
Cardiovascular Risk and Its Presentation in Chronic Kidney Disease

{kind=link}
{kind=link}
Abstract
1. Introduction
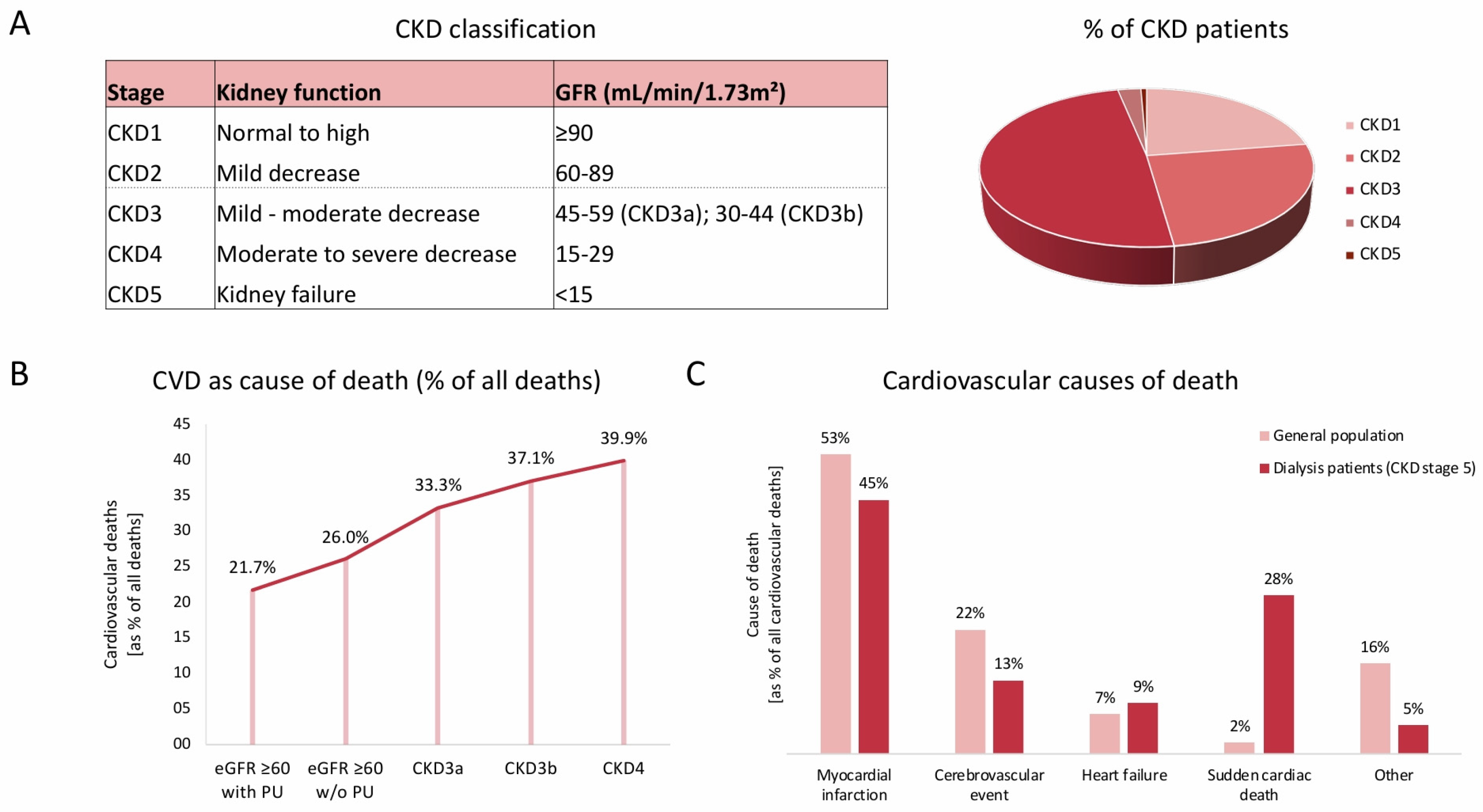
2. Cardiovascular Risk and Its Presentation in CKD
3. Traditional Risk Factors and Their Impact on Cardiovascular Risk in CKD Patients
3.1. Arterial Hypertension
3.2. Smoking
3.3. Dyslipidemia
3.4. Diabetes
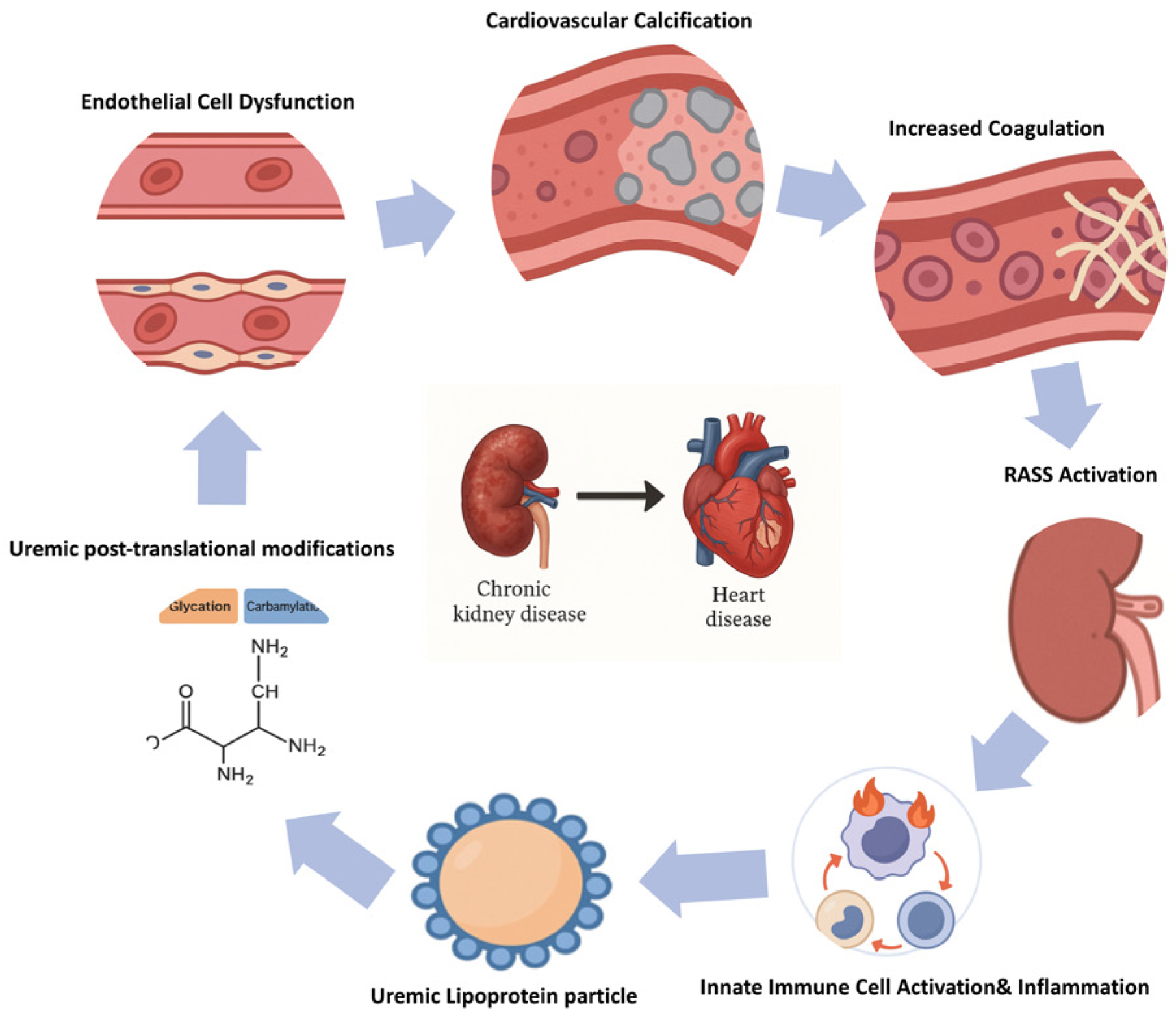
4. Pathophysiological Mechanisms Contributing to Increased Cardiovascular Risk in CKD
4.1. Uremic Toxins
4.2. Uremic Post-Translational Modifications and Uremic Lipoprotein Particles
4.3. Innate Immune Activation, Inflammation, and Oxidative Stress
4.4. Endothelial Dysfunction
4.5. Hypercoagulability and Altered Platelet Function
4.6. Cardiovascular Calcification
4.7. RAAS and SNS Activation
4.8. Anemia
5. Cardiovascular Biomarkers and Their Prognostic Impact on Cardiovascular Risk Assessment
5.1. N-Terminal Pro-BNP (NT-ProBNP)
5.2. High-Sensitivity Troponin T (Hs TnT)
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matsushita, K.; Ballew, S.H.; Wang, A.Y.-M.; Kalyesubula, R.; Schaeffner, E.; Agarwal, R. Epidemiology and risk of cardiovascular disease in populations with chronic kidney disease. Nat. Rev. Nephrol. 2022, 18, 696–707. [Google Scholar] [CrossRef]
- Vondenhoff, S.; Schunk, S.J.; Noels, H. Increased cardiovascular risk in patients with chronic kidney disease. Herz 2024, 49, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Coresh, J.; Sang, Y.; Chalmers, J.; Fox, C.; Guallar, E.; Jafar, T.; Jassal, S.K.; Landman, G.W.D.; Muntner, P.; et al. Estimated glomerular filtration rate and albuminuria for prediction of cardiovascular outcomes: A collaborative meta-analysis of individual participant data. Lancet Diabetes Endocrinol. 2015, 3, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolski, C.; Clark, E.G.; Sood, M.M. Venous thromboembolism in chronic kidney disease: Epidemiology, the role of proteinuria, CKD severity and therapeutics. J. Thromb. Thrombolysis 2017, 43, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.M.; Georgakis, M.K.; Franceschini, N.; Blacker, D.; Viswanathan, A.; Anderson, C.D. Interplay Between Chronic Kidney Disease, Hypertension, and Stroke: Insights From a Multivariable Mendelian Randomization Analysis. Neurology 2023, 101, e1960–e1969. [Google Scholar] [CrossRef]
- Lopau, K.; Wanner, C. Treatment rationale for coronary heart disease in advanced CKD. Herz 2021, 46, 221–227. [Google Scholar] [CrossRef]
- Sarnak, M.J.; Amann, K.; Bangalore, S.; Cavalcante, J.L.; Charytan, D.M.; Craig, J.C.; Gill, J.S.; Hlatky, M.A.; Jardine, A.G.; Landmesser, U.; et al. Chronic Kidney Disease and Coronary Artery Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 74, 1823–1838. [Google Scholar] [CrossRef]
- Manjunath, G.; Tighiouart, H.; Ibrahim, H.; MacLeod, B.; Salem, D.N.; Griffith, J.L.; Coresh, J.; Levey, A.S.; Sarnak, M.J. Level of kidney function as a risk factor for atherosclerotic cardiovascular outcomes in the community. J. Am. Coll. Cardiol. 2003, 41, 47–55. [Google Scholar] [CrossRef]
- Baaten, C.C.F.M.J.; Schröer, J.R.; Floege, J.; Marx, N.; Jankowski, J.; Berger, M.; Noels, H. Platelet Abnormalities in CKD and Their Implications for Antiplatelet Therapy. Clin. J. Am. Soc. Nephrol. 2022, 17, 155–170. [Google Scholar] [CrossRef]
- Babitt, J.L.; Lin, H.Y. Mechanisms of Anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Z.; Chen, H.; Huang, X.; Huang, X.; Lei, Y.; Liang, Q.; Wei, J.; Zhang, Q.; Guo, X.; et al. p53 SUMOylation Mediates AOPP-Induced Endothelial Senescence and Apoptosis Evasion. Front. Cardiovasc. Med. 2022, 8, 795747. [Google Scholar] [CrossRef]
