
Hypertrophic Cardiomyopathy and Phenocopies: New Therapies for Old Diseases—Current Evidence and Future Perspectives

 , , , , , and
, , , , , and
Abstract

1. Introduction
2. Hypertrophic Cardiomyopathy
2.1. Current and Emerging Therapies in oHCM
2.2. Practice Implications for oHCM
- HCM is the most common inherited cardiomyopathy with a worldwide prevalence of 1:500.
- Clinical presentation includes heart failure, atrial fibrillation, syncope, ventricular arrhythmias, and sudden death.
- Standard treatment of oHCM is based on non-vasodilating beta-blockers and non-dihydropyridine calcium channel blockers. If symptoms persist despite therapy with beta-blockers or calcium channel blockers titrated to the maximum dosage, disopyramide is added as second-line therapy.
- CMIs (mavacamten and aficamten) are a novel drug class that has been shown in recent trials to reduce LVOTO, symptoms and NYHA class and improve exercise tolerance in oHCM.
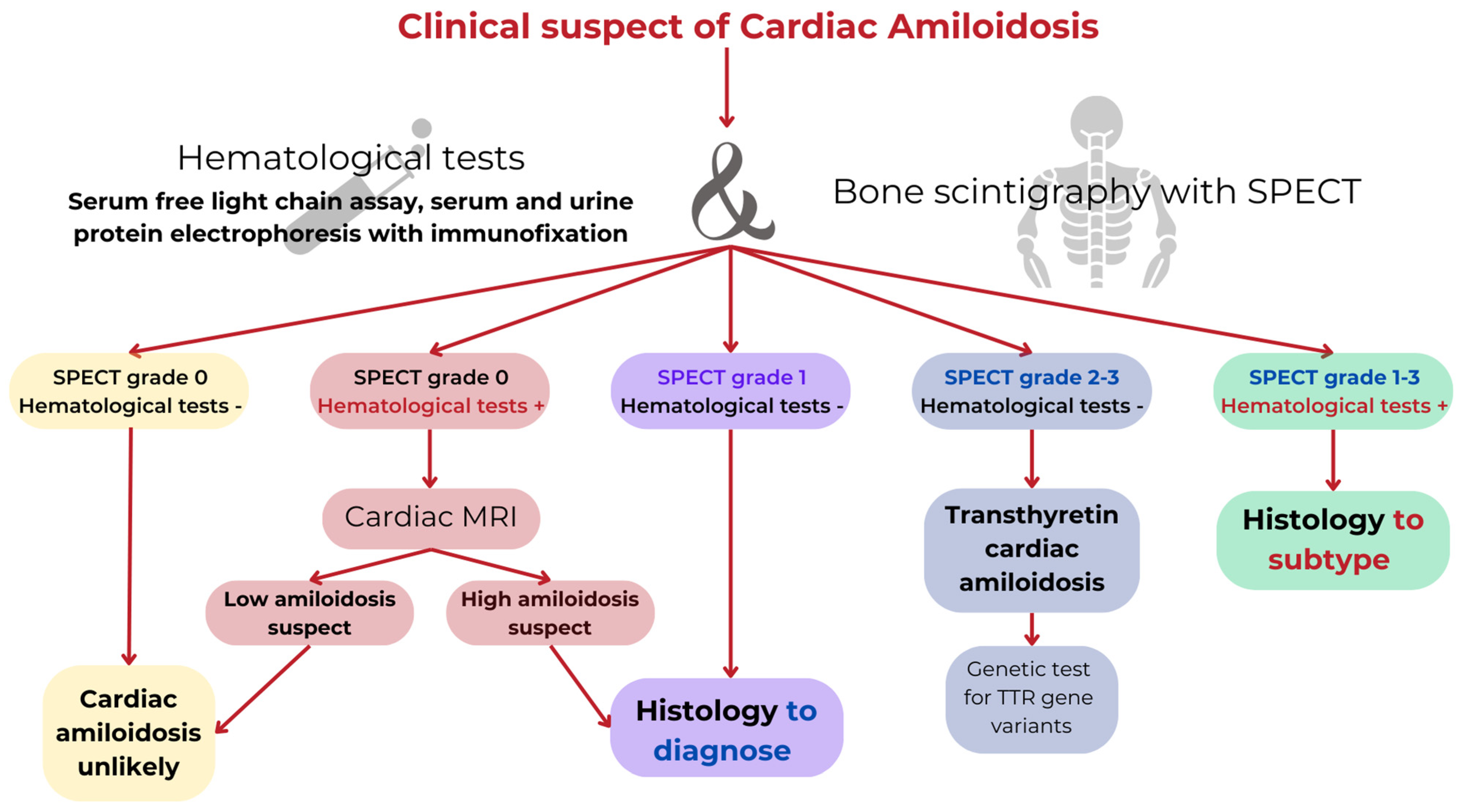
2.3. Cardiac Amyloidosis
2.4. Current and Emerging Therapies in CA
2.5. Stabilizers of TTR
2.6. TTR Suppressors: Gene Silencers
2.7. TTR Suppressors: (CRISPR)-Cas9 Technology
2.8. Degraders of Amyloid Fibrils
2.9. Practice Implications for CA
- CA is a restrictive cardiomyopathy caused by extracellular deposition of amyloid fibrils formed by misfolded proteins into the heart.
