction of Sirtuin 1 and Inhibiting HMGB1/Nf-ΚB Pathway. Front. Pharmacol. 2020, 11, 1073. [Google Scholar] [CrossRef]
- Liu, Z.; Gan, L.; Xu, Y.; Luo, D.; Ren, Q.; Wu, S.; Sun, C. Melatonin Alleviates Inflammasome-Induced Pyroptosis through Inhibiting NF-ΚB/GSDMD Signal in Mice Adipose Tissue. J. Pineal Res. 2017, 63, e12414. [Google Scholar] [CrossRef]
- Honda, H.; Nagai, Y.; Matsunaga, T.; Okamoto, N.; Watanabe, Y.; Tsuneyama, K.; Hayashi, H.; Fujii, I.; Ikutani, M.; Hirai, Y.; et al. Isoliquiritigenin Is a Potent Inhibitor of NLRP3 Inflammasome Activation and Diet-Induced Adipose Tissue Inflammation. J. Leukoc. Biol. 2014, 96, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Marchetti, C.; Mauro, A.G.; Chojnacki, J.; Mezzaroma, E.; Carbone, S.; Zhang, S.; Van Tassell, B.; Salloum, F.N.; Abbate, A. Inhibition of the NLRP3 Inflammasome Limits the Inflammatory Injury Following Myocardial Ischemia-Reperfusion in the Mouse. Int. J. Cardiol. 2016, 209, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, K.; Tousoulis, D. The Anti-Inflammatory Effect of Novel Antidiabetic Agents. Life 2022, 12, 1829. [Google Scholar] [CrossRef]
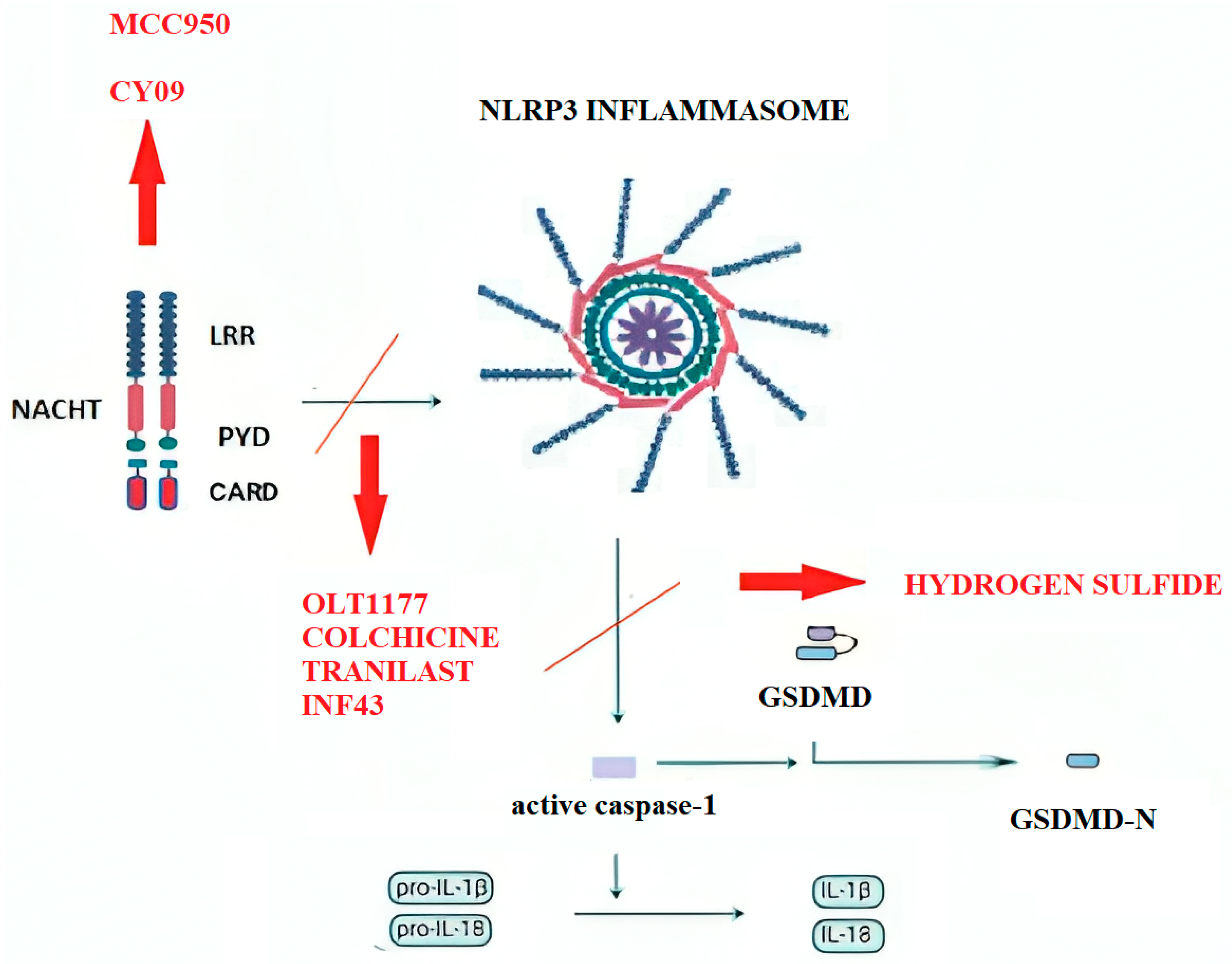

{kind=link}
| Authors | Year | Population | Study | Findings |
|---|---|---|---|---|
| Esser et al. [25] | 2013 | Human participants with different obesity phenotypes | Cross-sectional observational study | Increased expression of NLRP3 and IL1B in visceral adipose tissue from metabolically unhealthy obese patients |
| Yin et al. [26] | 2014 | Postmenopausal women, both lean and obese, undergoing elective abdominal surgery | Cross-sectional observational study | Genes associated with the NOD-like receptor pathway, including the NLRP3, were upregulated in adipocytes from obese individuals |
| Wang et al. [27] | 2015 | db/db mice | Pre-clinical experimental study (with in vivo and vitro methodologies) | NLRP3 and Caspase-1 expressions were increased in epididymal fat from db/db mice |
| Finucane et al. [28] | 2015 | C57BL/6 mice | Pre-clinical experimental study | NLRP3, Caspase-1, and IL1B expressions in adipose tissue were higher in mice treated for 6 months with a saturated fatty acid HFD in comparison with mice fed with a monounsaturated fatty acid HFD |
| Bitto et al. [29] | 2014 | db/db mice | Pre-clinical experimental study | NLRP3, ASC, caspase-1, IL-18, and IL-1 are upregulated during wound healing in animal models of T2DM in comparison with healthy animals |
| Coll et al. [30] | 2019 | Mouse bone marrow-derived macrophages and human monocyte-derived macrophages | Pre-clinical experimental study | MCC950, which inhibits the NLRP3 inflammasome, can be applied as a potential anti-inflammatory therapy in T2DM |
| Henriksbo et al. [31] | 2014 | ob/ob mice, 3T3-L1 adipocytes (murine adipocyte cell line) | Pre-clinical experimental study (with in vivo and vitro methodologies) | Fluvastatin provokes inflammation and insulin resistance in adipose tissue via the upregulation of NLRP3, which is consistent with the increased expression of NLRP3 in inflamed adipose tissues of T2DM patients |