- Claro, L.M.; Moreno-Amaral, A.N.; Gadotti, A.C.; Dolenga, C.J.; Nakao, L.S.; Azevedo, M.L.V.; De Noronha, L.; Olandoski, M.; De Moraes, T.P.; Stinghen, A.E.M.; et al. The Impact of Uremic Toxicity Induced Inflammatory Response on the Cardiovascular Burden in Chronic Kidney Disease. Toxins 2018, 10, 384. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, K.; Liu, Y.; Chen, J.; Cai, Q.; He, W.; Zhang, Y.; Wang, M.-H.; Wang, J.; Huang, H. Advanced oxidation protein products aggravate cardiac remodeling via cardiomyocyte apoptosis in chronic kidney disease. Am. J. Physiol.-Heart Circ. Physiol. 2018, 314, H475–H483. [Google Scholar] [CrossRef] [PubMed]
- Gajjala, P.R.; Fliser, D.; Speer, T.; Jankowski, V.; Jankowski, J. Emerging role of post-translational modifications in chronic kidney disease and cardiovascular disease. Nephrol. Dial. Transplant. 2015, 30, 1814–1824. [Google Scholar] [CrossRef] [PubMed]
- Harlacher, E.; Wollenhaupt, J.; Baaten, C.C.F.M.J.; Noels, H. Impact of Uremic Toxins on Endothelial Dysfunction in Chronic Kidney Disease: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 531. [Google Scholar] [CrossRef]
- Kaur, J.; Young, B.; Fadel, P. Sympathetic Overactivity in Chronic Kidney Disease: Consequences and Mechanisms. Int. J. Mol. Sci. 2017, 18, 1682. [Google Scholar] [CrossRef]
- Kim, Y.-H.; Nijst, P.; Kiefer, K.; Tang, W.H.W. Endothelial Glycocalyx as Biomarker for Cardiovascular Diseases: Mechanistic and Clinical Implications. Curr. Heart Fail. Rep. 2017, 14, 117–126. [Google Scholar] [CrossRef]
- Li, H.Y.; Hou, F.F.; Zhang, X.; Chen, P.Y.; Liu, S.X.; Feng, J.X.; Liu, Z.Q.; Shan, Y.X.; Wang, G.B.; Zhou, Z.M.; et al. Advanced Oxidation Protein Products Accelerate Renal Fibrosis in a Remnant Kidney Model. J. Am. Soc. Nephrol. 2007, 18, 528–538. [Google Scholar] [CrossRef]
- Portolés, J.; Martín, L.; Broseta, J.J.; Cases, A. Anemia in Chronic Kidney Disease: From Pathophysiology and Current Treatments, to Future Agents. Front. Med. 2021, 8, 642296. [Google Scholar] [CrossRef]
- Schunk, S.J.; Triem, S.; Schmit, D.; Zewinger, S.; Sarakpi, T.; Becker, E.; Hütter, G.; Wrublewsky, S.; Küting, F.; Hohl, M.; et al. Interleukin-1α Is a Central Regulator of Leukocyte-Endothelial Adhesion in Myocardial Infarction and in Chronic Kidney Disease. Circulation 2021, 144, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszek, E.; Oldakowska-Jedynak, U.; Kwiatkowska, M.; Glogowski, T.; Malyszko, J. Uremic Toxins, Oxidative Stress, Atherosclerosis in Chronic Kidney Disease, and Kidney Transplantation. Oxidative Med. Cell. Longev. 2021, 2021, 6651367. [Google Scholar] [CrossRef]
- Garikapati, K.; Goh, D.; Khanna, S.; Echampati, K. Uraemic Cardiomyopathy: A Review of Current Literature. Clin. Med. Insights Cardiol. 2021, 15, 1179546821998347. [Google Scholar] [CrossRef]
- Jankowski, J.; Floege, J.; Fliser, D.; Böhm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Valdivielso, J.M.; Rodríguez-Puyol, D.; Pascual, J.; Barrios, C.; Bermúdez-López, M.; Sánchez-Niño, M.D.; Pérez-Fernández, M.; Ortiz, A. Atherosclerosis in Chronic Kidney Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1938–1966. [Google Scholar] [CrossRef] [PubMed]