- In 95% of cases, CA is caused by the deposition of misfolded monoclonal immunoglobulin light chains derived from an abnormal clone of plasma cell proliferation (AL amyloidosis) or misfolded transthyretin (TTR amyloidosis).
- The improvement of diagnostic techniques and the development of ‘disease-modifying’ therapies have changed the natural course of CA.
- Chemotherapy agents (alkylating agents, proteasome inhibitors, immunomodulatory drugs, and monoclonal antibodies targeting clonal cells) are available in AL amyloidosis.
- In ATTR-CM, there are three types of therapies that block the amyloidogenic cascade at different points: TTR stabilizers (tafamidis, diflunisal, and acoramidis), TTR suppressors through gene silencing (patisiran, vutrisiran, eplontersen) and gene editing (NTLA-2001 based on CRISPR-Cas9 technology), and degraders of amyloid fibrils (monoclonal antibodies such as NI006/ALX2220, coramitug, and AT02).
3. Anderson–Fabry Disease
3.1. Current and Emerging Therapies in AFD
3.2. Practice Implications for AFD
- AFD is a rare X-linked lysosomal disorder caused by mutations in the GLA gene.
- The main goal of AFD treatment is to prevent disease progression.
- Specific approved treatments include ERT (agalsidase-alpha, agalsidase-beta, and the recently approved pegunigalsidase alfa) and chaperone therapy (migalastat).
- New therapeutic approaches including substrate reduction therapy, gene therapy, and mRNA therapies are under development.
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef]
- Rowin, E.J.; Maron, B.J.; Maron, M.S. The hypertrophic cardiomyopathy phenotype viewed through the prism of multimodality imaging: Clinical and etiologic implications. JACC Cardiovasc. Imaging 2020, 13, 2002–2016. [Google Scholar] [CrossRef] [PubMed]
- Teare, D. Asymmetrical hypertrophy of the heart in young adults. Br. Heart J. 1958, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Rowin, E.J.; Maron, M.S. Global burden of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2018, 6, 376–378. [Google Scholar] [CrossRef]
- Lehman, S.J.; Crocini, C.; Leinwand, L.A. Targeting the sarcomere in inherited cardiomyopathies. Nat. Rev. Cardiol. 2022, 19, 353–363. [Google Scholar] [CrossRef]
- Gerull, B.; Klaassen, S.; Brodehl, A. The Genetic Landscape of Cardiomyopathies. In Genetic Causes of Cardiac Disease; Erdmann, J., Moretti, A., Eds.; Spinger: Berlin/Heidelberg, Germany, 2019. [Google Scholar] [CrossRef]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 372–389. [Google Scholar] [CrossRef]
- Zaiser, E.; Sehnert, A.J.; Duenas, A.; Saberi, S.; Brookes, E.; Reaney, M. Patient experiences with hypertrophic cardiomyopathy: A conceptual model of symptoms and impacts on quality of life. J. Patient Rep. Outcomes 2020, 4, 102. [Google Scholar] [CrossRef]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.S.; et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: Insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef]
- Maron, M.S.; Olivotto, I.; Zenovich, A.G.; Link, M.S.; Pandian, N.G.; Kuvin, J.T.; Nistri, S.; Cecchi, F.; Udelson, J.E.; Maron, B.J. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006, 114, 2232–2239. [Google Scholar] [CrossRef]
- Dybro, A.M.; Rasmussen, T.B.; Nielsen, R.R.; Andersen, M.J.; Jensen, M.K.; Poulsen, S.H. Randomized trial of metoprolol in patients with obstructive hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2021, 78, 2505–2517. [Google Scholar] [CrossRef] [PubMed]
- Toshima, H.; Koga, Y.; Nagata, H.; Toyomasu, K.; Itaya, K.; Matoba, T. Comparable effects of oral diltiazem and verapamil in the treatment of hypertrophic cardiomyopathy. Double-blind crossover study. Jpn Heart J. 1986, 27, 701–715. [Google Scholar] [CrossRef]
- Sherrid, M.V.; Barac, I.; McKenna, W.J.; Elliott, P.M.; Dickie, S.; Chojnowska, L.; Casey, S.; Maron, B.J. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2005, 45, 1251–1258. [Google Scholar] [CrossRef]