| Kim et al. [32] | 2016 | Murine macrophage cell lines(iJ774) and bone marrow-derived macrophages | Pre-clinical experimental study (with in vivo and vitro methodologies) | NLRP3 can be suppressed by γ-tocotrienol, delaying the progression of T2DM |
| Authors | Year | Population | Study | Findings |
|---|---|---|---|---|
| Ridker et al. CANTOS trial [45] | 2017 | Patients with history of myocardial infarction and elevated hsCRP levels | Randomized, double-blind, placebo controlled, multicenter clinical trial | In total, 150 mg of Canakinumab significantly reduced cardiovascular death, providing the first definitive clinical evidence that reducing inflammation can lower CVD event risk |
| Yin Jin et al. [46] | 2022 | ApoE-/– mice | Pre-clinical experimental study | Targeting caspase-1 and the NLRP3 assembly may offer therapeutic potential in atherosclerotic cardiovascular diseases. |
| Lee et al. [47] | 2013 | Patients with untreated T2DM | Comparative experimental study | Increased expression of the inflammasome components NLRP3 and ASC was found in monocytes from newly identified, untreated type 2 DM subjects |
| Luo et al. [48] | 2014 | HFD and STZ induced rat models | Pre-clinical experimental study | Diabetic rats showed severe metabolic disorder, cardiac inflammation, cell death, disorganized ultrastructure, fibrosis, and excessive activation of NLRP3 |
| Wan et al. [49] | 2019 | Humans and ApoE-/– mice | Pre-clinical experimental study (with in vivo and vitro methodologies) | NLRP3 was involved in hyperglycemia-induced endothelial inflammation, both in vitro and in vivo |
| Xiao-Xue Li et al. [50] | 2019 | Diabetic rats | Pre-clinical experimental study | High glucose induced the assembly and activation of NLRP3 inflammasome in endothelial cells |
| Feng et al. [39] | 2016 | Rat glomerular mesangial cells | Pre-clinical experimental study | High glucose levels and LPS exposure prime the NRLP3 inflammasome in mesangial cells through the ROS/TXNIP signaling pathway, leading to diabetic nephropathy |
| Sun et al. [51] | 2019 | STZ-induced diabetic rat model | Pre-clinical experimental study | Suppression of TXNIP/NLRP3 activation ameliorates diabetic peripheral neuropathy |
| Yu Li et al. [52] | 2013 | Porcine model of atherosclerosis and DM | Pre-clinical experimental in vivo study | In vivo evidence that the dysregulation of SIRT1-AMPK-SREBP and stimulation of NLRP3 inflammasome may contribute to vascular lipid deposition and inflammation in atherosclerosis |
| Duewell et al. [53] | 2010 | Mice deficient in components of the NLRP3 inflammasome | Pre-clinical experimental study (with in vivo and vitro methodologies) | Crystalline cholesterol acts as an endogenous danger signal and its deposition in arteries or elsewhere is an early cause rather than a late consequence of NLRP3 activation and inflammation |