- Abd Alamir, M.; Radulescu, V.; Goyfman, M.; Mohler, E.R.; Gao, Y.L.; Budoff, M.J.; CRIC Study Investigators. Prevalence and correlates of mitral annular calcification in adults with chronic kidney disease: Results from CRIC study. Atherosclerosis 2015, 242, 117–122. [Google Scholar] [CrossRef]
- Alhaj, E.; Alhaj, N.; Rahman, I.; Niazi, T.O.; Berkowitz, R.; Klapholz, M. Uremic cardiomyopathy: An underdiagnosed disease. Congest. Heart Fail. 2013, 19, E40–E45. [Google Scholar] [CrossRef]
- Agharazii, M.; St-Louis, R.; Gautier-Bastien, A.; Ung, R.-V.; Mokas, S.; Larivière, R.; Richard, D.E. Inflammatory cytokines and reactive oxygen species as mediators of chronic kidney disease-related vascular calcification. Am. J. Hypertens. 2015, 28, 746–755. [Google Scholar] [CrossRef]
- Briet, M.; Barhoumi, T.; Mian, M.O.R.; Sierra, C.; Boutouyrie, P.; Davidman, M.; Bercovitch, D.; Nessim, S.J.; Frisch, G.; Paradis, P.; et al. Effects of recombinant human erythropoietin on resistance artery endothelial function in stage 4 chronic kidney disease. J. Am. Heart Assoc. 2013, 2, e000128. [Google Scholar] [CrossRef]
- Buglioni, A.; Burnett, J.C. Pathophysiology and the cardiorenal connection in heart failure. Circulating hormones: Biomarkers or mediators. Clin. Chim. Acta Int. J. Clin. Chem. 2015, 443, 3–8. [Google Scholar] [CrossRef]
- Nasrallah, R.; Hassouneh, R.; Hébert, R.L. PGE2, Kidney Disease, and Cardiovascular Risk: Beyond Hypertension and Diabetes. J. Am. Soc. Nephrol. JASN 2016, 27, 666–676. [Google Scholar] [CrossRef]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical Renin-Angiotensin system in kidney physiology. Compr. Physiol. 2014, 4, 1201–1228. [Google Scholar] [CrossRef]
- Tan, K.; Sethi, S.K. Biomarkers in cardiorenal syndromes. Transl. Res. J. Lab. Clin. Med. 2014, 164, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Goto, S.; Fukagawa, M. Role of Uremic Toxins for Kidney, Cardiovascular, and Bone Dysfunction. Toxins 2018, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Kumar, U.; Wettersten, N.; Garimella, P.S. Cardiorenal Syndrome: Pathophysiology. Cardiol. Clin. 2019, 37, 251–265. [Google Scholar] [CrossRef]
- Abramson, J.L.; Jurkovitz, C.T.; Vaccarino, V.; Weintraub, W.S.; McClellan, W. Chronic kidney disease, anemia, and incident stroke in a middle-aged, community-based population: The ARIC Study. Kidney Int. 2003, 64, 610–615. [Google Scholar] [CrossRef]
- Lau, W.L. Controversies: Stroke Prevention in Chronic Kidney Disease. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2021, 30, 105679. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.M.; Seliger, S.; Lakshminarayan, K.; Herzog, C.A.; Solid, C.A. Incidence of stroke before and after dialysis initiation in older patients. J. Am. Soc. Nephrol. JASN 2013, 24, 1166–1173. [Google Scholar] [CrossRef]
- Masson, P.; Webster, A.C.; Hong, M.; Turner, R.; Lindley, R.I.; Craig, J.C. Chronic kidney disease and the risk of stroke: A systematic review and meta-analysis. Nephrol. Dial. Transplant. 2015, 30, 1162–1169. [Google Scholar] [CrossRef]
- Major, R.W.; Cheng, M.R.I.; Grant, R.A.; Shantikumar, S.; Xu, G.; Oozeerally, I.; Brunskill, N.J.; Gray, L.J. Cardiovascular disease risk factors in chronic kidney disease: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0192895. [Google Scholar] [CrossRef]
- Ortiz, A.; Covic, A.; Fliser, D.; Fouque, D.; Goldsmith, D.; Kanbay, M.; Mallamaci, F.; Massy, Z.A.; Rossignol, P.; Vanholder, R.; et al. Epidemiology, contributors to, and clinical trials of mortality risk in chronic kidney failure. Lancet 2014, 383, 1831–1843. [Google Scholar] [CrossRef]