- Ommen, S.R.; Ho, C.Y.; Asif, I.M.; Balaji, S.; Burke, M.A.; Day, S.M.; Dearani, J.A.; Epps, K.C.; Evanovich, L.; Ferrari, V.A.; et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2024, 149, e1239–e1311. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.L.; Trivedi, D.V.; Sarkar, S.S.; Henze, M.; Ma, W.; Gong, H.; Rogers, C.S.; Gorham, J.M.; Wong, F.L.; Morck, M.M.; et al. Deciphering the super relaxed state of human beta-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc. Natl. Acad. Sci. USA 2018, 115, E8143–E8152. [Google Scholar] [CrossRef]
- Chuang, C.; Collibee, S.; Ashcraft, L.; Wang, W.; Vander Wal, M.; Wang, X.; Hwee, D.T.; Wu, Y.; Wang, J.; Chin, E.R.; et al. Discovery of aficamten (CK-274), a next-generation cardiac myosin inhibitor for the treatment of hypertrophic cardiomyopathy. J. Med. Chem. 2021, 64, 14142–14152. [Google Scholar] [CrossRef]
- Heitner, S.B.; Jacoby, D.; Lester, S.J.; Owens, A.; Wang, A.; Zhang, D.; Lambing, J.; Lee, J.; Semigran, M.; Sehnert, A.J. Mavacamten treatment for obstructive hypertrophic cardiomyopathy: A clinical trial. Ann. Intern Med. 2019, 170, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Masri, A.; Lester, S.J.; Stendahl, J.C.; Hegde, S.M.; Sehnert, A.J.; Balaratnam, G.; Shah, A.; Fox, S.; Wang, A. Long-Term Safety and Efficacy of Mavacamten in Symptomatic Obstructive Hypertrophic Cardiomyopathy: Interim Results of the PIONEER-OLE Study. J. Am. Heart Assoc. 2024, 13, e030607. [Google Scholar] [CrossRef]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef]
- Rader, F.; Oręziak, A.; Choudhury, L.; Saberi, S.; Fermin, D.; Wheeler, M.T.; Abraham, T.P.; Garcia-Pavia, P.; Zwas, D.R.; Masri, A.; et al. Mavacamten Treatment for Symptomatic Obstructive Hypertrophic Cardiomyopathy: Interim Results from the MAVA-LTE Study, EXPLORER-LTE Cohort. JACC Heart Fail. 2024, 12, 164–177. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Oręziak, A.; Masri, A.; Barriales-Villa, R.; Abraham, T.P.; Owens, A.T.; Jensen, M.K.; Wojakowski, W.; Seidler, T.; Hagege, A.; et al. Long-term effect of mavacamten in obstructive hypertrophic cardiomyopathy. Eur. Heart J. 2024, 45, 5071–5083. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.Y.; Owens, A.; Geske, J.B.; Wolski, K.; Naidu, S.S.; Smedira, N.G.; Cremer, P.C.; Schaff, H.; McErlean, E.; Sewell, C.; et al. Myosin Inhibition in Patients with Obstructive Hypertrophic Cardiomyopathy Referred for Septal Reduction Therapy. J. Am. Coll. Cardiol. 2022, 80, 95–108. [Google Scholar] [CrossRef]
- Desai, M.Y.; Owens, A.; Wolski, K.; Geske, J.B.; Saberi, S.; Wang, A.; Sherrid, M.; Cremer, P.C.; Lakdawala, N.K.; Tower-Rader, A.; et al. Mavacamten in Patients with Hypertrophic Cardiomyopathy Referred for Septal Reduction: Week 56 Results from the VALOR-HCM Randomized Clinical Trial. JAMA Cardiol. 2023, 8, 968–977. [Google Scholar] [CrossRef]
- Maron, M.S.; Masri, A.; Choudhury, L.; Olivotto, I.; Saberi, S.; Wang, A.; Garcia-Pavia, P.; Lakdawala, N.K.; Nagueh, S.F.; Rader, F.; et al. Phase 2 Study of Aficamten in Patients with Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2023, 81, 34–45. [Google Scholar] [CrossRef]
- Maron, M.S.; Masri, A.; Nassif, M.E.; Barriales-Villa, R.; Arad, M.; Cardim, N.; Choudhury, L.; Claggett, B.; Coats, C.J.; Düngen, H.D.; et al. Aficamten for Symptomatic Obstructive Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2024, 390, 1849–1861. [Google Scholar] [CrossRef]
- Maron, B.J.; Dearani, J.A.; Smedira, N.G.; Schaff, H.V.; Wang, S.; Rastegar, H.; Ralph-Edwards, A.; Ferrazzi, P.; Swistel, D.; Shemin, R.J.; et al. Ventricular septal myectomy for obstructive hypertrophic cardiomyopathy (Analysis Spanning 60 Years of Practice): AJC expert panel. Am. J. Cardiol. 2022, 180, 124–139. [Google Scholar] [CrossRef]