| Kirii et al. [54] | 2003 | apoE-/– and IL-1β-/– mice | Pre-clinical experimental in vivo study | IL-1β deficiency significantly reduced atherosclerotic lesion size in the aorta, suggesting that IL-1β promotes atherogenesis through both immune cell recruitment and endothelial activation |
| Qian An et al. [55] | 2017 | STZ-induced diabetic rats | Pre-clinical experimental in vivo study | Suppression of the NLRP3 inflammasome pathway via oleanolic acid attenuates carotid artery injury in diabetic rats |
| Song et al. [56] | 2015 | Cultured endothelial cells | Experimental in vitro cellular study | Inhibition of ER stress-associated TXNIP/NLRP3 inflammasome activation in endothelial cells improves endothelial homeostasis |
| Drugs | Mechanism of Action | Studies | Findings | Status | |
|---|---|---|---|---|---|
| NLRP3 inhibitors | MCC950 [67,68,69] | Non-covalent bonding to the NACHT domain | Many murine models (HFD, streptozotocin-induced ApoE-/– mice, etc.) and Humans | Reduced atherosclerotic plaque development, decreased the expression of adhesion molecules within the plaque, and lowered the number of macrophages present in the plaque | Clinical development was discontinued due to excessive renal inflammation and hepatic toxicity |
| Glyburide [70,71,72] | Inhibition of ATP-dependent potassium channels | Murine and humans models | Suppressed cardiac caspase-1 activity and minimized infarct size in mice undergoing myocardial ischemia followed by 24 h of reperfusion | Limited clinical use due to frequent hypoglycemia | |
| Bay 11-7082 [73,74] | NF-κΒ pathway inhibition | Myocardial ischemia–reperfusion murine models | Decreases leukocyte infiltration in the infarcted area and enhances cardiomyocyte survival, reducing infarct size | Pre-clinical studies | |
| OLT1177 [75,76,77] | Impairs ATPase activity of NLRP3 | Animal models of myocardial ischemia–reperfusion | Dose-dependent reduction in infarct size, and also improved ventricular function in a model of permanent coronary artery occlusion | Pre-clinical studies | |
| Colchicine [78,79,80,81] | Interferes with the NLRP3 complex by disrupting microtubule action | Human studies (COLCOT, LoDoCo) and mouse models of permanent cardiac ligation | Decreased the infiltration of neutrophils and macrophages, as well as the mRNA expression of pro-inflammatory cytokines and NLRP3 inflammasome components 24 h after myocardial infarction | FDA-approved for inflammatory diseases | |
| CY-09 [12,82] | Inhibition of the NLRP3 complex by binding directly to the ATP-binding motif of the NACHT domain | Murine models of type 2 Diabetes Mellitus | Prevented cardiac dysfunction linked to diabetic ischemic stroke | Pre-clinical studies | |