- Roehm, B.; Weiner, D.E. Blood pressure targets and kidney and cardiovascular disease: Same data but discordant guidelines. Curr. Opin. Nephrol. Hypertens. 2019, 28, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Zewinger, S.; Kleber, M.E.; Rohrer, L.; Lehmann, M.; Triem, S.; Jennings, R.T.; Petrakis, I.; Dressel, A.; Lepper, P.M.; Scharnagl, H.; et al. Symmetric dimethylarginine, high-density lipoproteins and cardiovascular disease. Eur. Heart J. 2017, 38, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Denic, A.; Glassock, R.J.; Rule, A.D. Structural and Functional Changes With the Aging Kidney. Adv. Chronic Kidney Dis. 2016, 23, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Mallappallil, M.; Friedman, E.A.; Delano, B.G.; McFarlane, S.I.; Salifu, M.O. Chronic kidney disease in the elderly: Evaluation and management. Clin. Pract. 2014, 11, 525–535. [Google Scholar] [CrossRef]
- Gupta, A.; Nagaraju, S.P.; Bhojaraja, M.V.; Swaminathan, S.M.; Mohan, P.B. Hypertension in Chronic Kidney Disease: An Update on Diagnosis and Management. South. Med. J. 2023, 116, 237–244. [Google Scholar] [CrossRef]
- Pugh, D.; Gallacher, P.J.; Dhaun, N. Management of Hypertension in Chronic Kidney Disease. Drugs 2019, 79, 365–379. [Google Scholar] [CrossRef]
- Ruiz-Hurtado, G.; Banegas, J.R.; Sarafidis, P.A.; Volpe, M.; Williams, B.; Ruilope, L.M. Has the SPRINT trial introduced a new blood-pressure goal in hypertension? Nat. Rev. Cardiol. 2017, 14, 560–565. [Google Scholar] [CrossRef]
- Taler, S.J.; Agarwal, R.; Bakris, G.L.; Flynn, J.T.; Nilsson, P.M.; Rahman, M.; Sanders, P.W.; Textor, S.C.; Weir, M.R.; Townsend, R.R. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for management of blood pressure in CKD. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2013, 62, 201–213. [Google Scholar] [CrossRef]
- ADVANCE Collaborative Group; Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008, 358, 2560–2572. [Google Scholar] [CrossRef]
- Rossing, P.; Caramori, M.L.; Chan, J.C.N.; Heerspink, H.J.L.; Hurst, C.; Khunti, K.; Liew, A.; Michos, E.D.; Navaneethan, S.D.; Olowu, W.A.; et al. KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2022, 102, S1–S127. [Google Scholar] [CrossRef] [PubMed]
- Cedillo-Couvert, E.; Ricardo, A.C. Smoking, Vascular Events, and ESRD in Patients With CKD. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2016, 68, 338–340. [Google Scholar] [CrossRef]
- Ali, S.; Dave, N.; Virani, S.S.; Navaneethan, S.D. Primary and Secondary Prevention of Cardiovascular Disease in Patients with Chronic Kidney Disease. Curr. Atheroscler. Rep. 2019, 21, 32. [Google Scholar] [CrossRef]
- Pirillo, A.; Casula, M.; Catapano, A.L. European guidelines for the treatment of dyslipidaemias: New concepts and future challenges. Pharmacol. Res. 2023, 196, 106936. [Google Scholar] [CrossRef]
- Tonelli, M.; Wanner, C.; Kidney Disease: Improving Global Outcomes Lipid Guideline Development Work Group Members. Lipid management in chronic kidney disease: Synopsis of the Kidney Disease: Improving Global Outcomes 2013 clinical practice guideline. Ann. Intern. Med. 2014, 160, 182. [Google Scholar] [CrossRef] [PubMed]
- Yeang, C.; Karwatowska-Prokopczuk, E.; Su, F.; Dinh, B.; Xia, S.; Witztum, J.L.; Tsimikas, S. Effect of Pelacarsen on Lipoprotein(a) Cholesterol and Corrected Low-Density Lipoprotein Cholesterol. J. Am. Coll. Cardiol. 2022, 79, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J. Therapeutic Potential of Lipoprotein(a) Inhibitors. Drugs 2024, 84, 637–643. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170000 participants in 26 randomised trials. Lancet 2010, 376, 1670–1681. [Google Scholar] [CrossRef]