- Maurizi, N.; Antiochos, P.; Owens, A.; Lakdwala, N.; Saberi, S.; Russell, M.W.; Fumagalli, C.; Skalidis, I.; Lin, K.Y.; Nathan, A.S.; et al. Long-Term Outcomes After Septal Reduction Therapies in Obstructive Hypertrophic Cardiomyopathy: Insights from the SHARE Registry. Circulation 2024, 150, 1377–1390. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.Y.; Nissen, S.E.; Abraham, T.; Olivotto, I.; Garcia-Pavia, P.; Lopes, R.D.; Verheyen, N.; Wever-Pinzon, O.; Wolski, K.; Jaber, W.; et al. Mavacamten in Symptomatic Nonobstructive Hypertrophic Cardiomyopathy: Design, Rationale, and Baseline Characteristics of ODYSSEY-HCM. JACC Heart Fail. 2025, 13, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, A.; Iversen, K.; Vejlstrup, N.; Ho, C.Y.; Havndrup, O.; Kofoed, K.F.; Norsk, J.; Jensen, M.; Bundgaard, H. Functional effects of losartan in hypertrophic cardiomyopathy-a randomised clinical trial. Heart 2016, 102, 285–291. [Google Scholar] [CrossRef]
- Olivotto, I.; Camici, P.G.; Merlini, P.A.; Rapezzi, C.; Patten, M.; Climent, V.; Sinagra, G.; Tomberli, B.; Marin, F.; Ehlermann, P.; et al. Efficacy of Ranolazine in Patients with Symptomatic Hypertrophic Cardiomyopathy: The RESTYLE-HCM Randomized, Double-Blind, Placebo-Controlled Study. Circ. Heart Fail. 2018, 11, e004124. [Google Scholar] [CrossRef]
- Maron, M.S.; Chan, R.H.; Kapur, N.K.; Jaffe, I.Z.; McGraw, A.P.; Kerur, R.; Maron, B.J.; Udelson, J.E. Effect of Spironolactone on Myocardial Fibrosis and Other Clinical Variables in Patients with Hypertrophic Cardiomyopathy. Am. J. Med. 2018, 131, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Velicki, L.; Popovic, D.; Okwose, N.C.; Preveden, A.; Tesic, M.; Tafelmeier, M.; Charman, S.J.; Barlocco, F.; MacGowan, G.A.; Seferovic, P.M.; et al. SILICOFCM Investigators. Sacubitril/valsartan for the treatment of non-obstructive hypertrophic cardiomyopathy: An open label randomized controlled trial (SILICOFCM). Eur. J. Heart Fail. 2024, 26, 1361–1368. [Google Scholar] [CrossRef]
- Aglan, A.; Fath, A.R.; Eldaly, A.S.; Anderson, A.S.; Phillips, J.S.; Maron, B.J.; Maron, M.S.; Rowin, E.J. Impact of Sodium-Glucose Cotransporter 2 Inhibitors on Mortality in Hypertrophic Cardiomyopathy. JACC Adv. 2024, 3, 100843. [Google Scholar] [CrossRef]
- Soyka, J. Uber amyloide degeneration. Prag. Med. Wochenschr. 1876, 1, 165–171. [Google Scholar]
- Pomerance, A. Senile cardiac amyloidosis. Br. Heart J. 1965, 27, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.H.; Alexander, K.M.; Liao, R.; Dorbala, S. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J. Am. Coll. Cardiol. 2016, 68, 1323–1341. [Google Scholar] [CrossRef]
- Gertz, M.A.; Benson, M.D.; Dyck, P.J.; Grogan, M.; Coelho, T.; Cruz, M.; Berk, J.L.; Plante-Bordeneuve, V.; Schmidt, H.H.J.; Merlini, G. Diagnosis, Prognosis, and Therapy of Transthyretin Amyloidosis. J. Am. Coll. Cardiol. 2015, 66, 2451–2466. [Google Scholar] [CrossRef] [PubMed]
- Grogan, M.; Scott, C.G.; Kyle, R.A.; Zeldenrust, S.R.; Gertz, M.A.; Lin, G.; Klarich, K.W.; Miller, W.L.; Maleszewski, J.J.; Dispenzieri, A. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J. Am. Coll. Cardiol. 2016, 68, 1014–1020. [Google Scholar] [CrossRef]
- Quock, T.P.; Yan, T.; Chang, E.; Guthrie, S.; Broder, M.S. Epidemiology of AL amyloidosis: A real-world study using US claims data. Blood Adv. 2018, 2, 1046–1053. [Google Scholar] [CrossRef]
- Schmidt, H.H.; Waddington-Cruz, M.; Botteman, M.F.; Carter, J.A.; Chopra, A.S.; Hopps, M.; Stewart, M.; Fallet, S.; Amass, L. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve 2018, 57, 829–837. [Google Scholar] [CrossRef]
- Ruberg, F.L.; Maurer, M.S. Cardiac Amyloidosis Due to Transthyretin Protein: A Review. JAMA 2024, 331, 778–791. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and treatment of cardiac amyloidosis: A position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2021, 42, 1554–1568. [Google Scholar] [CrossRef] [PubMed]
- Palladini, G.; Milani, P. Diagnosis and Treatment of AL Amyloidosis. Drugs 2023, 83, 203–216. [Google Scholar] [CrossRef]
- Wechalekar, A.D.; Fontana, M.; Quarta, C.C.; Liedtke, M. AL Amyloidosis for Cardiologists: Awareness, Diagnosis, and Future Prospects: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2022, 4, 427–441. [Google Scholar] [CrossRef]