| Tranilast [71,83,84] | Blocks the direct NLRP3-NLRP3 and NLRP3–ASC interaction | Mouse models of atherosclerosis and several animal models of hypertension, diabetic cardiomyopathy, and myocardial infarction | Enhanced NLRP3 ubiquitination, restricting NLRP3 inflammasome assembly and thereby reducing the initiation and progression of atherosclerotic plaques | Pre-clinical studies | |
| INF4E [85,86] | Inhibition of the NLRP3 ATPase activity | Murine models of myocardial ischemia | Reduced infarct size and improved left ventricular pressure | Clinical development was discontinued due to cytotoxic properties | |
| Hydrogen Sulfide [14,87] | Reduces NLRP3-dependent caspase-1 activation | Murine specimen undergoing ischemia–reperfusion injury | Diminished the IKKβ/NF-κB signaling pathway introducing cardioprotective properties in a hemorrhagic shock model | Pre-clinical studies | |
| Anti-Diabetic Drugs | Metformin [8,47] | Activates AMPK that reduces ER stress and mitochondrial fission leading to inhibition of caspase-1 | Studies in Monocyte-derived macrophages isolated from type 2 diabetic subjects | Protective properties against cell pyroptosis and myocardial ischemia–reperfusion injury by interfering with the AMPK/TOR signaling pathway | FDA-approved for Type-2 diabetes mellitus |
| SGLT2 inhibitors [88,89] | Modulatory effects on the AMPK/TOR pathway | Eight-week-old BTBR and wild-type mice | Improved left ventricular end-systolic and end-diastolic volumes, as well as the left ventricular ejection fraction by modulating the AMPK/TOR pathway | FDA-approved for Type-2 diabetes mellitus and heart failure | |
| Pioglitazone [90] | Downregulation of NF-κB | apoE (-/–) mice | Reduced ROS releases and attenuated renal damage | FDA-approved for Type-2 diabetes mellitus | |
| Acarbose [91] | Inhibition of NOX4-depedant superoxide production | Rats with T2D | Enhanced endothelial function in the aorta of diabetic rats | FDA-approved for Type-2 diabetes mellitus | |
| Saxagliptin [8] | AMPK-dependent caspase-1 inhibition | Type 2 diabetic (BTBR ob/ob) and wild-type (WT) mice | Mitigate the advancement of diabetic cardiomyopathy | FDA-approved for Type-2 diabetes mellitus | |
| Other pharmaceutical options | Eplerenone [92] | Inhibits phosphorylation of NF-κB and ROS production | C57BL/6 mice fed a high-fat diet (HFD) | Exhibited robust anti-inflammatory properties | FDA-approved drug for hypertension and heart failure |
| Verapamil [8] | Inhibits the assembly of NLRP3, reduces the release of IL-1β | Mouse models with diabetic retinopathy | Attenuated pathological neo-angiogenesis | FDA-approved drug for hypertension and angina pectoris | |
| Fenofibrate [93] | Unidentified mechanism of NRLP3 inhibition | Mouse models with Diabetic Retinopathy | Attenuated retinal leukostasis, vascular leakage and the progression of DR | FDA-approved for hypertriglyceridemia | |