- Robinson, J.G.; Jayanna, M.B.; Bairey Merz, C.N.; Stone, N.J. Clinical implications of the log linear association between LDL-C lowering and cardiovascular risk reduction: Greatest benefits when LDL-C > 100 mg/dL. PLoS ONE 2020, 15, e0240166. [Google Scholar] [CrossRef]
- SEARCH Collaborative Group; Link, E.; Parish, S.; Armitage, J.; Bowman, L.; Heath, S.; Matsuda, F.; Gut, I.; Lathrop, M.; Collins, R. SLCO1B1 Variants and Statin-Induced Myopathy—A Genomewide Study. N. Engl. J. Med. 2008, 359, 789–799. [Google Scholar] [CrossRef]
- Fellström, B.C.; Jardine, A.G.; Schmieder, R.E.; Holdaas, H.; Bannister, K.; Beutler, J.; Chae, D.-W.; Chevaile, A.; Cobbe, S.M.; Grönhagen-Riska, C.; et al. Rosuvastatin and Cardiovascular Events in Patients Undergoing Hemodialysis. N. Engl. J. Med. 2009, 360, 1395–1407. [Google Scholar] [CrossRef]
- Wanner, C.; Krane, V.; März, W.; Olschewski, M.; Mann, J.F.E.; Ruf, G.; Ritz, E. Atorvastatin in Patients with Type 2 Diabetes Mellitus Undergoing Hemodialysis. N. Engl. J. Med. 2005, 353, 238–248. [Google Scholar] [CrossRef]
- Kuznik, A.; Mardekian, J.; Tarasenko, L. Evaluation of cardiovascular disease burden and therapeutic goal attainment in US adults with chronic kidney disease: An analysis of national health and nutritional examination survey data, 2001–2010. BMC Nephrol. 2013, 14, 132. [Google Scholar] [CrossRef]
- Suh, S.H.; Kim, S.W. Dyslipidemia in Patients with Chronic Kidney Disease: An Updated Overview. Diabetes Metab. J. 2023, 47, 612–629. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.M.; Zoungas, S.; Rossing, P.; Groop, P.-H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Primers 2015, 1, 15018. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Cooper, M.E.; Zimmet, P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Alalawi, F.; Bashier, A. Management of diabetes mellitus in dialysis patients: Obstacles and challenges. Diabetes Metab. Syndr. 2021, 15, 1025–1036. [Google Scholar] [CrossRef]
- ACCORD Study Group; Buse, J.B.; Bigger, J.T.; Byington, R.P.; Cooper, L.S.; Cushman, W.C.; Friedewald, W.T.; Genuth, S.; Gerstein, H.C.; Ginsberg, H.N.; et al. Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial: Design and methods. Am. J. Cardiol. 2007, 99, 21i–33i. [Google Scholar] [CrossRef]
- Simpson, S.H.; Lee, J.; Choi, S.; Vandermeer, B.; Abdelmoneim, A.S.; Featherstone, T.R. Mortality risk among sulfonylureas: A systematic review and network meta-analysis. Lancet Diabetes Endocrinol. 2015, 3, 43–51. [Google Scholar] [CrossRef]
- Verma, A.; Patel, A.B.; Upadhyay, A.; Waikar, S.S. CREDENCE: Significant Victory for Diabetic Kidney Disease. Trends Endocrinol. Metab. TEM 2020, 31, 391–393. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.C.; Stefánsson, B.V.; Jongs, N.; Chertow, G.M.; Greene, T.; Hou, F.F.; McMurray, J.J.V.; Correa-Rotter, R.; Rossing, P.; Toto, R.D.; et al. Effects of dapagliflozin on major adverse kidney and cardiovascular events in patients with diabetic and non-diabetic chronic kidney disease: A prespecified analysis from the DAPA-CKD trial. Lancet Diabetes Endocrinol. 2021, 9, 22–31. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Armillotta, M.; Angeli, F.; Paolisso, P.; Belmonte, M.; Raschi, E.; Di Dalmazi, G.; Amicone, S.; Canton, L.; Fedele, D.; Suma, N.; et al. Cardiovascular therapeutic targets of sodium-glucose co-transporter 2 (SGLT2) inhibitors beyond heart failure. Pharmacol. Ther. 2025, 270, 108861. [Google Scholar] [CrossRef] [PubMed]