- Gonzalez-Lopez, E.; Maurer, M.S.; Garcia-Pavia, P. Transthyretin amyloid cardiomyopathy: A paradigm for advancing precision medicine. Eur. Heart J. 2025, 46, 999–1013. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Drachman, B.M.; Gottlieb, S.S.; Hoffman, J.E.; Hummel, S.L.; Lenihan, D.J.; Ebede, B.; Gundapaneni, B.; Li, B.; Sultan, M.B.; et al. Long-term survival with tafamidis in patients with transthyretin amyloid cardiomyopathy. Circ. Heart Fail. 2022, 15, e008193. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Sultan, M.B.; Gundapaneni, B.; Sekijima, Y.; Perfetto, F.; Hanna, M.; Witteles, R. Tafamidis efficacy among octogenarian patients in the phase 3 ATTR-ACT and ongoing long-term extension study. JACC Heart Fail. 2024, 12, 150–160. [Google Scholar] [CrossRef]
- Debonnaire, P.; Dujardin, K.; Verheyen, N.; Pouleur, A.C.; Droogmans, S.; Claeys, M.; Bohyn, A.; Bogaerts, K.; El Haddad, M.; Christiaen, E.; et al. Tafamidis in octogenarians with wild-type transthyretin cardiac amyloidosis: An international cohort study. Eur. Heart J. 2025, 46, 1057–1070. [Google Scholar] [CrossRef]
- Peiró-Aventín, B.; Cabrera-Romero, E.; Mora-Ayestarán, N.; Domínguez, F.; González-López, E.; García-Pavía, P. Safety and efficacy of diflunisal in transthyretin cardiac amyloidosis. Rev. Esp. Cardiol. 2024, 77, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.T.; Paxman, R.J.; Xu, J.; Webb, B.; Powers, E.T.; Kelly, J.W. Blinded potency comparison of transthyretin kinetic stabilisers by subunit exchange in human plasma. Amyloid 2021, 28, 24–29. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Judge, D.P.; Cappelli, F.; Fontana, M.; Garcia-Pavia, P.; Gibbs, S.; Grogan, M.; Hanna, M.; Hoffman, J.; Masri, A.; et al. Efficacy and Safety of Acoramidis in Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2024, 390, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Judge, D.P.; Gillmore, J.D.; Alexander, K.M.; Ambardekar, A.V.; Cappelli, F.; Fontana, M.; García-Pavía, P.; Grodin, J.L.; Grogan, M.; Hanna, M.; et al. Long-Term Efficacy and Safety of Acoramidis in ATTR-CM: Initial Report From the Open-Label Extension of the ATTRibute-CM Trial. Circulation 2025, 151, 601–611. [Google Scholar] [CrossRef]
- Solomon, S.D.; Adams, D.; Kristen, A.; Grogan, M.; González-Duarte, A.; Maurer, M.S.; Merlini, G.; Damy, T.; Slama, M.S.; Brannagan, T.H., 3rd; et al. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation 2019, 139, 431–443. [Google Scholar] [CrossRef]
- Maurer, M.S.; Kale, P.; Fontana, M.; Berk, J.L.; Grogan, M.; Gustafsson, F.; Hung, R.R.; Gottlieb, R.L.; Damy, T.; González-Duarte, A.; et al. Patisiran Treatment in Patients with Transthyretin Cardiac Amyloidosis. N. Engl. J. Med. 2023, 389, 1553–1565. [Google Scholar] [CrossRef]
- Fontana, M.; Berk, J.L.; Gillmore, J.D.; Witteles, R.M.; Grogan, M.; Drachman, B.; Damy, T.; Garcia-Pavia, P.; Taubel, J.; Solomon, S.D.; et al. Vutrisiran in Patients with Transthyretin Amyloidosis with Cardiomyopathy. N. Engl. J. Med. 2025, 392, 33–44. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 2021, 385, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Michalon, A.; Hagenbuch, A.; Huy, C.; Varela, E.; Combaluzier, B.; Damy, T.; Suhr, O.B.; Saraiva, M.J.; Hock, C.; Nitsch, R.M.; et al. A human antibody selective for transthyretin amyloid removes cardiac amyloid through phagocytic immune cells. Nat. Commun. 2021, 12, 3142. [Google Scholar] [CrossRef]
- Suhr, O.B.; Grogan, M.; Silva, A.M.D.; Karam, C.; Garcia-Pavia, P.; Drachman, B.; Zago, W.; Tripuraneni, R.; Kinney, G.G. PRX004 in variant amyloid transthyretin (ATTRv) amyloidosis: Results of a phase 1, open-label, dose-escalation study. Amyloid 2024, 32, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.B.; Stuckey, A.; Powell, D.; Lands, R.; Whittle, B.; Wooliver, C.; Macy, S.; Foster, J.S.; Guthrie, S.; Kennel, S.J.; et al. Clinical confirmation of pan-amyloid reactivity of radioiodinated peptide 124I-p5+14 (AT-01) in patients with diverse types of systemic amyloidosis demonstrated by PET/CT imaging. Pharmaceuticals 2023, 16, 629. [Google Scholar] [CrossRef] [PubMed]
- Desnick, R.J.; Ioannou, Y.A.; Eng, C.M. Alpha-galactosidase A deficiency: Fabry disease. In The Metabolic and Molecular Basis of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3733–3774. [Google Scholar]