| Atorvastatin [8,94] | Inhibition of NLRP3 inflammasome via TXNIP | Murine models of diabetic cardiomyopathy | Ameliorated diastolic dysfunction and cardiac fibrosis | FDA-approved lipid-lowering agent | |
| β-hydroxybutyrate [95] | Abolishes K+ efflux and reduces ASC oligomerization and speck formation via unknown mechanism | Mouse models of ketogenic diet | Inhibited caspase-1 activation, and reduced neutrophil count and hyperglycemia | Pre-clinical studies | |
| Natural Substances | Resveratrol [96,97,98] | Modulation of AMPK signaling pathway | Diabetic murine models | Restriction of inflammation and adipose dysfunction | Pre-clinical studies |
| Berberine [99] | Enhances AMPK-dependent autophagy | HFD-fed murine models | Improved insulin sensitivity and glucose tolerance | ||
| Parthenolide [17,96] | Impairs ATPase activity of NLRP3, suppresses IκB kinase, and NF-κB | mouse ASC (polyclonal anti-mouse ASC), mouse NLRP3 (polyclonal anti-NLRP3 PYD), mouse caspase-1 p20 (monoclonal anti-mouse caspase-1 p20) | Exhibited anti-inflammatory properties via macrophage blockage | ||
| Melatonin [100] | suppresses NF-κB signaling by decreasing NF-κB and p65 protein levels in the cytoplasm and nucleus | HFD-fed murine models | Profound decrease in adipose tissue pyroptosis | ||
| Glycyrrhizin (GL) and Isoliquiritigenin (ILG) [96,101] | Inhibits mitogen-activated protein kinase (MAPK) activation | HFD-fed murine models | Diminished Il-1β production and adipose tissue inflammation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karamitsos, K.; Oikonomou, E.; Theofilis, P.; Ikonomidis, I.; Kassi, E.; Lambadiari, V.; Gialafos, E.; Tsatsaragkou, A.; Mystakidi, V.-C.; Zisimos, K.; et al. The Role of NLRP3 Inflammasome in Type 2 Diabetes Mellitus and Its Macrovascular Complications. J. Clin. Med. 2025, 14, 4606. https://doi.org/10.3390/jcm14134606
Karamitsos K, Oikonomou E, Theofilis P, Ikonomidis I, Kassi E, Lambadiari V, Gialafos E, Tsatsaragkou A, Mystakidi V-C, Zisimos K, et al. The Role of NLRP3 Inflammasome in Type 2 Diabetes Mellitus and Its Macrovascular Complications. Journal of Clinical Medicine. 2025; 14(13):4606. https://doi.org/10.3390/jcm14134606
Chicago/Turabian StyleKaramitsos, Konstantinos, Evangelos Oikonomou, Panagiotis Theofilis, Ignatios Ikonomidis, Eva Kassi, Vaia Lambadiari, Elias Gialafos, Aikaterini Tsatsaragkou, Vasiliki-Chara Mystakidi, Konstantinos Zisimos, and et al. 2025. "The Role of NLRP3 Inflammasome in Type 2 Diabetes Mellitus and Its Macrovascular Complications" Journal of Clinical Medicine 14, no. 13: 4606. https://doi.org/10.3390/jcm14134606
APA StyleKaramitsos, K., Oikonomou, E., Theofilis, P., Ikonomidis, I., Kassi, E., Lambadiari, V., Gialafos, E., Tsatsaragkou, A., Mystakidi, V.-C., Zisimos, K., Dimitriadis, K., Tousoulis, D., & Siasos, G. (2025). The Role of NLRP3 Inflammasome in Type 2 Diabetes Mellitus and Its Macrovascular Complications. Journal of Clinical Medicine, 14(13), 4606. https://doi.org/10.3390/jcm14134606