- Michos, E.D.; Bakris, G.L.; Rodbard, H.W.; Tuttle, K.R. Glucagon-like peptide-1 receptor agonists in diabetic kidney disease: A review of their kidney and heart protection. Am. J. Prev. Cardiol. 2023, 14, 100502. [Google Scholar] [CrossRef]
- Eutox Database. Available online: https://database.uremic-toxins.org/home.php (accessed on 24 June 2025).
- Bansal, N.; Katz, R.; Dalrymple, L.; de Boer, I.; DeFilippi, C.; Kestenbaum, B.; Park, M.; Sarnak, M.; Seliger, S.; Shlipak, M. NT-ProBNP and Troponin T and Risk of Rapid Kidney Function Decline and Incident CKD in Elderly Adults. Clin. J. Am. Soc. Nephrol. 2015, 10, 205–214. [Google Scholar] [CrossRef]
- Li, X.-T.; Zhang, M.-W.; Zhang, Z.-Z.; Cao, Y.-D.; Liu, X.-Y.; Miao, R.; Xu, Y.; Song, X.-F.; Song, J.-W.; Liu, Y.; et al. Abnormal apelin-ACE2 and SGLT2 signaling contribute to adverse cardiorenal injury in patients with COVID-19. Int. J. Cardiol. 2021, 336, 123–129. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Koenig, W.; Libby, P.; Everett, B.M.; Lefkowitz, M.; Thuren, T.; Cornel, J.H. Inhibition of Interleukin-1β by Canakinumab and Cardiovascular Outcomes in Patients With Chronic Kidney Disease. J. Am. Coll. Cardiol. 2018, 71, 2405–2414. [Google Scholar] [CrossRef]
- Ridker, P.M.; Devalaraja, M.; Baeres, F.M.M.; Engelmann, M.D.M.; Hovingh, G.K.; Ivkovic, M.; Lo, L.; Kling, D.; Pergola, P.; Raj, D.; et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 2060–2069. [Google Scholar] [CrossRef]
- Vallance, P.; Calver, A.; Collier, J. The vascular endothelium in diabetes and hypertension. J. Hypertension. Suppl. Off. J. Int. Soc. Hypertens. 1992, 10, S25–S29. [Google Scholar] [CrossRef]
- Carrero, J.J.; Yilmaz, M.I.; Lindholm, B.; Stenvinkel, P. Cytokine dysregulation in chronic kidney disease: How can we treat it? Blood Purif. 2008, 26, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef]
- Klinger, M.; Madziarska, K. Mortality predictor pattern in hemodialysis and peritoneal dialysis in diabetic patients. Adv. Clin. Exp. Med. 2018, 28, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- London, G.M. THE CLINICAL EPIDEMIOLOGY OF CARDIOVASCULAR DISEASES IN CHRONIC KIDNEY DISEASE: Cardiovascular Disease in Chronic Renal Failure: Pathophysiologic Aspects. Semin. Dial. 2003, 16, 85–94. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial Calcification in Chronic Kidney Disease: Key Roles for Calcium and Phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef]
- Giachelli, C.M.; Jono, S.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H. Vascular calcification and inorganic phosphate. Am. J. Kidney Dis. 2001, 38, S34–S37. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Uitto, J.; Reutelingsperger, C.P. Vitamin K-dependent carboxylation of matrix Gla-protein: A crucial switch to control ectopic mineralization. Trends Mol. Med. 2013, 19, 217–226. [Google Scholar] [CrossRef]
- Barreto, D.V.; Barreto, F.C.; Carvalho, A.B.; Cuppari, L.; Cendoroglo, M.; Draibe, S.A.; Moyses, R.M.A.; Neves, K.R.; Jorgetti, V.; Blair, A.; et al. Coronary calcification in hemodialysis patients: The contribution of traditional and uremia-related risk factors. Kidney Int. 2005, 67, 1576–1582. [Google Scholar] [CrossRef]