- Germain, D.P. Fabry disease. Orphanet. J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef]
- Fabry, J. Ein Beitrag zur Kenntniss der Purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae). Arch. Dermatol. Syph. 1898, 43, 187–200. [Google Scholar] [CrossRef]
- Anderson, W. A case of “Angeio-Keratoma”. Br. J. Dermatol. 1898, 10, 113–117. [Google Scholar] [CrossRef]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Pieroni, M.; Namdar, M.; Olivotto, I.; Desnick, R.J. Anderson-Fabry disease management: Role of the cardiologist. Eur. Heart J. 2024, 45, 1395–1409. [Google Scholar] [CrossRef]
- Linhart, A.; Germain, D.P.; Olivotto, I.; Akhtar, M.M.; Anastasakis, A.; Hughes, D.; Namdar, M.; Pieroni, M.; Hagège, A.; Cecchi, F.; et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur. J. Heart Fail. 2020, 22, 1076–1096. [Google Scholar] [CrossRef]
- Meucci, M.C.; Lillo, R.; Del Franco, A.; Monda, E.; Iannaccone, G.; Baldassarre, R.; Di Nicola, F.; Parisi, V.; Lombardo, A.; Spinelli, L.; et al. Prognostic Implications of the Extent of Cardiac Damage in Patients with Fabry Disease. J. Am. Coll. Cardiol. 2023, 82, 1524–1534. [Google Scholar] [CrossRef]
- Del Franco, A.; Iannaccone, G.; Meucci, M.C.; Lillo, R.; Cappelli, F.; Zocchi, C.; Pieroni, M.; Graziani, F.; Olivotto, I. Clinical staging of Anderson-Fabry cardiomyopathy: An operative proposal. Heart Fail. Rev. 2024, 29, 431–444. [Google Scholar] [CrossRef]
- Garman, S.C.; Garboczi, D.N. The molecular defect leading to Fabry disease: Structure of human alpha-galactosidase. J. Mol. Biol. 2004, 337, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Vedder, A.C.; Breunig, F.; Donker-Koopman, W.E.; Mills, K.; Young, E.; Winchester, B.; Ten Berge, I.J.; Groener, J.E.; Aerts, J.M.; Wanner, C.; et al. Treatment of Fabry disease with different dosing regimens of agalsidase: Effects on antibody formation and GL-3. Mol. Genet. Metab. 2008, 94, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Niemann, M.; Störk, S.; Breunig, F.; Beer, M.; Sommer, C.; Herrmann, S.; Ertl, G.; Wanner, C. Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: Evidence for disease progression towards serious complications. J. Intern Med. 2013, 274, 331–341. [Google Scholar] [CrossRef]
- Azevedo, O.; Gago, M.F.; Miltenberger-Miltenyi, G.; Sousa, N.; Cunha, D. Fabry Disease Therapy: State-of-the-Art and Current Challenges. Int. J. Mol. Sci. 2020, 22, 206. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Ramaswami, U.; Hernberg-Ståhl, E.; Hughes, D.A.; Kampmann, C.; Mehta, A.B.; Nicholls, K.; Niu, D.M.; Pintos-Morell, G.; Reisin, R.; et al. Twenty years of the Fabry Outcome Survey (FOS): Insights, achievements, and lessons learned from a global patient registry. Orphanet. J. Rare Dis. 2022, 17, 238. [Google Scholar] [CrossRef]
- Frustaci, A.; Verardo, R.; Galea, N.; Alfarano, M.; Magnocavallo, M.; Marchitelli, L.; Sansone, L.; Belli, M.; Cristina, M.; Frustaci, E.; et al. Long-Term Clinical-Pathologic Results of Enzyme Replacement Therapy in Prehypertrophic Fabry Disease Cardiomyopathy. J. Am. Heart Assoc. 2024, 13, e032734. [Google Scholar] [CrossRef]
- Pieroni, M.; Moon, J.C.; Arbustini, E.; Barriales-Villa, R.; Camporeale, A.; Vujkovac, A.C.; Elliott, P.M.; Hagege, A.; Kuusisto, J.; Linhart, A.; et al. Cardiac Involvement in Fabry Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 922–936. [Google Scholar] [CrossRef]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef]
- Lenders, M.; Nordbeck, P.; Kurschat, C.; Eveslage, M.; Karabul, N.; Kaufeld, J.; Hennermann, J.B.; Patten, M.; Cybulla, M.; Müntze, J.; et al. Treatment of Fabry Disease management with migalastat-outcome from a prospective 24 months observational multicenter study (FAMOUS). Eur. Heart J. Cardiovasc. Pharmacother. 2022, 8, 272–281. [Google Scholar] [CrossRef]