- Zoccali, C.; Mallamaci, F. Arterial Stiffness as a Cardiovascular Risk Factor in Stage 5D Chronic Kidney Disease Patients: An Age Affair. Am. J. Nephrol. 2017, 45, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Converse, R.L.; Jacobsen, T.N.; Toto, R.D.; Jost, C.M.; Cosentino, F.; Fouad-Tarazi, F.; Victor, R.G. Sympathetic overactivity in patients with chronic renal failure. N. Engl. J. Med. 1992, 327, 1912–1918. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guan, X.; Shao, Y.; Zhou, J.; Huang, Y. The Molecular Mechanism and Therapeutic Strategy of Cardiorenal Syndrome Type 3. Rev. Cardiovasc. Med. 2023, 24, 52. [Google Scholar] [CrossRef]
- Weiss, G.; Goodnough, L.T. Anemia of chronic disease. N. Engl. J. Med. 2005, 352, 1011–1023. [Google Scholar] [CrossRef]
- Singh, A.K. The controversy surrounding hemoglobin and erythropoiesis-stimulating agents: What should we do now? Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2008, 52 (Suppl. S6), S5–S13. [Google Scholar] [CrossRef]
- Singh, A.K.; Szczech, L.; Tang, K.L.; Barnhart, H.; Sapp, S.; Wolfson, M.; Reddan, D. Anaemia of CKD—the CHOIR study revisited. Nephrol. Dial. Transplant. 2007, 22, 1806–1810. [Google Scholar] [CrossRef]
- Haase, V.H. Hypoxia-inducible factor-prolyl hydroxylase inhibitors in the treatment of anemia of chronic kidney disease. Kidney Int. Suppl. 2021, 11, 8–25. [Google Scholar] [CrossRef]
- Sanghani, N.S.; Haase, V.H. Hypoxia-Inducible Factor Activators in Renal Anemia: Current Clinical Experience. Adv. Chronic Kidney Dis. 2019, 26, 253–266. [Google Scholar] [CrossRef]
- Han, X.; Zhang, S.; Chen, Z.; Adhikari, B.K.; Zhang, Y.; Zhang, J.; Sun, J.; Wang, Y. Cardiac biomarkers of heart failure in chronic kidney disease. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 510, 298–310. [Google Scholar] [CrossRef]
- Castiglione, V.; Aimo, A.; Vergaro, G.; Saccaro, L.; Passino, C.; Emdin, M. Biomarkers for the diagnosis and management of heart failure. Heart Fail. Rev. 2022, 27, 625–643. [Google Scholar] [CrossRef] [PubMed]
- Metzinger-Le Meuth, V.; Burtey, S.; Maitrias, P.; Massy, Z.A.; Metzinger, L. MicroRNAs in the pathophysiology of CKD-MBD: Biomarkers and innovative drugs. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 337–345. [Google Scholar] [CrossRef]
- Paloian, N.J.; Giachelli, C.M. A current understanding of vascular calcification in CKD. Am. J. Physiol. Ren. Physiol. 2014, 307, F891–F900. [Google Scholar] [CrossRef]
- Motshwari, D.D.; Matshazi, D.M.; Erasmus, R.T.; Kengne, A.P.; Matsha, T.E.; George, C. MicroRNAs Associated with Chronic Kidney Disease in the General Population and High-Risk Subgroups-A Systematic Review. Int. J. Mol. Sci. 2023, 24, 1792. [Google Scholar] [CrossRef]
- DeFilippi, C.; van Kimmenade, R.R.J.; Pinto, Y.M. Amino-terminal pro-B-type natriuretic peptide testing in renal disease. Am. J. Cardiol. 2008, 101, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Yasue, H.; Yoshimura, M.; Sumida, H.; Kikuta, K.; Kugiyama, K.; Jougasaki, M.; Ogawa, H.; Okumura, K.; Mukoyama, M.; Nakao, K. Localization and mechanism of secretion of B-type natriuretic peptide in comparison with those of A-type natriuretic peptide in normal subjects and patients with heart failure. Circulation 1994, 90, 195–203. [Google Scholar] [CrossRef]
- van Kimmenade, R.R.J.; Januzzi, J.L.; Bakker, J.A.; Houben, A.J.; Rennenberg, R.; Kroon, A.A.; Crijns, H.J.G.M.; van Dieijen-Visser, M.P.; de Leeuw, P.W.; Pinto, Y.M. Renal clearance of B-type natriuretic peptide and amino terminal pro-B-type natriuretic peptide a mechanistic study in hypertensive subjects. J. Am. Coll. Cardiol. 2009, 53, 884–890. [Google Scholar] [CrossRef]