- Camporeale, A.; Bandera, F.; Pieroni, M.; Pieruzzi, F.; Spada, M.; Bersano, A.; Econimo, L.; Lanzillo, C.; Rubino, M.; Mignani, R.; et al. Effect of Migalastat on cArdiac InvOlvement in FabRry DiseAse: MAIORA study. J. Med. Genet. 2023, 60, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Kizhner, T.; Azulay, Y.; Hainrichson, M.; Tekoah, Y.; Arvatz, G.; Shulman, A.; Ruderfer, I.; Aviezer, D.; Shaaltiel, Y. Characterization of a chemically modified plant cell culture expressed human alpha-Galactosidase-A enzyme for treatment of Fabry disease. Mol. Genet. Metab. 2015, 114, 259–267. [Google Scholar] [CrossRef]
- Wallace, E.L.; Goker-Alpan, O.; Wilcox, W.R.; Holida, M.; Bernat, J.; Longo, N.; Linhart, A.; Hughes, D.A.; Hopkin, R.J.; Tøndel, C.; et al. Head-to-head trial of pegunigalsidase alfa versus agalsidase beta in patients with Fabry disease and deteriorating renal function: Results from the 2-year randomised phase III BALANCE study. J. Med. Genet. 2024, 61, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Guérard, N.; Oder, D.; Nordbeck, P.; Zwingelstein, C.; Morand, O.; Welford, R.W.D.; Dingemanse, J.; Wanner, C. Lucerastat, an Iminosugar for Substrate Reduction Therapy: Tolerability, Pharmacodynamics, and Pharmacokinetics in Patients With Fabry Disease on Enzyme Replacement. Clin. Pharmacol. Ther. 2018, 103, 703–711. [Google Scholar] [CrossRef]
- Deegan, P.B.; Goker-Alpan, O.; Geberhiwot, T.; Hopkin, R.J.; Lukina, E.; Tylki-Szymanska, A.; Zaher, A.; Sensinger, C.; Gaemers, S.J.M.; Modur, V.; et al. Venglustat, an orally administered glucosylceramide synthase inhibitor: Assessment over 3 years in adult males with classic Fabry disease in an open-label phase 2 study and its extension study. Mol. Genet. Metab. 2023, 138, 106963. [Google Scholar] [CrossRef] [PubMed]
- Jeyakumar, J.M.; Kia, A.; Tam, L.C.S.; McIntosh, J.; Spiewak, J.; Mills, K.; Heywood, W.; Chisari, E.; Castaldo, N.; Verhoef, D.; et al. Preclinical evaluation of FLT190, a liver-directed AAV gene therapy for Fabry disease. Gene Ther. 2023, 30, 487–502. [Google Scholar] [CrossRef]
- Yasuda, M.; Huston, M.W.; Pagant, S.; Gan, L.; St Martin, S.; Sproul, S.; Richards, D.; Ballaron, S.; Hettini, K.; Ledeboer, A.; et al. AAV2/6 gene therapy in a murine model of Fabry disease results in supraphysiological enzyme activity and effective substrate reduction. Mol. Ther. Methods Clin. Dev. 2020, 18, 607–619. [Google Scholar] [CrossRef]
- DeRosa, F.; Smith, L.; Shen, Y.; Huang, Y.; Pan, J.; Xie, H.; Yahalom, B.; Heartlein, M.W. Improved Efficacy in a Fabry Disease Model Using a Systemic mRNA Liver Depot System as Compared to Enzyme Replacement Therapy. Mol. Ther. 2019, 27, 878–889. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy (Category) | oHCM * | Non-oHCM | Notes |
|---|---|---|---|
| Non-vasodilating Beta-Blockers | First-line therapy for symptom relief and gradient reduction | Used for angina or arrhythmias | |
| Calcium Channel Blockers (non-DHP **) | Commonly used as alternative to beta-blockers. Avoid in case of severe LVOTO # | Used for diastolic dysfunction and rate control | Use verapamil or diltiazem; avoid dihydropyridines in oHCM * |
| Disopyramide | Used as add-on therapy for drug-refractory obstructive symptoms | Not used. | Positive effect as rhythm control strategy in AF ##; anticholinergic effects limit use. QT interval monitoring is required. |
| Myosin Inhibitor—Mavacamten and Aficamten | Second-line for symptomatic oHCM * | Not approved | Requires LVEF § monitoring. Excellent response and safety as stand-alone therapy |
| Ranolazine | Not routinely used—off-label in selected cases. | Reduce symptoms linked to microvascular angina | RESTYLE-HCM trial suggests possible benefit on diastolic function. Antiarrhythmic effect is beneficial |
| Valsartan | Use with caution | Investigational—potential disease-modifying role | VANISH trial: slowed progression in early-stage sarcomeric HCM |
| Septal Myectomy | First line for drug-refractory LVOTO # or LVOTO # obstruction primarily caused by mitral valve alterations | Not applicable | Excellent outcomes in high-volume centers |
| Alcohol Septal Ablation | Yes—alternative to surgery for selected patients | Not applicable | Minimally invasive; risk of AV §§ block |
| Heart Transplantation | For end-stage, drug-refractory cases | Considered—for restrictive/dilated progression | Early candidacy is key |
| Amyloid Type | Therapy | Mechanism/Class | Status and Key Clinical Evidence |
|---|---|---|---|
| A-TTR * | Tafamidis | TTR ** stabilizer | Approved for hereditary and wild-type forms. ATTR-ACT trial: reduced mortality and hospitalization. |
| Acoramidis | TTR ** stabilizer | Approved 2024. ATTRibute-CM trial showed functional and biomarker benefit. | |
| Patisiran | siRNA # TTR ** silencer | Approved for hATTR-PN ##; APOLLO-B showed preserved functional capacity. | |
| Vutrisiran | siRNA # TTR ** silencer | Approved 2024 for ATTR-CM §. HELIOS-B showed reduced CV §§ events. | |
| Inotersen | ASO // TTR ** silencer | Approved for hATTR-PN ##. | |
| Eplontersen | ASO // TTR ** silencer | Approved for hATTR-PN #. CARDIO-TTRansform trial for isolated cardiac involvement is ongoing. | |
| NTLA-2001 | CRISPR-Cas9 *** gene editing | Investigational. Phase I showed 90% TTR ** knockdown. | |
| AL ^ | Daratumumab + CyBorD ### | Anti-CD38 + chemotherapy | Approved. ANDROMEDA trial showed deep hematologic and organ responses. |
| ASCT §§§ + Melphalan | High-dose chemo + stem cell | Standard in eligible patients. Durable remissions. | |
| CAEL-101 | Anti-amyloid monoclonal antibody | Investigational. Aims to clear cardiac AL ^ deposits. |
| Therapy | Mechanism/Class | Target | Status and Key Clinical Evidence |
|---|---|---|---|
| Agalsidase beta (Fabrazyme®) | Enzyme replacement | Systemic | Approved. Reduces Gb3 *; long-term studies show stabilization of renal and cardiac function. |
| Agalsidase alfa (Replagal®) | Enzyme replacement | Systemic | Approved in EU **/Canada. Stabilizes LV *** mass and kidney function. |
| Pegunigalsidase alfa (Elfabrio®) | Enzyme replacement (PEGylated) | Systemic | Approved in 2023. Longer half-life and reduced immunogenicity. |
| Migalastat (Galafold®) | Pharmacologic chaperone | Systemic | Approved for amenable mutations. Oral route. |
| Venglustat | Substrate reduction therapy | Systemic | Investigational. Reduces plasma Gb3 *; ongoing trials. |
| Lucerastat | Substrate reduction therapy | Systemic | Investigational. Reduced Gb3 *; failed pain endpoint. |
| ST-920 | Gene therapy (liver-directed) | Systemic | Investigational. Sustained enzyme activity post-single dose. |
| 4D-310 | Gene therapy (cardiac-directed) | Cardiac | Investigational. Early studies show effective cardiac gene uptake. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfarano, M.; Ciccarelli, F.; Marchionni, G.; Ballatore, F.; Costantino, J.; Lattanzio, A.; Pecci, G.; Stavagna, S.; Iannelli, L.; Galardo, G.; et al. Hypertrophic Cardiomyopathy and Phenocopies: New Therapies for Old Diseases—Current Evidence and Future Perspectives. J. Clin. Med. 2025, 14, 4228. https://doi.org/10.3390/jcm14124228
Alfarano M, Ciccarelli F, Marchionni G, Ballatore F, Costantino J, Lattanzio A, Pecci G, Stavagna S, Iannelli L, Galardo G, et al. Hypertrophic Cardiomyopathy and Phenocopies: New Therapies for Old Diseases—Current Evidence and Future Perspectives. Journal of Clinical Medicine. 2025; 14(12):4228. https://doi.org/10.3390/jcm14124228
Chicago/Turabian StyleAlfarano, Maria, Federico Ciccarelli, Giulia Marchionni, Federico Ballatore, Jacopo Costantino, Antonio Lattanzio, Giulia Pecci, Silvia Stavagna, Leonardo Iannelli, Gioacchino Galardo, and et al. 2025. "Hypertrophic Cardiomyopathy and Phenocopies: New Therapies for Old Diseases—Current Evidence and Future Perspectives" Journal of Clinical Medicine 14, no. 12: 4228. https://doi.org/10.3390/jcm14124228
APA StyleAlfarano, M., Ciccarelli, F., Marchionni, G., Ballatore, F., Costantino, J., Lattanzio, A., Pecci, G., Stavagna, S., Iannelli, L., Galardo, G., Lavalle, C., Miraldi, F., Vizza, C. D., & Chimenti, C. (2025). Hypertrophic Cardiomyopathy and Phenocopies: New Therapies for Old Diseases—Current Evidence and Future Perspectives. Journal of Clinical Medicine, 14(12), 4228. https://doi.org/10.3390/jcm14124228