- Vickery, S.; Price, C.P.; John, R.I.; Abbas, N.A.; Webb, M.C.; Kempson, M.E.; Lamb, E.J. B-type natriuretic peptide (BNP) and amino-terminal proBNP in patients with CKD: Relationship to renal function and left ventricular hypertrophy. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2005, 46, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.S.; Toto, R.; Jarolim, P.; Uno, H.; Eckardt, K.-U.; Kewalramani, R.; Levey, A.S.; Lewis, E.F.; McMurray, J.J.V.; Parving, H.-H.; et al. Association between cardiac biomarkers and the development of ESRD in patients with type 2 diabetes mellitus, anemia, and CKD. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2011, 58, 717–728. [Google Scholar] [CrossRef]
- Park, M.; Shlipak, M.G.; Katz, R.; Agarwal, S.; Ix, J.H.; Hsu, C.-Y.; Peralta, C.A. Subclinical cardiac abnormalities and kidney function decline: The multi-ethnic study of atherosclerosis. Clin. J. Am. Soc. Nephrol. CJASN 2012, 7, 1137–1144. [Google Scholar] [CrossRef]
- Shlipak, M.G.; Katz, R.; Kestenbaum, B.; Fried, L.F.; Siscovick, D.; Sarnak, M.J. Clinical and subclinical cardiovascular disease and kidney function decline in the elderly. Atherosclerosis 2009, 204, 298–303. [Google Scholar] [CrossRef]
- Patel, U.D.; Garg, A.X.; Krumholz, H.M.; Shlipak, M.G.; Coca, S.G.; Sint, K.; Thiessen-Philbrook, H.; Koyner, J.L.; Swaminathan, M.; Passik, C.S.; et al. Preoperative serum brain natriuretic peptide and risk of acute kidney injury after cardiac surgery. Circulation 2012, 125, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Zelnick, L.R.; Ballantyne, C.M.; Chaves, P.H.M.; Christenson, R.H.; Coresh, J.; de Filippi, C.R.; de Lemos, J.A.; Daniels, L.B.; Go, A.S.; et al. Upper Reference Limits for High-Sensitivity Cardiac Troponin T and N-Terminal Fragment of the Prohormone Brain Natriuretic Peptide in Patients With CKD. Am. J. Kidney Dis. 2022, 79, 383–392. [Google Scholar] [CrossRef]
- Long, B.; Belcher, C.N.; Koyfman, A.; Bronner, J.M. Interpreting troponin in renal disease: A narrative review for emergency clinicians. Am. J. Emerg. Med. 2020, 38, 990–997. [Google Scholar] [CrossRef]
- Stacy, S.R.; Suarez-Cuervo, C.; Berger, Z.; Wilson, L.M.; Yeh, H.-C.; Bass, E.B.; Michos, E.D. Role of troponin in patients with chronic kidney disease and suspected acute coronary syndrome: A systematic review. Ann. Intern. Med. 2014, 161, 502–512. [Google Scholar] [CrossRef]
- Tarapan, T.; Musikatavorn, K.; Phairatwet, P.; Takkavatakarn, K.; Susantitaphong, P.; Eiam-Ong, S.; Tiranathanagul, K. High sensitivity Troponin-I levels in asymptomatic hemodialysis patients. Ren. Fail. 2019, 41, 393–400. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schunk, S.J.; Zimmermann, P. Cardiovascular Risk and Its Presentation in Chronic Kidney Disease. J. Clin. Med. 2025, 14, 4567. https://doi.org/10.3390/jcm14134567
Schunk SJ, Zimmermann P. Cardiovascular Risk and Its Presentation in Chronic Kidney Disease. Journal of Clinical Medicine. 2025; 14(13):4567. https://doi.org/10.3390/jcm14134567
Chicago/Turabian StyleSchunk, Stefan J., and Paul Zimmermann. 2025. "Cardiovascular Risk and Its Presentation in Chronic Kidney Disease" Journal of Clinical Medicine 14, no. 13: 4567. https://doi.org/10.3390/jcm14134567
APA StyleSchunk, S. J., & Zimmermann, P. (2025). Cardiovascular Risk and Its Presentation in Chronic Kidney Disease. Journal of Clinical Medicine, 14(13), 4567. https://doi.org/10.3390/jcm14134567